
CO2 Promoting Polymorphic Transformation of Clarithromycin: Polymorph Characterization, Pathway Design, and Mechanism Study


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Preparation of Solvates and Form 0′
2.3. Single Crystals Preparation of Solvates and Form 0′
2.4. Phase Transformation Experiments
2.4.1. Phase Transformation Experiments in Air and Vacuum
2.4.2. Phase Transformation Experiments in CO2 Atmosphere
2.5. Characterization
2.5.1. Single Crystal X-ray Diffraction (SCXRD)
2.5.2. Powder X-ray Diffraction (PXRD)
2.5.3. Thermal Analysis
2.5.4. Hot Stage Microscopy (HSM)
2.5.5. Measurement of the Physical Adsorption Capacity
2.5.6. Measurement of Chemical Adsorption Capacity
2.6. Computational Method
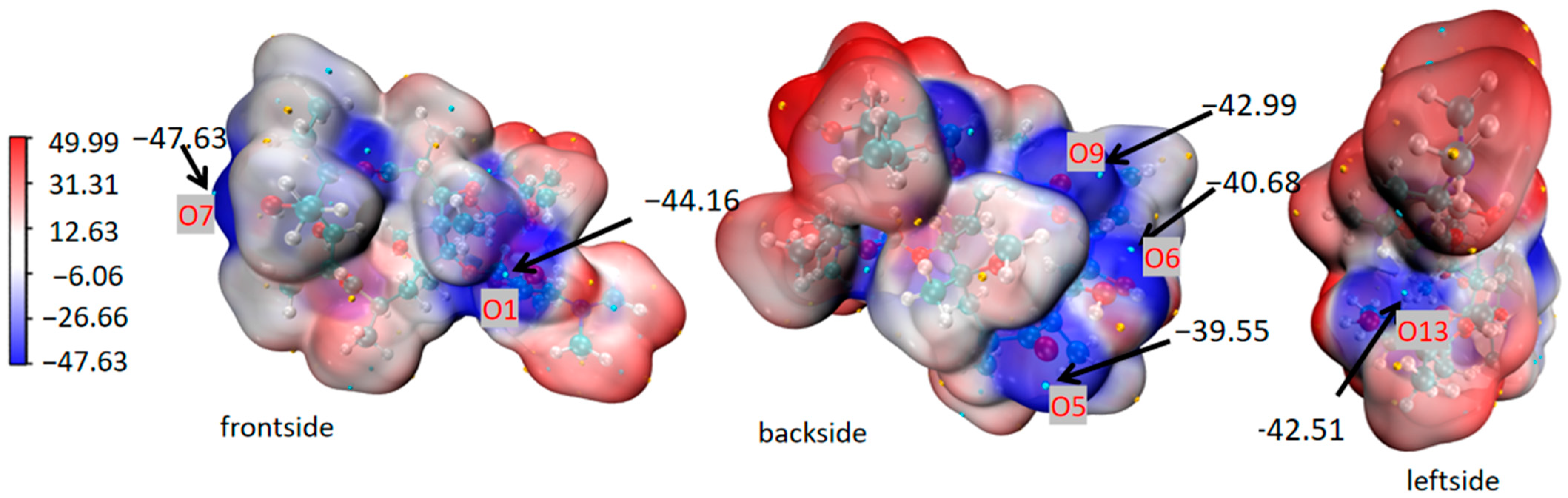
2.6.1. Molecular Electrostatic Potential Surface (MEPS)
2.6.2. Adsorption Energy Calculation
2.6.3. Crystal Habit Prediction
3. Results and Discussion
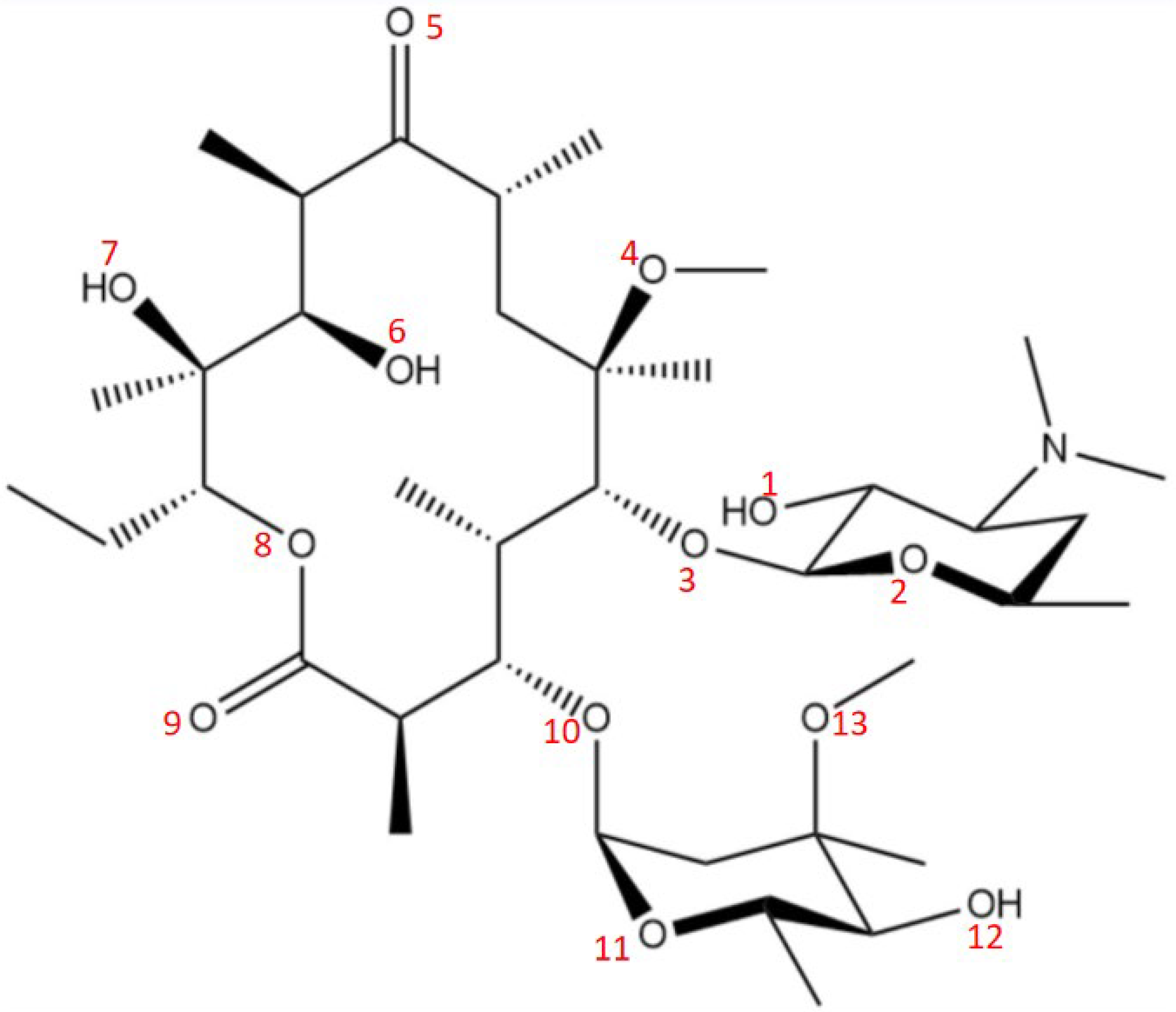
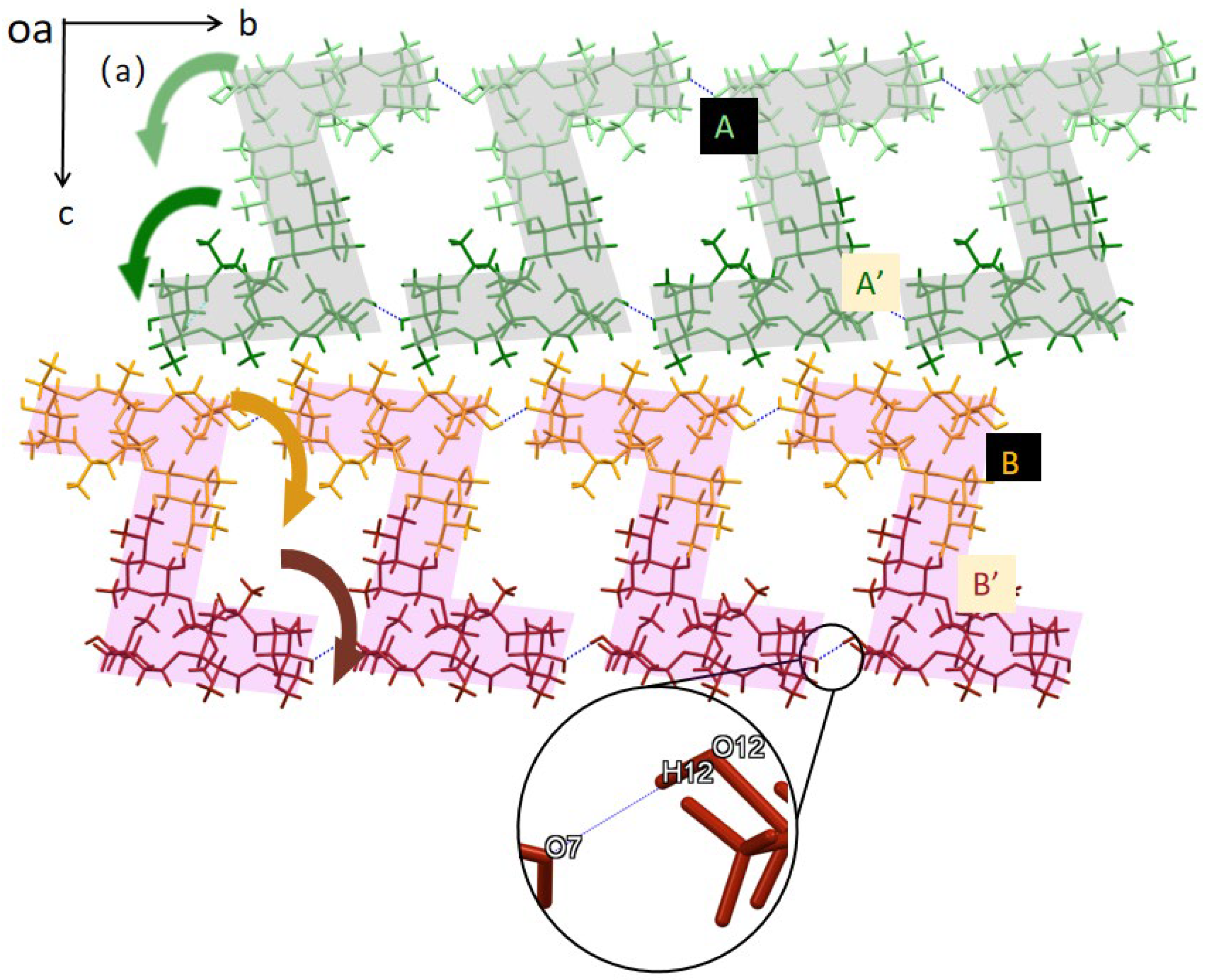
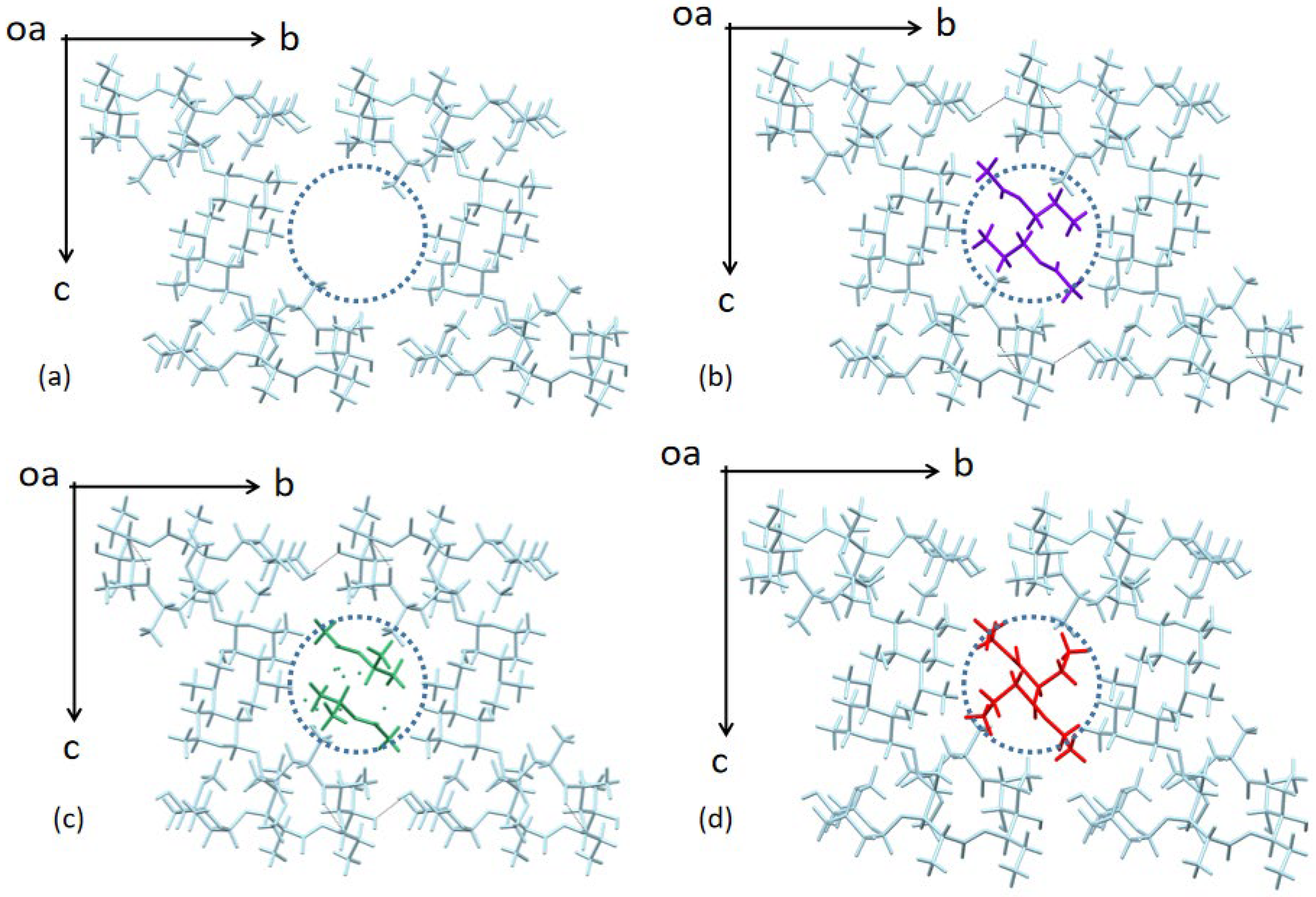
3.1. Crystal Structure Analysis of Form 0′ and Solvates
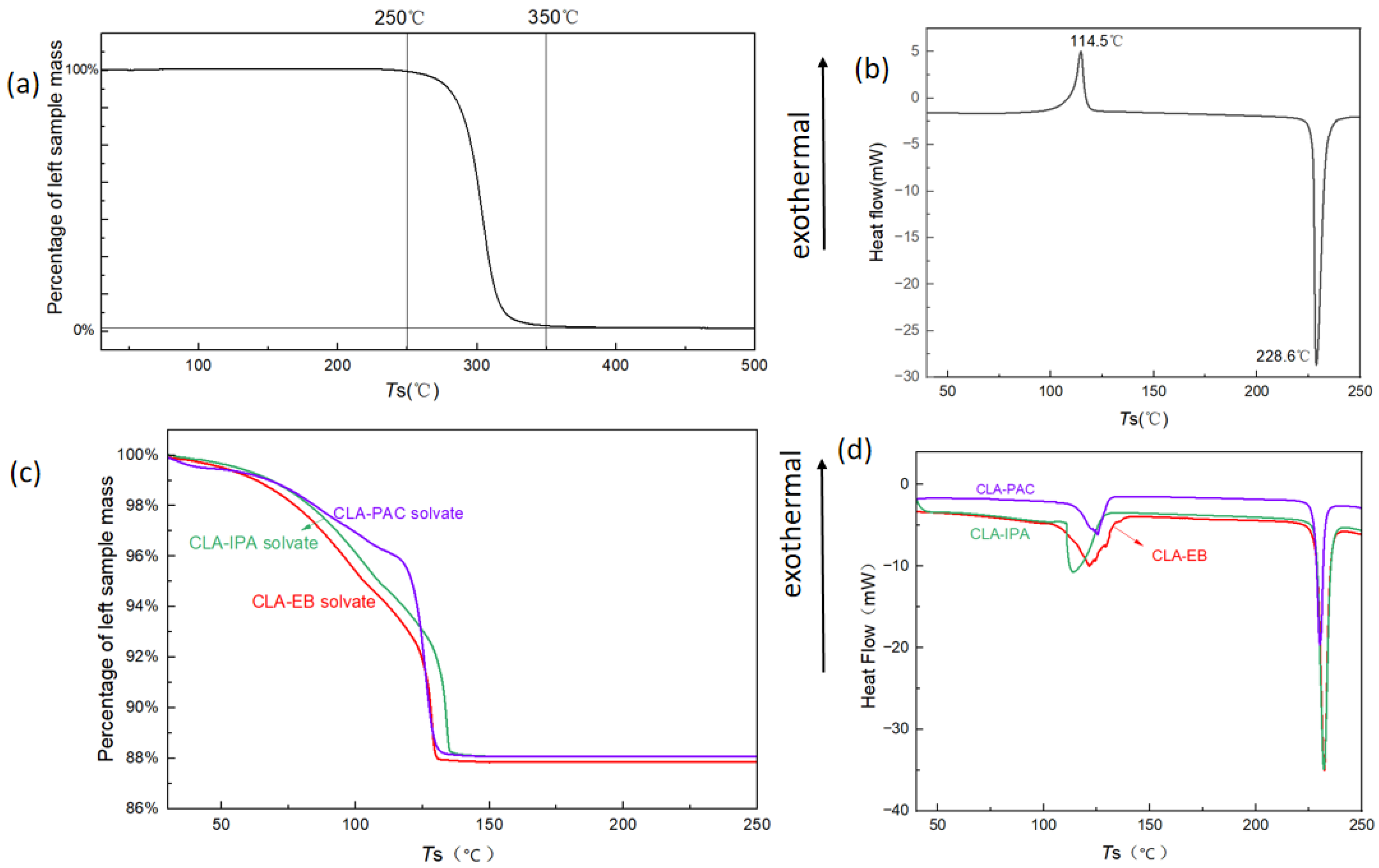
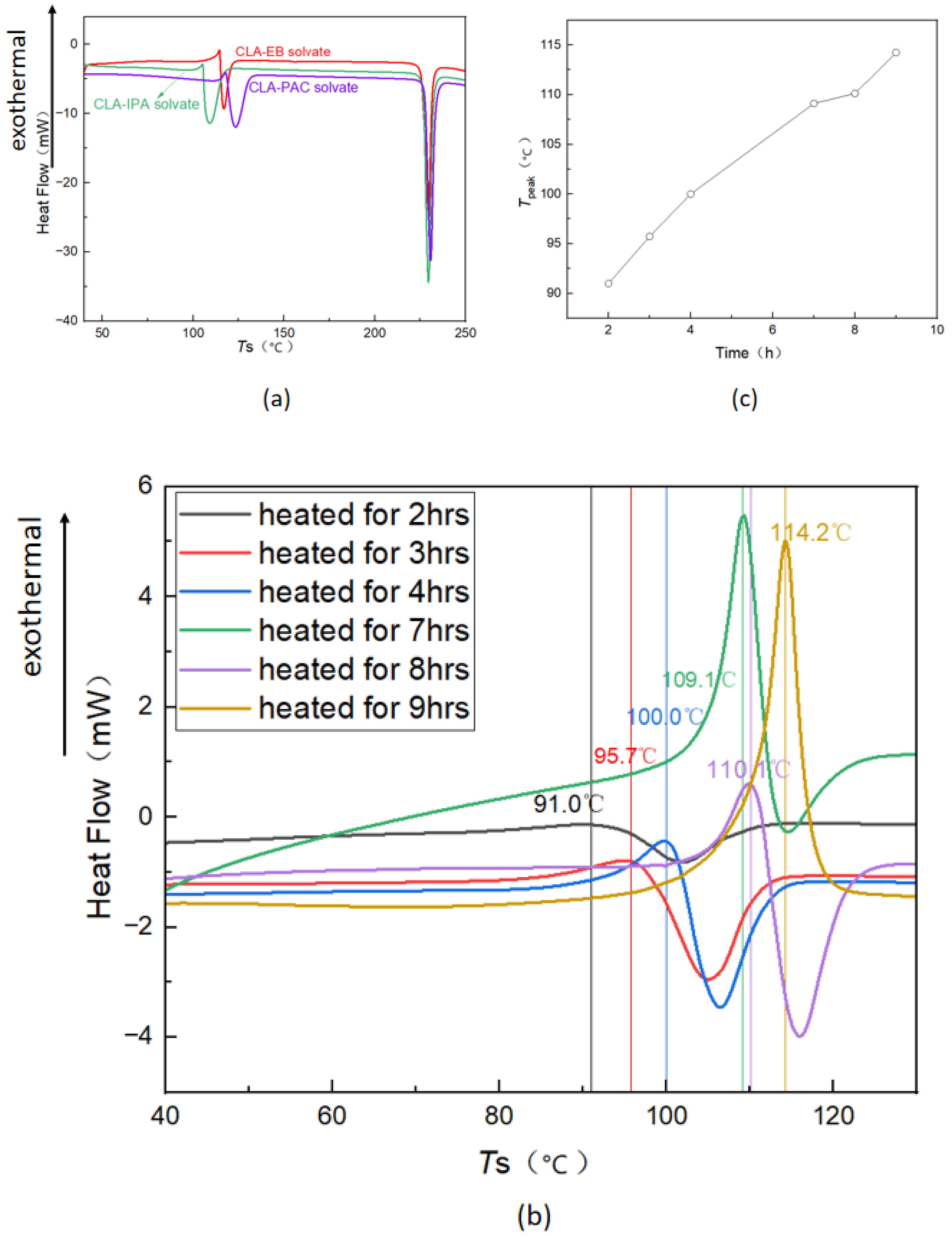
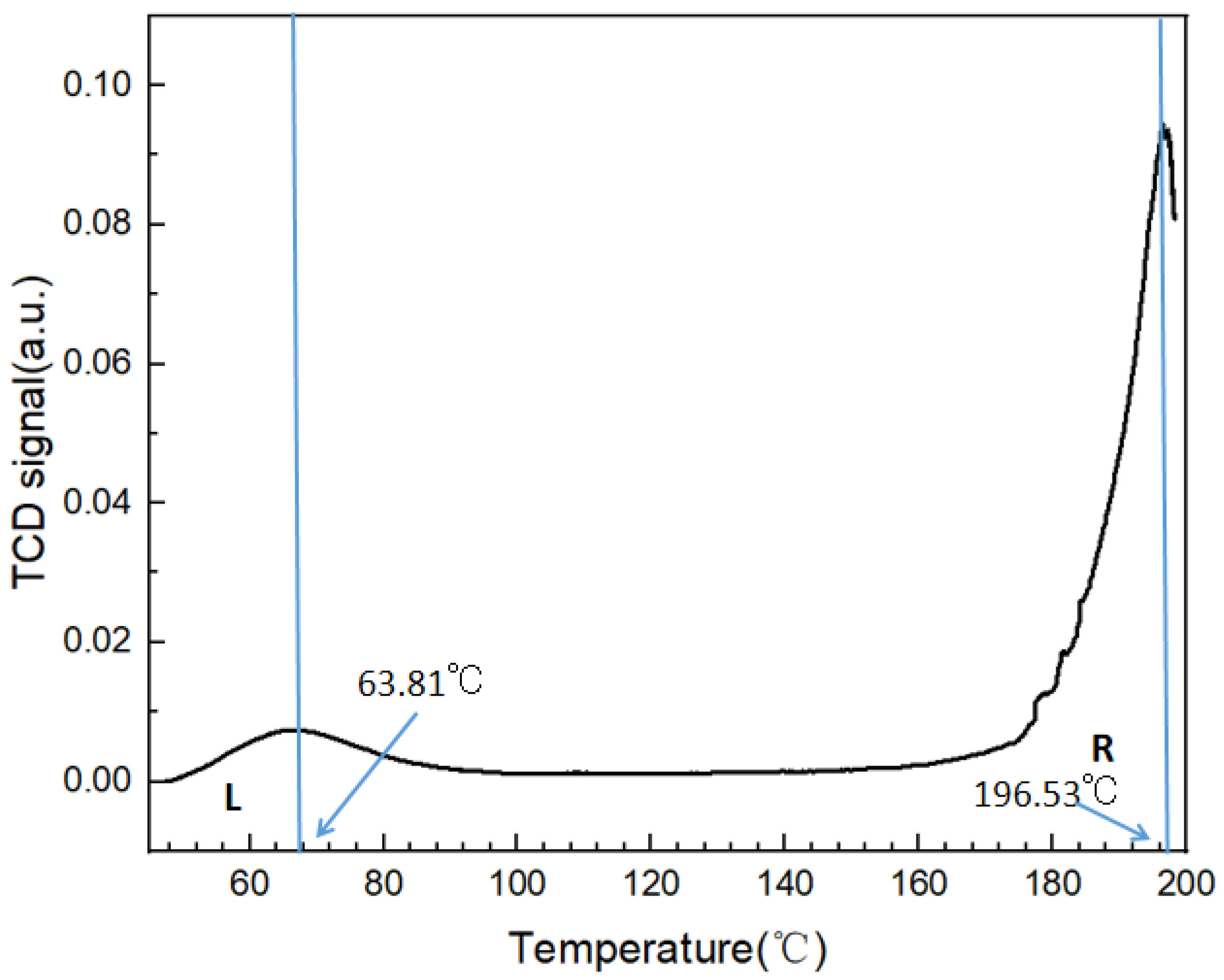
3.2. Thermal Analysis of Form 0′ and Solvates
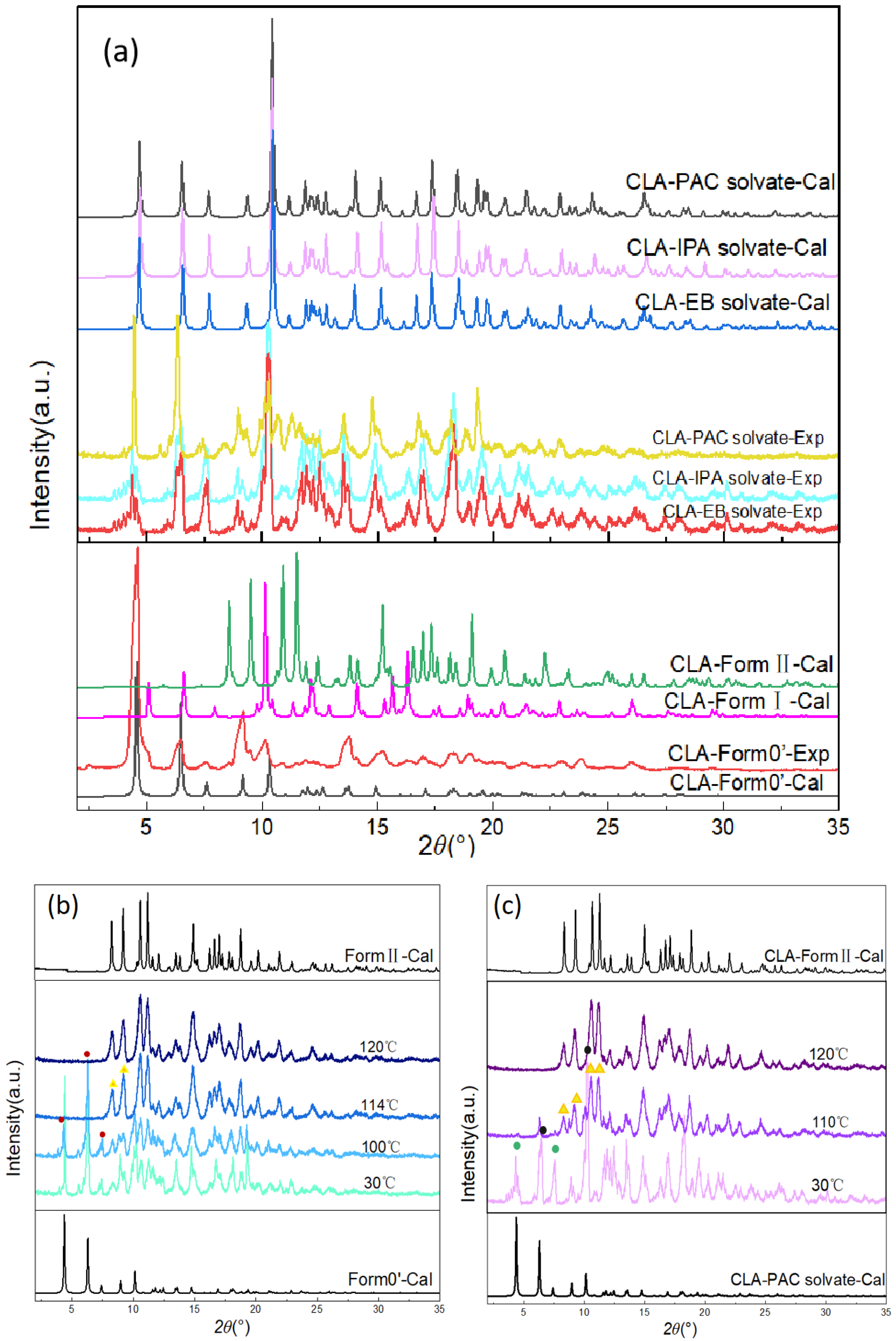
3.3. PXRD Analysis of Form 0′ and Solvates
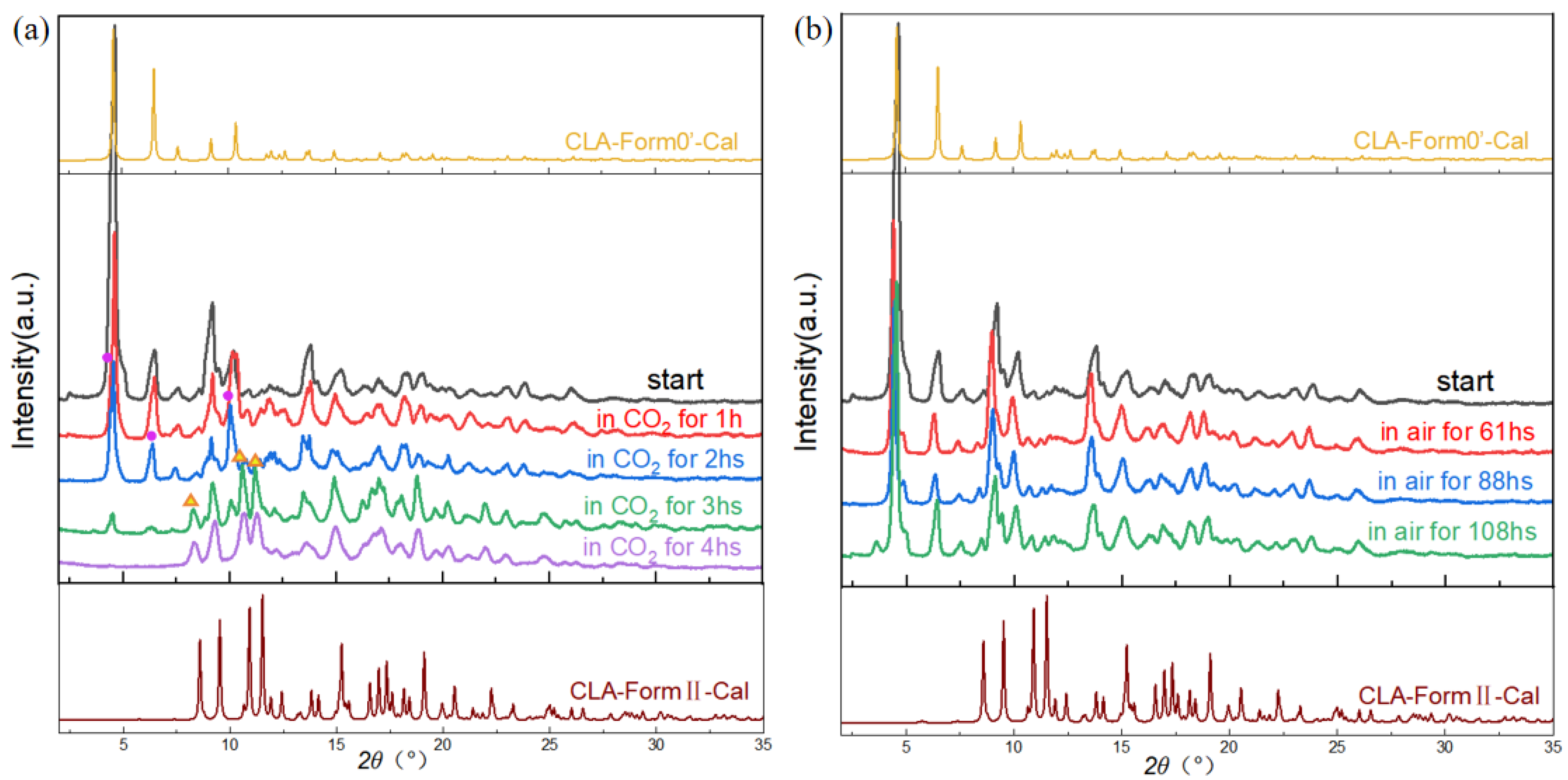
3.4. Phase Transformation of Solvates and Form 0′
3.5. Mechanism of CO2 Promoting Polymorphic Transformation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Du, D.; Shi, Z.P.; Ren, G.B.; Qi, M.H.; Li, Z.; Xu, X.Y. Preparation and characterization of several azoxystrobin channel solvates. J. Mol. Struct. 2019, 1189, 40–50. [Google Scholar] [CrossRef]
- Bērziņš, A.; Trimdale, A.; Kons, A.; Zvanina, D. On the Formation and Desolvation Mechanism of Organic Molecule Solvates: A Structural Study of Methyl Cholate Solvates. Cryst. Growth Des. 2017, 17, 5712–5724. [Google Scholar] [CrossRef]
- Yuan, W.; Yu, C.; Xu, S.; Ni, L.; Xu, W.; Shan, G.; Bao, Y.; Pan, P. Self-evolving materials based on metastable-tostable crystal transition of a polymorphic. Mater. Horiz. 2022, 9, 756–763. [Google Scholar] [CrossRef]
- Jin, M. Mechanical-Stimulation-Triggered and Solvent-Vapor-Induced Reverse Single-Crystal-to-Single-Crystal Phase Transitions with Alterations of the Luminescence Color. J. Am. Chem. Soc. 2018, 140, 2875–2879. [Google Scholar] [CrossRef]
- Hanna, S.L.; Zhang, X.; Otake, K.I.; Drout, R.J.; Li, P.; Islamoglu, T.; Farha, O.K. Guest-Dependent Single-Crystal-to-Single-Crystal Phase Transitions in a Two-Dimensional Uranyl-Based Metal−Organic Framework. Cryst. Growth Des. 2019, 19, 506–512. [Google Scholar] [CrossRef]
- Liao, X.; He, J.; Yu, J. Process analysis of phase transformation of a to b-form crystal of syndiotactic polystyrene investigated in supercritical CO2. Polymer 2005, 46, 5789–5796. [Google Scholar] [CrossRef]
- Ramachandran, J.P.; Antony, A.; Ramakrishnan, R.M.; Wallen, S.L.; Raveendran, P. CO2-solvated liquefaction of polyethylene glycol (PEG): A novel, green process for the preparation of drug-excipient composites at low temperatures. J. CO2 Util. 2022, 59, 101971. [Google Scholar]
- Ramachandran, J.P.; Kottammal, A.P.; Antony, A.; Ramakrishnan, R.M.; Wallen, S.L.; Raveendran, P. Green processing: CO2-induced glassification of sucrose octaacetate and its implications in the spontaneous release of drug from drug-excipient composites. J. CO2 Util. 2021, 47, 101472. [Google Scholar] [CrossRef]
- Hu, D.; Li, W.; Wu, K.; Cui, L.; Xu, Z.; Zhao, L. Utilization of supercritical CO2 for controlling the crystal phase transition and cell morphology of isotactic polybutene-1 foams. J. CO2 Util. 2022, 66, 102265. [Google Scholar] [CrossRef]
- Tian, J.; Thallapally, P.K.; Dalgarno, S.J.; Atwood, J.L. Free Transport of Water and CO2 in Nonporous Hydrophobic Clarithromycin Form II Crystals. J. Am. Chem. Soc. 2009, 131, 13216–13217. [Google Scholar] [CrossRef]
- Sohn, Y.-T.; Rhee, J.-K.; Im, W.-B. Polymorphism of Clarithromycin. Pharmacol. Toxicol. Pharm. 2000, 4, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.H.; Yao, G.W. A New Crystal Structure of Clarithromycin. J. Chem. Crystallogr. 2008, 38, 61–64. [Google Scholar] [CrossRef]
- Ito, M.; Shiba, R.; Watanabe, M.; Iwao, Y.; Itai, S.; Noguchi, S. Phase transitions of antibiotic clarithromycin forms I, IV and new form VII crystals. Int. J. Pharm. 2018, 547, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, H.; Sugawara, Y.; Adachi, T.; Morimoto, S.; Watanabe, Y. Structure of 6-O-methylerythromycin A (clarithromycin). Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1993, 49, 1227–1230. [Google Scholar] [CrossRef]
- Tian, J.; Dalgarno, S.J.; Atwood, J.L. A New Strategy of Transforming Pharmaceutical Crystal Forms. J. Am. Chem. Soc. 2011, 133, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.; Zhang, G.G. Crystallographic characterization of several erythromycin A solvates: The environment of the solvent molecules in the crystal lattice. J. Pharm. Sci. 2007, 96, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Baronsky, J.; Preu, M.; Traeubel, M.; Urbanetz, N.A. Perfusion calorimetry in the characterization of solvates forming isomorphic desolvates. Eur. J. Pharm. Sci. 2011, 44, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Kuncham, S.; Shete, G.; Bansal, A.K. Quantification of clarithromycin polymorphs in presence of tablet excipients. J. Excip. Food Chem. 2000, 5, 65–78. [Google Scholar]
- Miñambres, G.G.; Aiassa, V.; Longhi, M.R.; Chattah, A.K.; Garnero, C. Insights into the ethanol solvate form of clarithromycin. J. Mol. Struct. 2022, 1264, 133170. [Google Scholar] [CrossRef]
- Rajbhar, P.; Sahu, A.K.; Gautam, S.S.; Prasad, R.K.; Singh, V.; Nair, S.K. Formulation and Evaluation of Clarithromycin CoCrystals Tablets Dosage Forms to Enhance the Bioavailability. Pharma Innov. J. 2016, 5, 5–13. [Google Scholar]
- Liu, J.-H.; Riley, D.A. Herstellung von Kristallinen Form II von Clarithromycin. WO1998004574A1, 5 February 1998. [Google Scholar]
- Gildea, R.J.; Bourhis, L.J.; Dolomanov, O.V.; Grosse-Kunstleve, R.W.; Puschmann, H.; Adams, P.D.; Howard, J.A. iotbx.cif: A comprehensive CIF toolbox. J. Appl. Crystallogr. 2011, 44 Pt 6, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71 Pt 1, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71 Pt 1, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Scalmani, G.; Frisch, M.J. Comment on “A smooth, nonsingular, and faithful discretization scheme for polarizable continuum models: The switching/gaussian approach” [J. Chem. Phys. 133, 244111 (2010)]. J. Chem. Phys. 2011, 134, 117101. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent molecular orbital methods. XII. Further extensions of Gaussian—Type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 5, 2257–2261. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 2021, 23, 20323–20328. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 38, 7338–7364. [Google Scholar] [CrossRef]
- Hartman, P.; Bennema, P. The attachment energy as a habit controlling factor: I. Theoretical considerations. J. Cryst. Growth 1980, 1, 145–156. [Google Scholar] [CrossRef]
- Li, W. Studies on thermal decomposition mechanism of clarithromycin and determination of the kinetic parameters. Chin. J. Antibiot. 2009, 34, 419–421. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | CLA-Form 0′ | CLA-PAC Solvate | CLA-IPA Solvate | CLA-EB Solvate |
|---|---|---|---|---|
| Empirical formula | C38H69NO13 | C38H69NO13·C5H10O2 | C38H69NO13·C5H10O2 | C38H69NO13·C6H12O2 |
| Formula weight | 747.94 | 850.07 | 850.07 | 864.09 |
| Crystal system | orthorhombic | orthorhombic | orthorhombic | orthorhombic |
| Space group | P212121 | P212121 | P212121 | P212121 |
| Temperature (°C) | 25 | −165 | −165 | −165 |
| a (Å) | 8.7700 (4) | 8.7114 (2) | 8.6827 (2) | 8.7063 (3) |
| b (Å) | 14.5393 (6) | 14.5078 (3) | 14.4474 (3) | 14.5448 (5) |
| c (Å) | 38.5455 (16) | 37.6484 (12) | 37.9694 (8) | 37.8502 (11) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 90 | 90 | 90 | 90 |
| γ (°) | 90 | 90 | 90 | 90 |
| Cell volume (Å3) | 4914.90 | 4758.10 | 4762.97 | 4793.02 |
| ρ, kg·m3 | 1.011 × 103 | 1.187 × 103 | 1.185 × 103 | 1.197 × 103 |
| Z | 4 | 4 | 4 | 4 |
| Rint | 0.0801 | 0.0683 | 0.0566 | 0.0853 |
| R1 (I > 2σ(I)) | 0.0671 | 0.0526 | 0.0490 | 0.0614 |
| wR2 | 0.1640 | 0.1035 | 0.1040 | 0.1326 |
| CCDC NO. | 2,339,166 | 2,339,163 | 2,339,164 | 2,339,165 |
| Adsorption Capacity (mol·g−1) | 25 (°C) | 50 (°C) |
|---|---|---|
| Form 0′ | 0.31240 | 0.12914 |
| Form II | 0.24877 | 0.11153 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, L.; Jing, D.; Wang, Y.; Bao, Y. CO2 Promoting Polymorphic Transformation of Clarithromycin: Polymorph Characterization, Pathway Design, and Mechanism Study. Crystals 2024, 14, 394. https://doi.org/10.3390/cryst14050394
Hou L, Jing D, Wang Y, Bao Y. CO2 Promoting Polymorphic Transformation of Clarithromycin: Polymorph Characterization, Pathway Design, and Mechanism Study. Crystals. 2024; 14(5):394. https://doi.org/10.3390/cryst14050394
Chicago/Turabian StyleHou, Lixin, Dingding Jing, Yanfeng Wang, and Ying Bao. 2024. "CO2 Promoting Polymorphic Transformation of Clarithromycin: Polymorph Characterization, Pathway Design, and Mechanism Study" Crystals 14, no. 5: 394. https://doi.org/10.3390/cryst14050394
APA StyleHou, L., Jing, D., Wang, Y., & Bao, Y. (2024). CO2 Promoting Polymorphic Transformation of Clarithromycin: Polymorph Characterization, Pathway Design, and Mechanism Study. Crystals, 14(5), 394. https://doi.org/10.3390/cryst14050394