3.1. Intrinsic Point Defects in Pristine MgO
Pristine crystalline MgO possesses a typical rock-salt structure (NaCl type) with a space group of
(No. 225), as shown in
Figure 1. Given the relative ionic radius difference between O
2− and Mg
2+, respectively, 1.21 Å and 0.86 Å [
49], the structure can be described as the cations occupying the octahedral coordination site within cubic close-packed anion arrays. Ideally, both Mg
2+ and O
2− ions are sitting in a perfect octahedral coordination environment. Each octahedron is connected by edge-sharing and corner-sharing with other octahedra of the same ion species. The bond length after bulk optimisation was calculated to be 2.105 Å.
For undoped MgO, the intrinsic defects considered include Schottky defect and Frenkel defects, in which four basic point defects are involved. Using Kröger–Vink notation, intrinsic point defects include Mg vacancy
, Mg interstitial
, O vacancy
, and O interstitial
. In the Kröger–Vink notations, V and i indicate vacancy and interstitial sites, respectively. The superscript forward slash (/) and dot (
indicate a negative and positive effective charge possessed by the point defect, respectively. For example,
means a Mg interstitial point defect with two positive effective charges.
indicates a substitutional Nd (on Mg site) point defect with one positive effective charge. A superscript x means the defect has no effective charge. Schottky and Frenkel defects are expressed as
The calculated intrinsic defect formation energies per point defect are shown in
Table 4. The current results are in reasonable agreement with experimental and other simulations [
16,
50,
51,
52,
53]. The calculated lattice constant and Schottky energy are 4.210 Å and 6.22 eV, respectively, which agree well the experimental results of 4.207 Å [
50] and 5–7 eV [
53]. Here, Schottky and Frenkel defect energies are calculated using completely isolated point defects. Comparatively, the Schottky defect, which consists of one
and one
requires less formation energy. Thus, it is considered more favourable. A more favourable Schottky intrinsic defect indicates that when a vacancy is formed in pristine MgO another oppositely charged vacancy will be found with more probability for electrostatic compensation. As intrinsic vacancies are preferred, further speculation logically follows: substitution defects may be preferred in doped MgO. Within the cubic, close-packed anion array in the rock-salt structure, two types of interstitial sites are available: octahedral and tetrahedral. Classically, the ratio of ion size largely impacts which site is to be occupied. So, any dopant with a size slightly larger than Mg will be more likely to occupy the octahedral site and become a substitutional dopant on an Mg site. Fundamentally, which sites dopants will occupy depends on the characteristics of the introduced dopants, such as the effective charge or atomic size and the structure of the host material.
3.2. Point Defects and Defect Complexes in Neodymium Doped Magnesium
From the perspective of optical applications, the main purpose of doping active ions into a host material is, on the one hand, to take advantage of the unique physical properties of the host, such as thermal conductivity and intrinsic transparency; on the other hand, it is to take advantage of the crystal field of the host. The coordination environment (such as coordination number and coordination polyhedron) plays a significant role in doped optical materials as it had been observed and studied in crystal field theory [
54]. When it comes to optical centres of rare earth ions, it is known that their electronic structures are less perturbed by the structure of the host material. The 4f orbitals are often unfilled, and they are well sheltered by the outer 5s5p orbitals according to the Aufbau principle. However, the optical properties of the doped system, or the absorption and emission of dopants are still impacted by the local atomic structure. The nature of doping means that the induced distortions in the host should not be ignored: that is, it is necessary to focus on the point defect configurations. In this section, point defects and defect complexes induced by Nd are investigated, and coordination distortions are revealed. By introducing Nd, two possible types of point defects are present,
and
, and their defect formation energies were calculated to be −19.53 eV and −11.37 eV, respectively. These are the two forms in which Nd can be incorporated into MgO.
Two critical questions require consideration: From the point of energy, firstly, which is the more favourable way for Nd to be doped into MgO:
or
? Secondly, which is the more favoured defect that exists as the negative charge compensator for the Nd dopants:
or
? To answer these questions, 22 quasi-chemical reactions were formulated under conservation and electroneutrality rules and are listed in
Appendix A. The enthalpy per rare earth doping of the reaction is considered a benchmark for quantitative evaluations of the probability of obtaining certain defect configurations practically. For example, in reaction (8),
, where
is the defect formation energy of point defect,
is the lattice energy of crystalline oxide. Point defects are isolated from each other at this point in the analysis. The defect reaction with the lowest enthalpy was found to involve three-point defects, two Nd substitutions on Mg sites, and one Mg vacancy as a charge compensator, and the enthalpy was calculated to be 3.79 eV per Nd doping. Another possible charge compensator for
is
:
The enthalpies shown are normalised to per Nd atom doping. The magnesium vacancy is the favoured compensator for positively charged defects instead of oxygen interstitial since its enthalpy is lower, as shown in reactions (8) and (9). On the other hand, in the rock-salt structure of MgO, the available tetrahedral interstitial sites are the sites surrounded by four oxygen anions. So, when oxygen is placed on one of these interstitial sites, immense relaxation of the lattice must then be induced because of the large electrostatic repulsions among local anions; this is undoubtedly less favoured for a negatively charged compensator to exist naturally.
The result also indicates that Nd favours substitution on the Mg site instead of an interstitial site in the MgO host. The reactions with the lowest enthalpy changes where one and two Nd
i are involved were found to be
It is quite straightforward to see that the magnitude of the effective charge becomes higher when Ndi appears. The more Nd dopants that exist in the form of interstitials, the more magnesium vacancies are needed for charge compensation, which has a positive value of formation energy. It undoubtedly increases the enthalpy of the solution. Also, from the point of view of ionic size and effective charge, an Nd interstitial will result in more distortions to the host structure.
When point defects approach close to each other, the defect complexes can be formed. The defect complex must be considered to further determine the more favoured way to accommodate Nd dopants and the more favoured charge compensator. The mutual interactions between the point defects in a complex may influence the enthalpies of defect reactions. The way in which different configurations of defect complexes were tested involved using the Mg atom at the origin (0, 0, 0); either substitutional or interstitial point defects were placed on the available sites around it within a volume of 74.62
, which is the volume of one MgO unit cell. In the current context, the association energy is defined as the energy difference between the defect formation energy of a complex and the sum of the defect formation energies of those isolated point defects:
is the defect energy of the defect complex, is the defect energy of an isolated point defect, and is the total number of point defects in the defect complex. The clustering of point defects is called a complex normally when , but the formulation of for a pair of point defects is the same. A positive value of association energy is, as a result of this, considered unfavoured as it will increase the enthalpy when being taken into the quasi-chemical reaction. A negative value of association energy is considered to be favoured as it will decrease the enthalpy. It is determined mainly by the relative positions of point defects.
Defect pairs of two-point defects with oppositely effective charges, where Nd dopants are involved, were first considered. The lowest association energy (the highest absolute value) in each complex is listed in
Table 5.
It was found in the simulations that when
was initially placed at (
,
,
), close to the (0, 0, 0)
, the Nd atom moved into the vacancy position after relaxation, with a formation energy the same as
(−11.37 eV). If Nd favours an interstitial site in MgO, the association energy of the (
+
) pair is expected to be a positive value. The calculated negative value of the association energy of (
+
) shows that the favoured way to accommodate Nd in MgO is the substitution on the Mg site and not an interstitial site. Taking the association energies of the four defect pairs of
Table 5 into the quasi-chemical reactions, one of the lowest enthalpies was found to be
It is the same set of reaction as shown in reaction (8). The information that can be extracted, so far, according to those results is: There are great probabilities that dopant Nd exists in the form of , and that is more favoured as a charge compensator rather than .
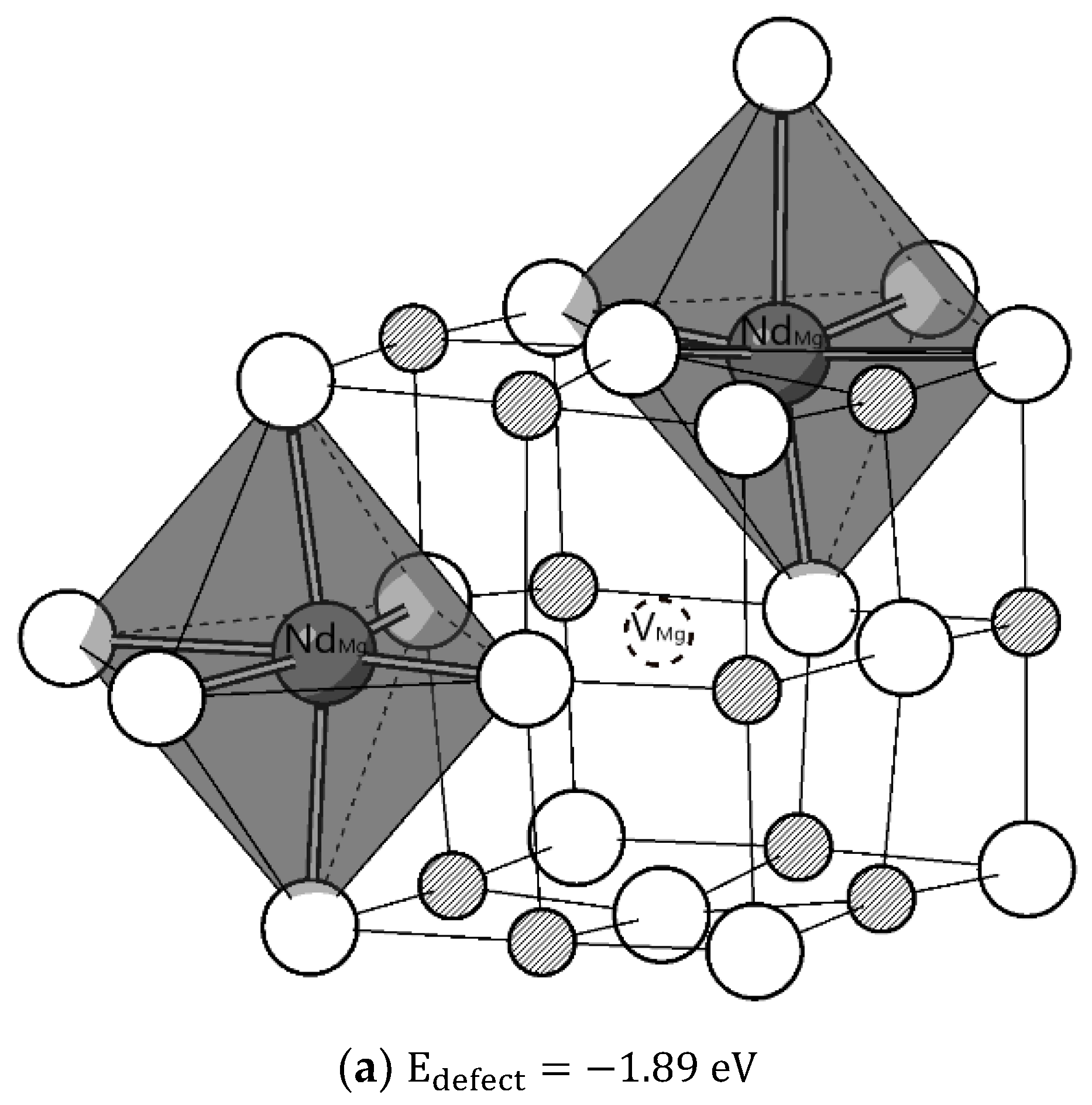
To further investigate the structures of defects in Nd:MgO, the defect complex containing three individual point defects
was considered. A total of 35 different configurations were tested to find the structure of the minimised formation energy. Initially, a magnesium vacancy was fixed at the origin, and configurations were tested by putting the other two Nd substitutions in other available Mg sites. It was found that associations between these three-point defects can further reduce the normalised reaction enthalpy to 2.72 eV, a reduction of 28% when all point defects are isolated as in reaction (8) and the relaxed structure of this defect complex is shown in
Figure 2a.
Figure 2a shows that both Nd substitutional defects remain in octahedral coordination with distortions to the polyhedron in bond angle and bond length.
Figure 2b shows the found configuration of the highest defect energy. The interatomic distances of Nd and the distances between point defects are listed in
Table 6.
Compared to undoped MgO, where the optimised Mg–O length is 2.105 Å, the mean Nd–O lengths are around 2.255 Å and 2.215 Å for configurations (a) and (b), respectively. In configuration (a), changes in Mg–O bond length can be due to the changes in the electrostatic interaction between the substitutional defect and the surrounding oxygen ions. Although one Nd substitutional defect possesses a positive effective charge compared to the original Mg ion, thus polarizing the neighbouring oxide ions, which could result in a shorter bond length, Nd has a larger ionic size than Mg. When placed at the Mg site, the bond length increases after relaxation to accommodate it and this effect dominates. Two Nd dopants in configuration (a) are closer to the Mg vacancy compared to configuration (b), and they experience more powerful electrostatic interactions with Mg vacancy, exerting greater distortions on octahedral coordination. The interdopant distance Nd–Nd in configuration (a) is 4.80 Å, which is shorter than 6.12 Å in configuration (b). Comparisons of these two configurations show that it is energetically favourable for Nd dopants to aggregate in Nd:MgO.
Above all, it is more probable that Nd dopants exist as substitution defects on the Mg site with Mg vacancy as the charge compensator. A defect complex is more favoured than isolated point defects. In solely Nd-doped MgO, one may expect the existence of a Mg vacancy near substitutional Nd dopants.
3.3. Influences of Lithium on Energetics and Defect Structures of Neodymium-Doped Magnesium Oxide
According to the experimental results obtained by Dorel [
12], ionic exchange between Nd and Mg was not observed when MgO was the host. So, Nd dopants may not be successfully doped into the MgO host, or the quantity doped may have been too low to remain detectable. The difficulty of doping rare earth elements into MgO was also stressed by Sannamyan [
13]. Li co-doping has been experimentally proven to improve the crystallinity and optical performance in rare-earth-doped MgO [
6,
9,
10]. It is important to ask the following questions: In which way is Li making an impact? Does Li make it easier for Nd to enter the MgO host? The standing points to view the questions here are structure and energy. A similar procedure as in Nd:MgO was conducted: defect and association energies were calculated for Nd, Li:MgO. Via Li co-doping, two extra possible point defects are introduced—
and
—and the defect energies were calculated to be −1.23 eV and 15.89 eV, respectively. The quasi-chemical reactions for these two-point defects in Li:MgO are as follows:
The most favourable way to accommodate Li in MgO is and compensating for each other, as shown in reaction (15). By comparing reactions (16) and (18), is relatively more favourable than in MgO.
Applying the point defects of Nd and Li to the quasi-chemical reactions (56 possible reactions are shown in
Appendix A), the one with the lowest enthalpy was found to contain two Nd substitutional and two Li substitutional point defects on Mg sites:
The defect complexes considered and their respective calculated association energies are listed in
Table 7.
Figure 3 shows the data points of simulation results regarding the relations between association energy and effective charge (
EC) or the number of point defects (
N) in defect complexes. Defect complexes containing more point defects with electroneutral effective charges are more favourable. The range of association energy is broadened by including more point defects in the complex. The ranges for a two-, three-, and four-point defect complex are 0.39 eV, 0.93 eV, and 1.60 eV. When more point defects join the complex, more configurations become possible, increasing the range. It shows that the range of association energy strongly depends on the relative atomic arrangements of included point defects. That is why it is necessary to conduct a systematic investigation to determine the most favourable defect structure. Depending on the specific atomic arrangement, associations can be favoured or unfavoured. This dependence on positions only occurs when the effective charge possessed by the defect complex is non-zero (−1 or +1 in the current case). The associations are always favoured when
EC = 0, in both
N = 2 and
N = 4. With an increased
N, it is more promising to find lower association energy. So far, the most potentially favourable defect complex in Nd, Li: MgO was found to be a
with zero effective charges. The relative positions of the point defects included do not change the sign of association energy, but the range of the association energy of four-point-defect complex is the largest compared to the complex with two- or three-point defects. The quasi-chemical reaction with the lowest enthalpy is
Remember that in Nd:MgO the Mg vacancy was found to be the favourable charge compensator for the Nd-substitution point defect. With Li co-doping, Li substitution point defects play the role of the charge compensator. Compared to the Mg vacancy with two negative effective charges and a vacant site within the crystalline lattice, the substitution by Li possesses one negative effective charge without leaving any charged vacant space, so causes less structural perturbation to the crystal structure of MgO compared to . This result shines light on why Li co-doped materials have better crystallinity.
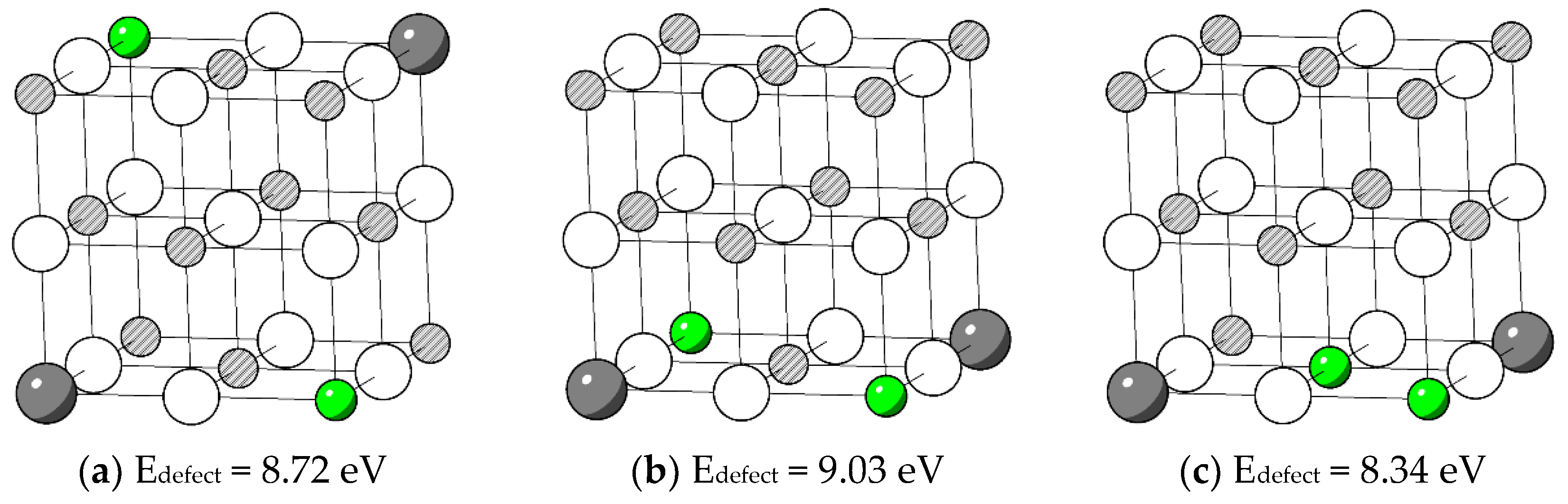
Even though the MgO in the rock-salt structure is of high symmetry, point defects break the perfect local symmetry and result in many possible configurations. One Nd substitution defect was first fixed at the origin position (0, 0, 0). The number of possible configurations of the was then , including some double counts. Thus, to put the other three cation substitutions on other available neighbouring Mg sites within the volume of one unit cell, 280 possible configurations were tested to find the favourable defect structure of .
The relaxed structure of the configuration with the lowest defect energy E
defect is shown in
Figure 4a.
Figure 4b shows the configuration that has the highest defect energy found. Interatomic distances of the two configurations are listed in
Table 8.
The configuration with the lower defect energy is more disordered in bond length bond angle. Li favours a position close to Nd. Compared to the enthalpy of 2.72 eV obtained in reaction (14) without the co-dopant, in reaction (21), incorporating Li reduces the enthalpy by 28%. This result indicates that Nd can be doped into MgO more easily with Li as a co-dopant. Structurally, firstly, the Li has a smaller ion size than Mg, which is beneficial for the additional doping of large rare earth elements. Secondly, the relaxation of the O atom, which is surrounded by four substitutional point defects, causes large displacements in its position, as shown in
Figure 5. Two Nd substitutional point defects are located at two sides of the oxygen atom, making it an equilibrium Coulombic attraction condition; this displacement of the O atom is mainly attributed to the two Li substitutional point defects, because of a repulsive electrostatic interaction. From the point of the doping objective, the displacement of oxygen atom makes more room for the accommodations of two Nd-substitution dopants.
The following question may be raised: What is the correlation between the defect energy of a defect complex and its configuration—the exact atomic arrangements? Remember that the association energy in the current context is defined as from Equation (12) (a negative value of is favoured as it means the has a lower defect energy than and it will decrease the reaction enthalpy). One might assume that the closer those point defects approach each other, the greater the association (the higher absolute value of association energy), the lower the defect energy they will have. But, the relationship between the configuration of defect complex and defect formation energy is not that simple, even in the MgO host, which has a relatively simple crystal structure.
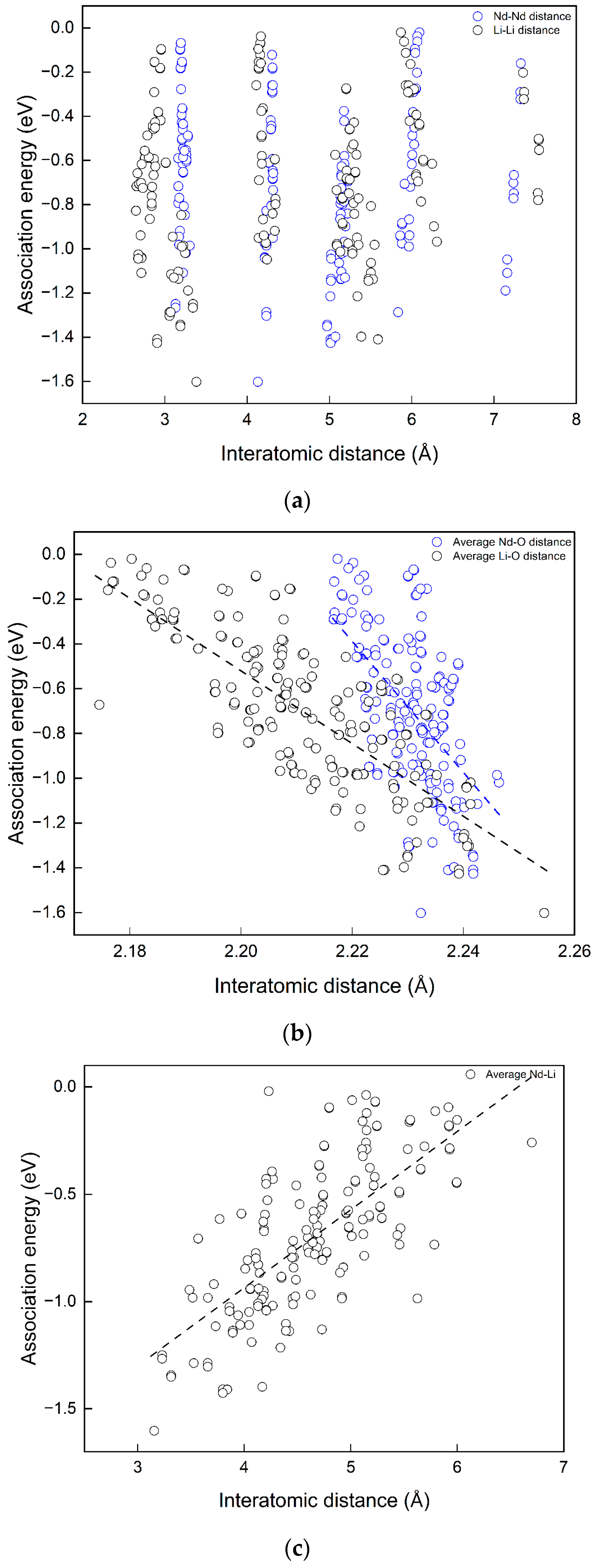
As shown in
Figure 6, in a single MgO unit cell, the arrangement for four substitutional point defects to stay as distant from each other as possible is to put them on four diagonal corners of the cubic as in (a). Nevertheless, this configuration is more favoured than the condition where all substitutions are located on four face diagonal sites, as in (b). Start from (b), moving one Li closer to the other three substitutional point defects as shown in (c), the defect energy decreases. From selected examples, it is not clear which interatomic distance (Nd–O, Nd–Nd, or Nd–Li) in the defect complexes is more significant for their defect energies (or association energies). To further investigate the correlations, interatomic distances were extracted from relaxed structures of defect complexes. The interatomic distances considered include Nd–Nd, Li–Li, average Nd–Li, average Nd–O bond length, and average Li–O distance (the distance between Li and the first nearest six O atoms). The scattered data points of E
association vs. interatomic distance are shown in
Figure 7.
According to
Figure 7a, there is no apparent dependency between E
association and interatomic distances between two Nd dopants or two Li co-dopants in the complexes, since for each considered Nd–Nd and Li–Li distance high or low association energies are possible as in (a). On the contrary, dependencies of E
asscociation on Nd–Li, Nd–O, and Li–O distances are clear in
Figure 7b,c. The trend lines are shown as black dashed lines. Larger average Nd–O and Li–O are favoured for associations in the cluster. A shorter average Nd–Li distance is favoured. The trend line of the average Nd–O bond length has the steepest gradient. The correlations are not simply linear: no single interatomic distance solely determines the magnitude of the association energies of the defect complex. This is the great difficulty in analysing the defect complexes. However, the results above can be used to estimate or compare association energies among defect complexes in different configurations. In other words, one may approximate the more energetically favourable defect structure by comparing average Nd–O, Li–O, or Nd–Li distances.
3.4. Comparisons with Erbium-Doped Magnesium Oxide
According to Sanamyan [
13], besides Nd: MgO, Li was also found to have a direct positive impact on the doping and optical properties of Er:MgO. Er, Li:MgO has a better visible transparency than Nd, Li:MgO. It is meaningful to compare the doping of Er and Nd into MgO from the point of energy. Intuitively, it might be easier to dope Er than Nd, as Er possesses a smaller ionic size. Calculations were performed to investigate Er doped MgO from a theoretical aspect. Because of the unavailability of Er–O Buckingham potential parameters with the same O–O potential parameters in the literature, Er–O Buckingham parameters were derived via relaxed fitting with GULP. The derivation methods of the potential parameters were outlined by Lewis and Gale [
37,
55,
56]. The sum of least squares method was utilized to measure the fitting quality. By fitting to the experimental results [
57,
58,
59], the derived Er–O Buckingham parameters are
A = 1381.518 eV,
= 0.349 Å, and C
6 = 0 eV·Å
6. The lattice energy of Er
2O
3 was calculated to be −134.97 eV, and the defect energy of
and
was calculated to be −14.817 eV and −23.244 eV, respectively. For Er:MgO and Er, Li:MgO, the following quasi-chemical reactions were found to possess the lowest enthalpy:
Furthermore, defects of the complexes of
and
were considered. A total of 35 configurations of
—the same as
—were tested and they were found to have the same lowest-formation-energy configuration. As for
, 22 configurations were selected from the 280 configurations that were tested in the
. The range of the association energies of the 280 tested configurations of
is from −1.6 to 0 eV, as shown in
Figure 7. Based on an energy interval of 0.2 eV, in total, 22 configurations of
were selected, whose association energies fell within the eight ranges from −1.6 to 0 eV. It was found that
has the same favourable configuration as
. The quasi-chemical reactions for Er:MgO and Er, Li:MgO of the lowest enthalpies were
It shows that associations are also favoured in Er:MgO and Er, Li:MgO. And, Li co-doping also lowers (by 30%) the enthalpy for Er doping into MgO, a similar effect as in Nd:MgO. Compared to reaction (14) of Nd:MgO ( = 2.72 eV) and (21) of Nd, Li:MgO ( = 1.96 eV), doping Er into the MgO host requires less energy: 2.26 eV for Er: MgO and 1.59 eV for Er, Li:MgO.
With respect to the coordination octahedra of the rare earth dopants in MgO,
Figure 8 shows the information of the dopant-oxygen bond lengths of two dopant-centred octahedra in each of the doped systems. The two dopants possess the same octahedron in their respective systems. The bond length of Mg–O in the pristine MgO 2.125 Å is indicated by the black dashed line. Comparing the systems with Li and without Li, it is apparent that in Li co-doped systems, the Nd–O and Er–O bond lengths—the maximum, the mean, and the minimum bond lengths—are shorter. Compared to Nd, Er has the shorter bond lengths. Besides the bond lengths, their distorted coordination octahedra also have distorted bond angles. A method to describe the distortions of a polyhedron systematically was proposed by Baur [
60], where three distortion indices (DI) were defined: bond-length distortion, bond-angle distortion, and edge-length distortion (edge of the polyhedron). These distortion indices were originally defined for tetrahedral polyhedra. For octahedral polyhedron, analogous expressions of three distortion indices are [
61]:
where
stands for the bond length between the cation atom
and oxygen atom
,
stands for bond angle,
stands for edge length of octahedral polyhedron, and
indicates the mean value of each quantity. The calculated DI of the dopant-centred octahedra are listed in
Table 9.
With the Baur method, RE:MgO and RE, Li:MgO have similar DIs for bond-length and edge-length distortions. The DIs of bond-angle distortions in RE:MgO are relatively higher than in RE, Li:MgO. This is due to the high effective charges (−2) possessed by the Mg vacancy, causing large repulsions of the O atoms near the Mg vacancy. The difference between each quantity and the respective mean value is in an order of magnitude of −1 (0.1), so the calculated indices are in an order of magnitude of −2 (0.01). So, it is sufficient to keep the third significant figure of the numerical value when comparing degrees of distortions between octahedra. This information on the bond lengths and the distortions described above may be useful for comparative spectroscopic studies of active RE ions in RE:MgO and RE, Li:MgO, where the local symmetries are decisive for the optical properties.
3.5. Incorporating Additional Li Co-Dopants
According to reaction (21), the defect complex discussed was
where the ratio between Nd and Li was 1:1. What will happen when more Li ions are introduced into the system? It is meaningful to consider the excess-Li condition? In the experimental practice, the mole ratio between doped Nd and Li is 1:3 [
6]. Based on the results obtained in the last section (reactions (15) and (21), and the most favourable configuration of
), the defect complex
was considered. Extra
and
were placed around the most favourable configuration of
. In this defect complex, there are two Nd atoms and six Li atoms (Nd:Li = 1:3 or Nd, 3Li:MgO). The excess
point defects were placed at the nearest neigbouring Mg sites to Nd dopants, and the excess
were tested for the eight nearest neighbouring interstitial sites to Nd dopants. The most favourable configuration of
that was found is shown in
Figure 9 (
is normalized to per Nd dopant). The bond length and inter-atomic distances are listed in
Table 10. Note that the average Nd–O bond length is 0.01 Å shorter than that of Nd, Li:MgO. The interdopant distance is 0.17 Å longer than that of Nd, Li:MgO. The quasi-chemical reaction after excess-Li co-doping is shown in (27).
Compared to reaction (21), co-doping excess Li into the MgO host requires extra energy of 1.42 eV per Nd dopant. Though the excess
and
point defects are not needed for the charge compensation of the Nd dopants, they were found to have an impact on the distortion of the octahedral coordination geometry of Nd dopants. The calculated Baur DIs are shown in
Table 11.
Three distortion indices of octahedral coordination geometries of Nd dopants were found to decrease significantly in the excess-Li co-doped condition compared to
Table 9. The reductions in the bond-length, bond-angle, and edge-length distortions are 52.2%, 76.5%, and 81.3%, respectively. The dramatic reductions of the distortion indices may be partially responsible for the experimentally observed increase in optical emission intensity via Li co-doping [
6], considering that a less-distorted coordination geometry for Nd might contribute to more localized Stark levels in the 4f manifolds, which is beneficial to obtain emissions with less bandwidth and increased intensity [
54,
62].















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}