
Preparation and Structural Variety of Neutral Heptaphospha-Nortricyclane Derivatives of Zinc and the Coinage Metals



Abstract

1. Introduction
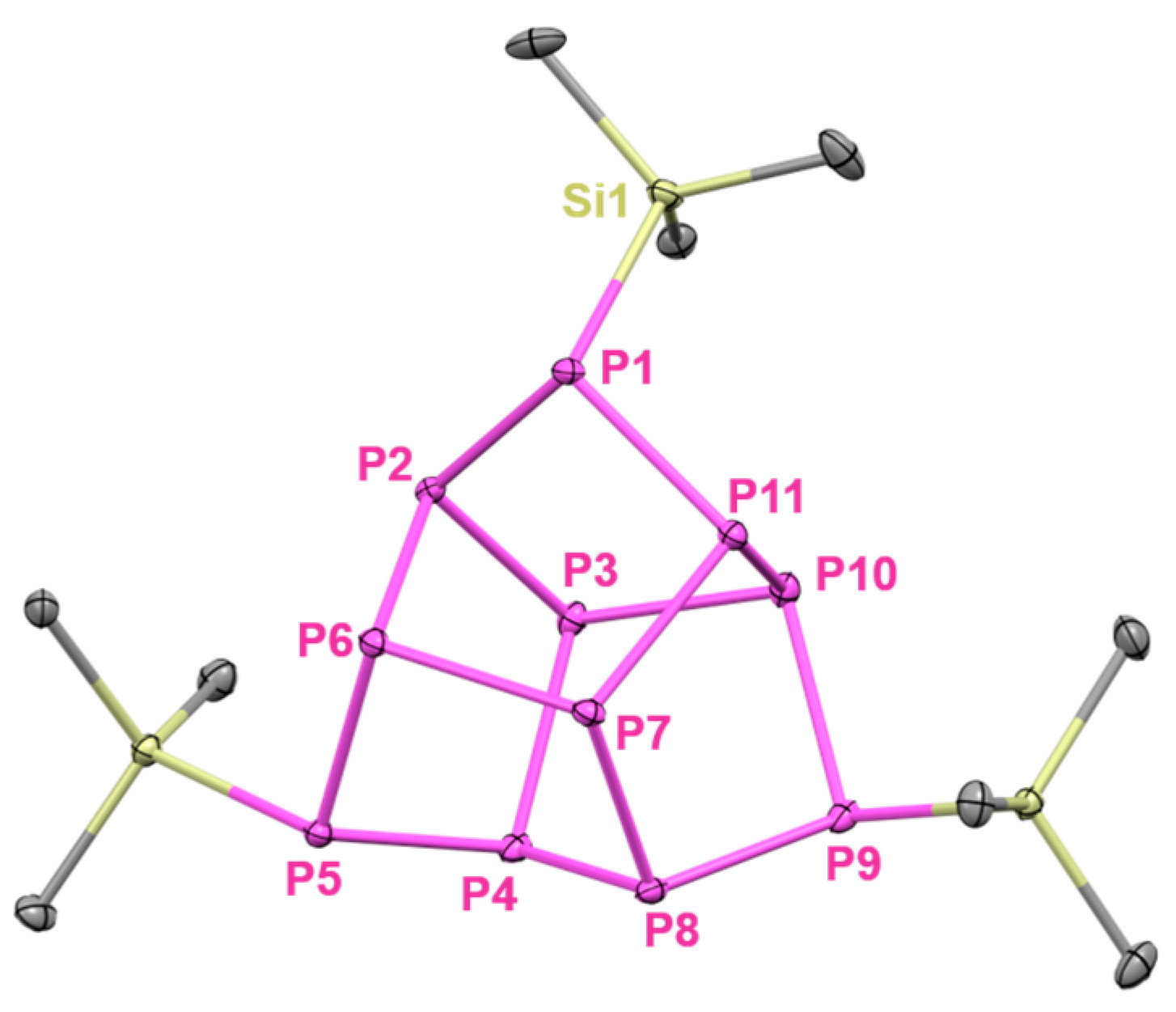
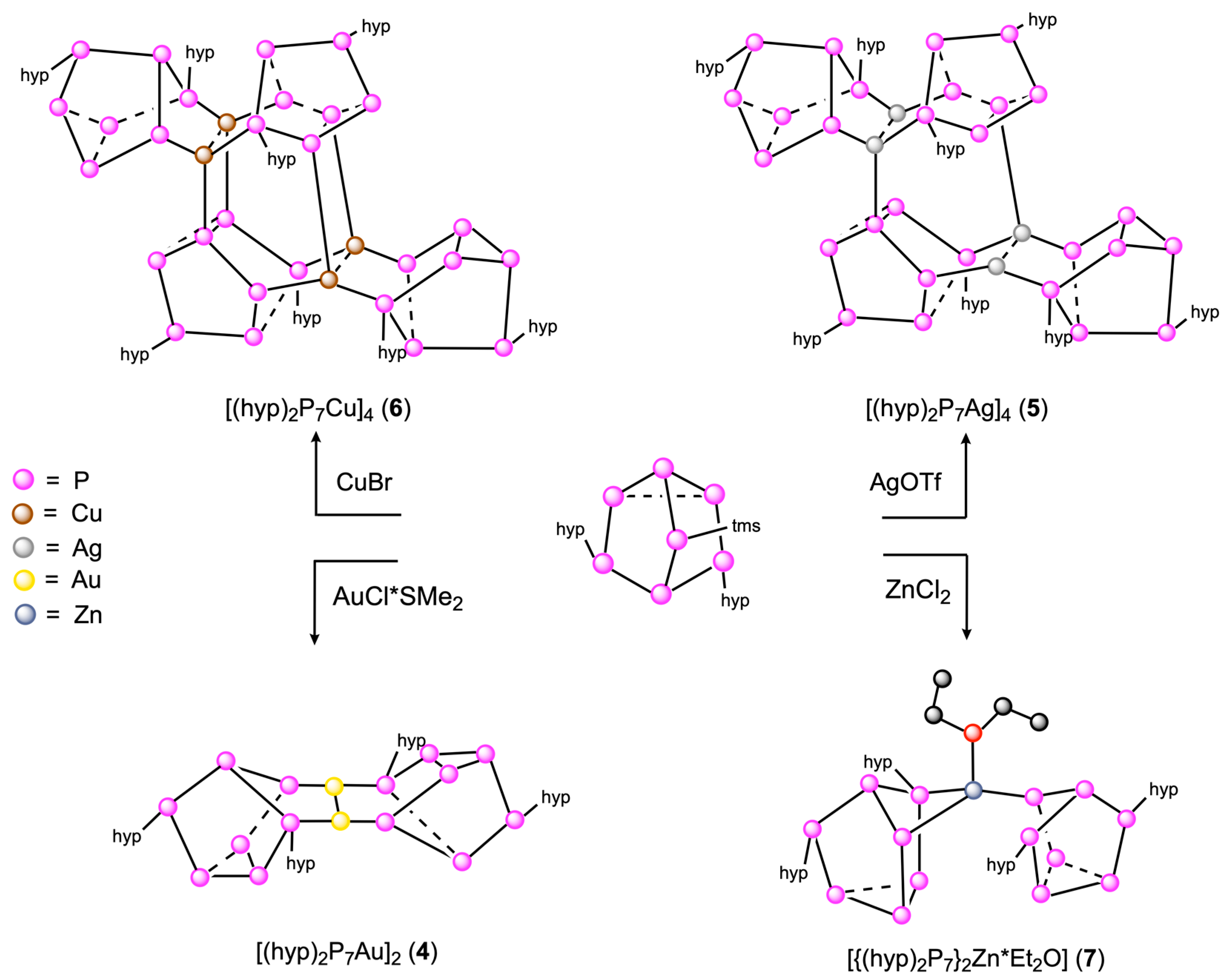
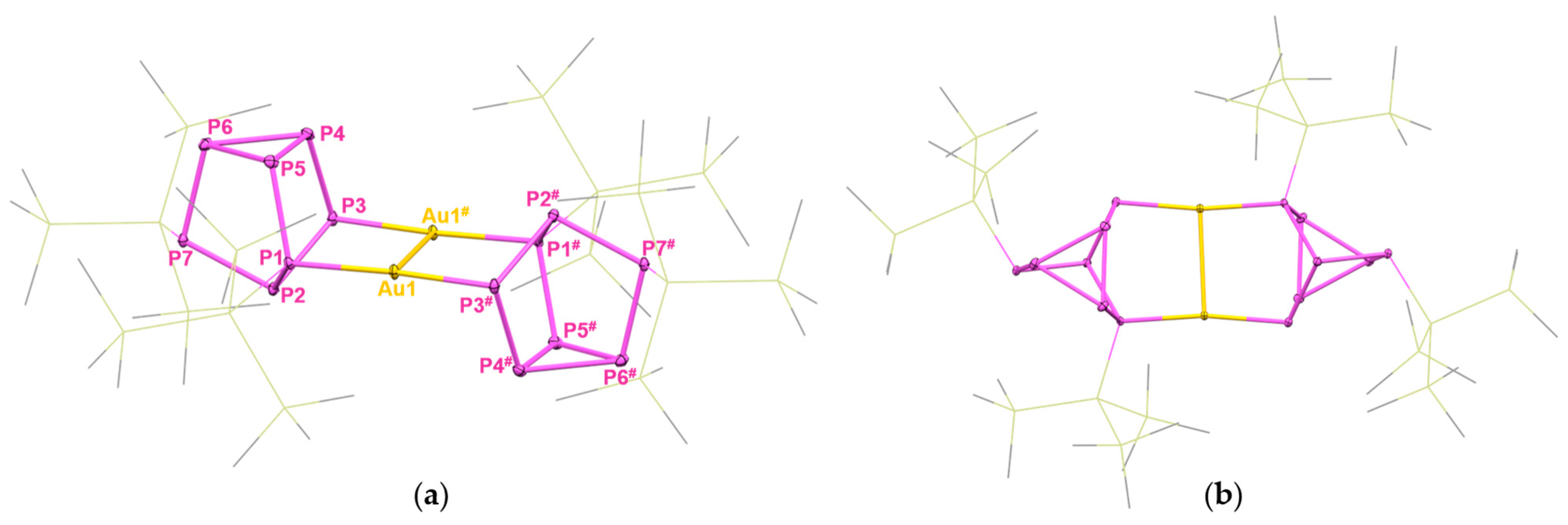
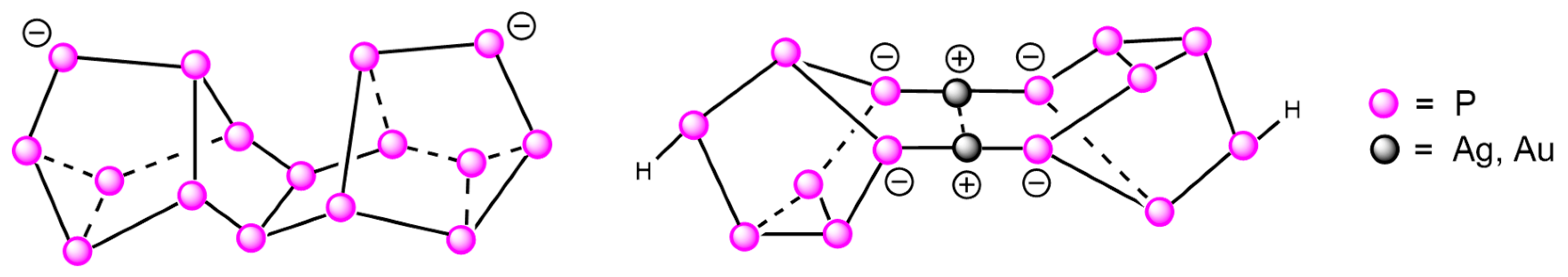
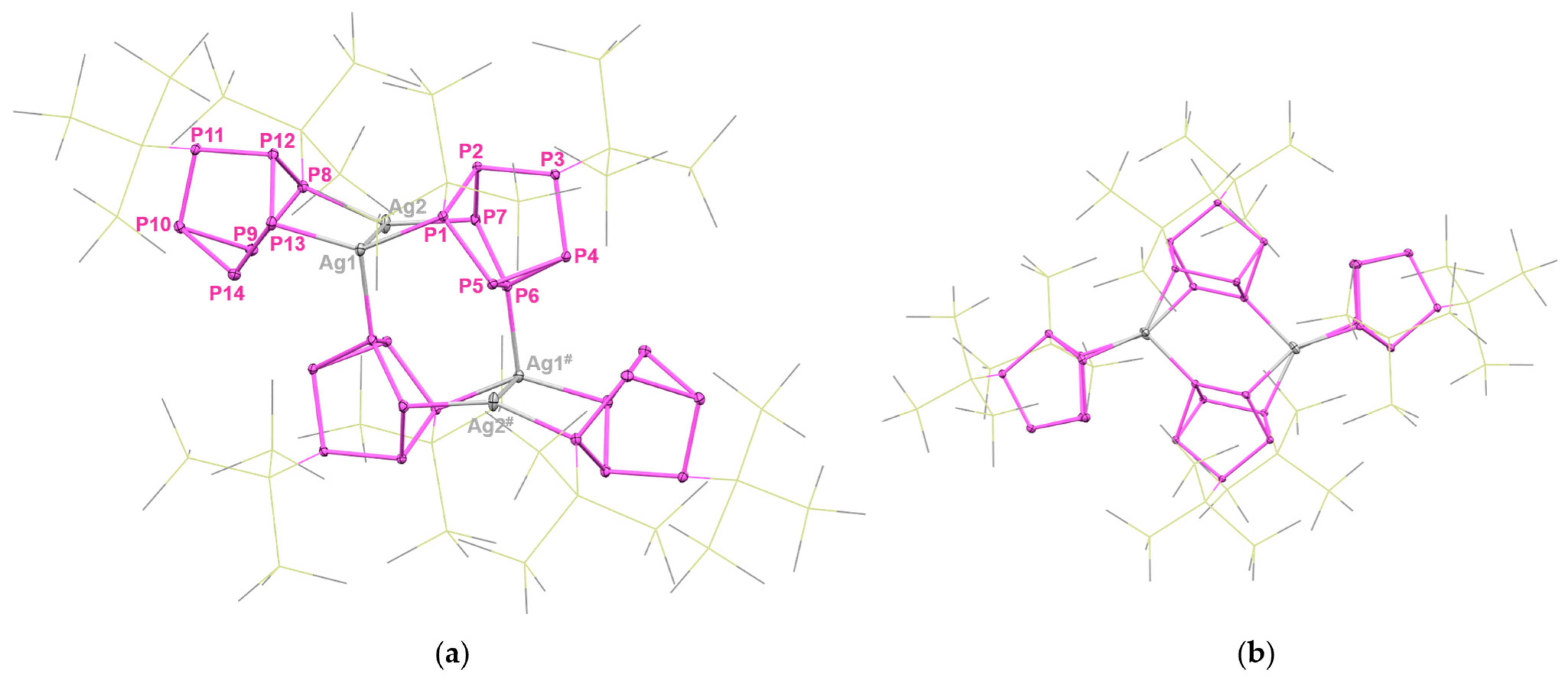
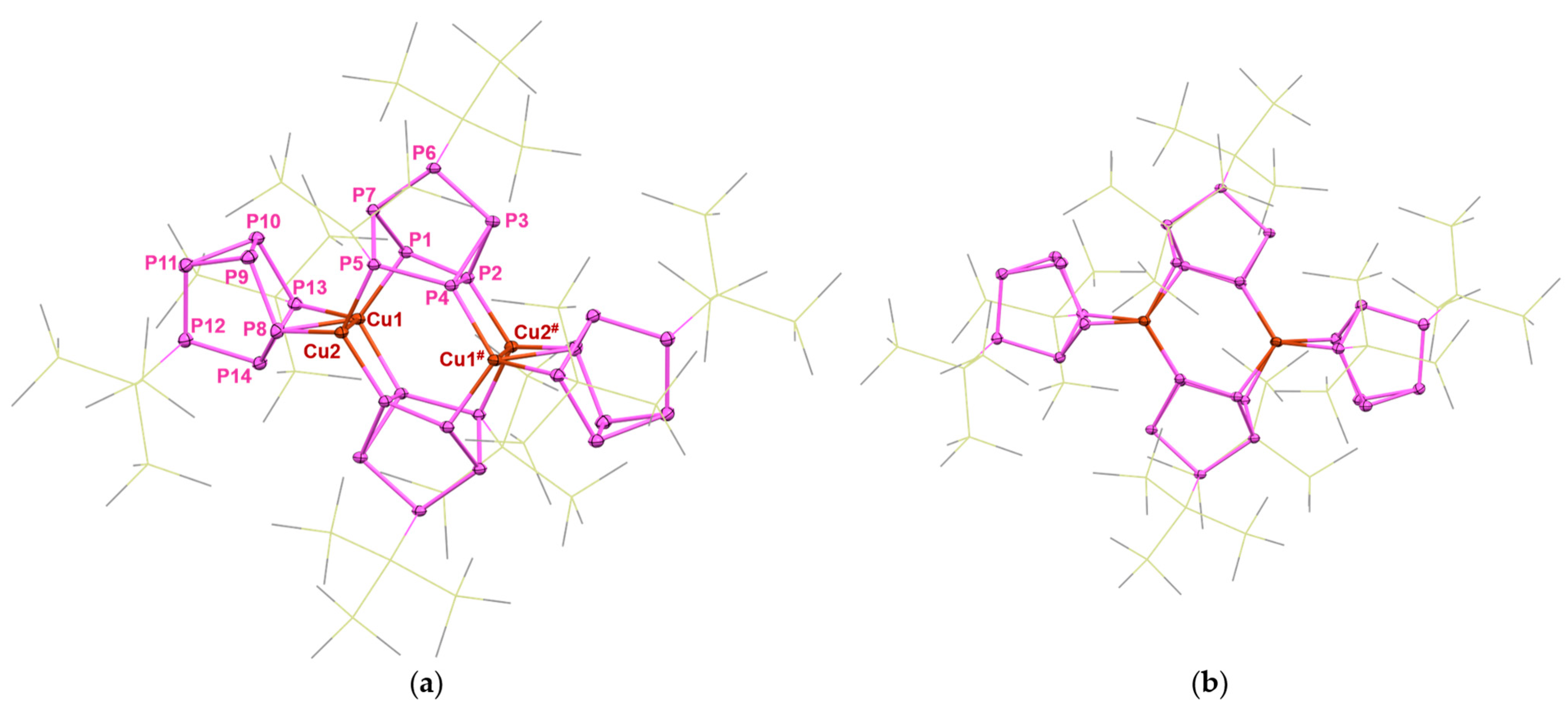
2. Results and Discussion
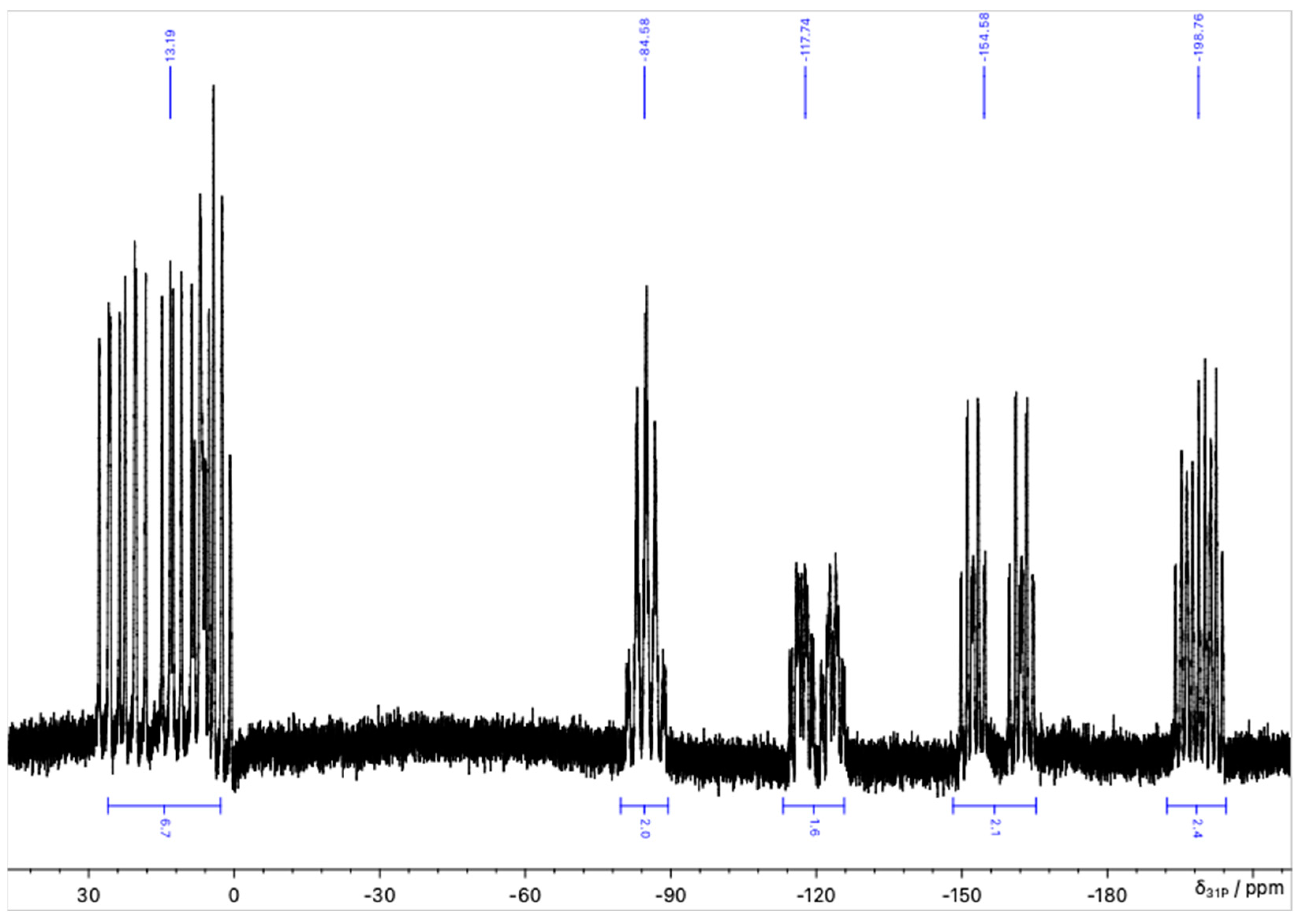
NMR Spectroscopy of 4 and 7
3. Conclusions
4. Experimental Section
4.1. Materials and Methods
4.1.1. NMR-Spectroscopy
4.1.2. Single Crystal X-ray Diffraction
4.2. Syntheses
4.2.1. Synthesis of 3,5,7-Tris(trimethylsilyl)tricyclo[2.2.1.02,6]heptaphosphane ((tms)3P7, 1)
4.2.2. Synthesis of 3,5-Bis(1,1,1,3,3,3-hexamethyl-2-(trimethylsilyl)trisilan-2-yl)-7-(trimethylsilyl)tricyclo[2.2.1.02,6]heptaphosphane ((hyp)2(tms)P7, 3)
4.2.3. Synthesis of [hyp2P7Au]2 (4)
4.2.4. Synthesis of [hyp2P7Ag]4 (5)
4.2.5. Synthesis of [hyp2P7Cu]4 (6)
4.2.6. Synthesis of [{(hyp)2P7}2Zn].OEt2 (7)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baudler, M.; Glinka, K. Monocyclic and Polycyclic Phosphanes. Chem. Rev. 1993, 93, 1623–1667. [Google Scholar] [CrossRef]
- Von Schnering, H.-G.; Honle, W. Bridging Chasms with Polyphosphides. Chem. Rev. 1988, 88, 243–273. [Google Scholar] [CrossRef]
- Baudler, M. Chain and Ring Phosphorus Compounds—Analogies between Phosphorus and Carbon Chemistry. Angew. Chem. Int. Ed. Engl. 1982, 21, 492–512. [Google Scholar] [CrossRef]
- Turbervill, R.S.P.; Goicoechea, J.M. From Clusters to Unorthodox Pnictogen Sources: Solution-Phase Reactivity of [E7]3− (E = P-Sb) Anions. Chem. Rev. 2014, 114, 10807–10828. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, W.; von Schnering, H.G. Sr3P14, Ein Phosphid Mit Isolierten P7(3−)Gruppen. Naturwissenschaften 1972, 59, 420. [Google Scholar] [CrossRef]
- Dahlmann, W.; von Schnering, H.G. Die Polyphosphide SrP3 Und Ba3P14. Naturwissenschaften 1973, 60, 429. [Google Scholar] [CrossRef]
- Siegl, H.; Krumlacher, W.; Hassler, K. Synthesis and Structure of P7[Si(SiMe3)3]3 (Tri(Hypersilyl)Heptaphosphanortricyclane). Monatshefte Fuer Chem. 1999, 130, 139–145. [Google Scholar]
- Noblet, P.; Dransfeld, A.; Fischer, R.; Flock, M.; Hassler, K. Derivatization of Tris(Trimethylsilyl)Heptaphosphane. J. Organomet. Chem. 2011, 696, 652–660. [Google Scholar] [CrossRef]
- Weber, D.; Mujica, C.; von Schnering, H.G. Tris-(Trimethyl-plumbyl)-heptaphospha-nortricyclen P7(PbMe3)3. Angew. Chem. Int. Ed. Engl. 1982, 21, 1801–1812. [Google Scholar] [CrossRef]
- Noblet, P.; Cappello, V.; Tekautz, G.; Baumgartner, J.; Hassler, K. Heptaphosphanortricyclenes with Oligosilyl Substituents: Syntheses and Reactions. Eur. J. Inorg. Chem. 2011, 2011, 101–109. [Google Scholar] [CrossRef]
- Fritz, G.; Schneider, H.W. Zur Synthese Der Heptaphospha-nortricyclane R3P7R = Et, i-Pr, n-Bu, i-Bu, SiH2Me, SiH3, Et2P–SiMe2. Z. Anorg. Allg. Chem. 1990, 584, 12–20. [Google Scholar] [CrossRef]
- Fritz, G.; Layher, E.; Goesmann, H.; Hanke, D.; Persau, C. [t-Bu2P]3P7 Und (t-Bu2Sb)3P7 Sowie Untersuchungen Zur Bildung von Heptaphosphanen(3) Mit PMe2-, PF2- Und P(CF3)2-Gruppen. Z. Allg. Anorg. Chem. 1991, 594, 36–46. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Fenske, D.; Fromm, K.; Krautscheid, H.; Krautscheid, U.; Treutler, O. Zintl Anions as Starting Compounds for the Synthesis of Polynuclear Transition Metal Complexes. Chem. Eur. J. 1996, 2, 238–244. [Google Scholar] [CrossRef]
- Jo, M.; Li, J.; Dragulescu-Andrasi, A.; Rogachev, A.Y.; Shatruk, M. Incorporation of Coinage Metal-NHC Complexes into Heptaphosphide Clusters. Dalton Trans. 2020, 49, 12955–12959. [Google Scholar] [CrossRef] [PubMed]
- Hönle, W.; von Schnering, H.G.; Fritz, G.; Schneider, H.W. Komplexchemie P-reicher Phosphane und Silylphosphane. II. Kristall- und Molekülstrukturen von Chromcarbonylkomplexen Mit Heptaphospha-nortricyclan Als Liganden. Z. Allg. Anorg. Chem. 1990, 584, 51–70. [Google Scholar] [CrossRef]
- Van IJzendoorn, B.; Vitorica-Yrezabal, I.J.; Whitehead, G.F.S.; Mehta, M. Heteroallene Capture and Exchange at Functionalised Heptaphosphane Clusters. Chem. Eur. J. 2022, 28, e202103737. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Hoelderich, W. Ein Neuer Typ Silylphosphane; (Me3Si)3P7, (Me3Si)4P14, (Me=CH3). Naturwissenschaften 1975, 62, 573–575. [Google Scholar] [CrossRef]
- Knapp, C.M.; Large, J.S.; Rees, N.H.; Goicoechea, J.M. The Bis(Hydrogenheptaphosphide)Iron(Ii) Dianion: A Zintl Ion Analogue of Ferrocene? Chem. Commun. 2011, 47, 4111–4113. [Google Scholar] [CrossRef] [PubMed]
- Muldoon, M.J. Modern Multiphase Catalysis: New Developments in the Separation of Homogeneous Catalysts. Dalton Trans. 2010, 39, 337–348. [Google Scholar] [CrossRef]
- Scott, C.; Eichhorn, B.W.; Rheingold, A.L.; Bott, S.G. Synthesis, Structure, and Properties of the [E7M(CO)3]3− Complexes Where E = P, As, Sb and M = Cr, Mo, W. J. Am. Chem. Soc. 1994, 116, 8077–8086. [Google Scholar]
- Charles, S.; Fettinger, J.C.; Eichhorn, B.W. Synthesis, Structure, and Reactivities of [η2-P7M(CO)4]3−, [η2-HP7M(CO)4]2−, and [η2-RP7M(CO)4]2− Zintl Ion Complexes Where M=Mo, W. Inorg. Chem. 1996, 35, 1540–1548. [Google Scholar] [CrossRef]
- Charles, S.; Fettinger, J.C.; Bott, S.G.; Eichhorn, B.W. Synthesis and Characterization of [η4-P7Ni(CO)]3−, [η4-HP7Ni(CO)]2−, and [η2-P7PtH(PPh3)]2−: Two Electronically Equivalent Protonated Zintl Ion Complexes with Markedly Different Structures. Chem. Commun. 1989, 28, 5837. [Google Scholar] [CrossRef]
- Von Schnering, H.G.; Fenske, D.; Hönle, W.; Binnewies, M.; Peters, K. Novel Polycyclic Phosphanes and Arsanes: Pi11(SiMe3)3 and As7(SiMe3)3. Angew. Chem. Int. Ed. Engl. 1979, 18, 679. [Google Scholar] [CrossRef]
- Knapp, C.M.; Jackson, C.S.; Large, J.S.; Thompson, A.L.; Goicoechea, J.M. Heteroatomic Molecular Clusters Derived from Group 15 Zintl Ion Cages: Synthesis and Isolation of [M2(HP7)2]2− (M = Ag, Au), Two Novel Cluster Anions Exhibiting Metallophilic Interactions. Inorg. Chem. 2011, 50, 4021–4028. [Google Scholar] [CrossRef]
- Von Schnering, H.G.; Manriquez, V.; Hönle, W. Bis(Tetraphenylphosphonium) Hexadecaphosphide, a Salt Containing the Novel Polycyclic Anion P2−16. Angew. Chem. Int. Ed. Engl. 1981, 20, 594–595. [Google Scholar] [CrossRef]
- Baudler, M.; Düster, D. Beiträge Zur Chemie Des Phosphors, 175 Dinatrium-Hexadecaphosphid: Darstellung Durch Spaltung von Weißem Phosphor Mit Natrium. Z. Naturforsch. 1987, 42, 335–336. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Scherbaum, F.; Huber, B.; Müller, G. Polyauriomethane Compounds. Angew. Chem. Int. Ed. Engl. 1988, 27, 419–421. [Google Scholar] [CrossRef]
- Schmidbaur, H. Ludwig Mond Lecture. High-Carat Gold Compounds. Chem. Soc. Rev. 1995, 24, 391–400. [Google Scholar] [CrossRef]
- Jaw, H.-R.C.; Meral Savas, M.; Rogers, R.D.; Mason, W.R. Crystal Structures and Solution Electronic Absorption and MCD Spectra for Perchlorate and Halide Salts of Binuclear Gold(I) Complexes Containing Bridging Me2PCH2PMe2(Dmpm) or Me2PCH2CH2PMe2 (Dmpe) Ligands. Inorg. Chem. 1989, 28, 1028–1037. [Google Scholar] [CrossRef]
- Pyykkö, P. Strong Closed-Shell Interactions in Inorganic Chemistry. Chem. Rev. 1997, 97, 597–636. [Google Scholar] [CrossRef]
- Scherbaum, F.; Grohmann, A.; Huber, B.; Krüger, C.; Schmidbaur, H. “Aurophilicity” as a Consequence of Relativistic Effects: The Hexakis(Triphenylphosphaneaurio)Methane Dication [(Ph3PAu)6C]2⊕. Angew. Chem. Int. Ed. Engl. 1988, 27, 1544–1546. [Google Scholar] [CrossRef]
- Pyykkö, P. Theoretical Chemistry of Gold. Angew. Chem. Int. Ed. 2004, 43, 4412–4456. [Google Scholar] [CrossRef] [PubMed]
- Dobrzańska, L.; Raubenheimer, H.G.; Barbour, L.J. Borromean Sheets Assembled by Self-Supporting Argentophilic Interactions. Chem. Commun. 2005, 5050–5052. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Zeng, J.Y.; Lee, H.M. Argentophilic Interaction and Anionic Control of Supramolecular Structures in Simple Silver Pyridine Complexes. Inorg. Chim. Acta 2007, 360, 21–30. [Google Scholar] [CrossRef]
- Liu, X.; Guo, G.C.; Fu, M.L.; Liu, X.H.; Wang, M.S.; Huang, J.S. Three Novel Silver Complexes with Ligand-Unsupported Argentophilic Interactions and Their Luminescent Properties. Inorg. Chem. 2006, 45, 3679–3685. [Google Scholar] [CrossRef]
- Mohamed, A.A.; Pérez, L.M.; Fackler, J.P. Unsupported Intermolecular Argentophilic Interaction in the Dimer of Trinuclear Silver(I) 3,5-Diphenylpyrazolates. Inorg. Chim. Acta 2005, 358, 1657–1662. [Google Scholar] [CrossRef]
- Blessing, R.H. An Empirical Correction for Absorption Anisotropy. Acta Crystallogr. A 1995, A51, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SADBS Version 2.10 Siemens Area Detector Correction; Universitaet Goettingen: Goettingen, Germany, 2003. [Google Scholar]
- Sheldrick, G.M. SHELXTL Version 6.1; Bruker AXS, Inc.: Madison, WI, USA, 2002. [Google Scholar]
- Sheldrick, G.M. GM SHELXS97 and SHELXL97; Universitaet Goettingen: Goettingen, Germany, 2002. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Single-Crystal Structure Validation with the Program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 4 | 5 | 6 | |||
|---|---|---|---|---|---|
| Au1-Au1 # | 2.9542(2) | Ag1-Ag2 | 2.8833(6) | Cu1-Cu2 | 2.654(1) |
| Au1-P1 | 2.322(1) | Ag1-P1 | 2.494(1) | Cu1-P1 | 2.287(3) |
| Au1-P3 # | 2.328(1) | Ag1-P13 | 2.450(1) | Cu1-P13 | 2.524(2) |
| P1-P2 | 2.192(1) | P1-P2 | 2.202(2) | P1-P2 | 2.151(2) |
| P1-P5 | 2.199(2) | P1-P5 | 2.214(2) | P1-P7 | 2.166(2) |
| P1∙∙∙P3 | 3.293(2) | P1∙∙∙P7 | 3.295(2) | P1∙∙∙P5 | 3.340(2) |
| P2-P3 | 2.170(2) | P2-P3 | 2.195(2) | P2-P3 | 2.218(3) |
| P2-P7 | 2.197(2) | P2-P7 | 2.159(2) | P2-P4 | 2.239(2) |
| P3-P4 | 2.197(2) | P3-P4 | 2.186(3) | P3-P4 | 2.230(3) |
| P4-P5 | 2.222(2) | P4-P5 | 2.228(2) | P3-P6 | 2.200(2) |
| P4-P6 | 2.216(2) | P4-P6 | 2.226(2) | P4-P5 | 2.225(2) |
| P5-P6 | 2.220(2) | P5-P6 | 2.218(2) | P5-P7 | 2.198(3) |
| P6-P7 | 2.201(2) | P6-P7 | 2.169(2) | P6-P7 | 2.196(3) |
| Ag1-P6dimer2 | 2.581(2) | Cu1-P4dimer2 | 2.411(2) | ||
| Cu2-P2 dimer2 | 2.252(2) | ||||
| P1-Au1-P3 # | 171.64(4) | P1-Ag1-P13 | 138.86(5) | P1-Cu1-P13 | 110.23(7) |
| P1-Au1-Au1 # | 91.55(3) | P1-Ag1-Ag2 | 91.37(4) | P1-Cu1-Cu2 | 95.88(6) |
| Papex-P1-Au1 | 110.96(6) | Papex-P1-Ag1 | 110.21(7) | Papex-P1-M1 | 105.78(9) |
| 7 | |
|---|---|
| Zn-P1 | 2.434(1) |
| Zn-P3 | 2.5839(9) |
| Zn-P1# | 2.259(1) |
| Zn∙∙∙P3 # | 3.4284(9) |
| Zn-O | 2.108(7) |
| P1-P2 | 2.1627(9) |
| P1-P5 | 2.175(1) |
| P1∙∙∙P3 | 3.1913(8) |
| P2-P3 | 2.1713(9) |
| P2-P7 | 2.162(1) |
| P3-P4 | 2.183(1) |
| P4-P5 | 2.2283(9) |
| P4-P6 | 2.211(1) |
| P5-P6 | 2.215(1) |
| P6-P7 | 2.179(1) |
| P1-Zn-P3 | 78.93(3) |
| P1-Zn-P1 # | 145.54(5) |
| P1-Zn-P3 # | 85.80(3) |
| P3-Zn-P3 # | 142.31(3) |
| P1-Zn-O | 103.1(2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roller, C.A.; Doler, B.; Fischer, R.C. Preparation and Structural Variety of Neutral Heptaphospha-Nortricyclane Derivatives of Zinc and the Coinage Metals. Crystals 2024, 14, 586. https://doi.org/10.3390/cryst14070586
Roller CA, Doler B, Fischer RC. Preparation and Structural Variety of Neutral Heptaphospha-Nortricyclane Derivatives of Zinc and the Coinage Metals. Crystals. 2024; 14(7):586. https://doi.org/10.3390/cryst14070586
Chicago/Turabian StyleRoller, Clara A., Berenike Doler, and Roland C. Fischer. 2024. "Preparation and Structural Variety of Neutral Heptaphospha-Nortricyclane Derivatives of Zinc and the Coinage Metals" Crystals 14, no. 7: 586. https://doi.org/10.3390/cryst14070586
APA StyleRoller, C. A., Doler, B., & Fischer, R. C. (2024). Preparation and Structural Variety of Neutral Heptaphospha-Nortricyclane Derivatives of Zinc and the Coinage Metals. Crystals, 14(7), 586. https://doi.org/10.3390/cryst14070586