
Exploring Methane Storage Capacities of M2(BDC)2(DABCO) Sorbents: A Multiscale Computational Study

 ,
,
Abstract
1. Introduction
2. Methods and Modeling
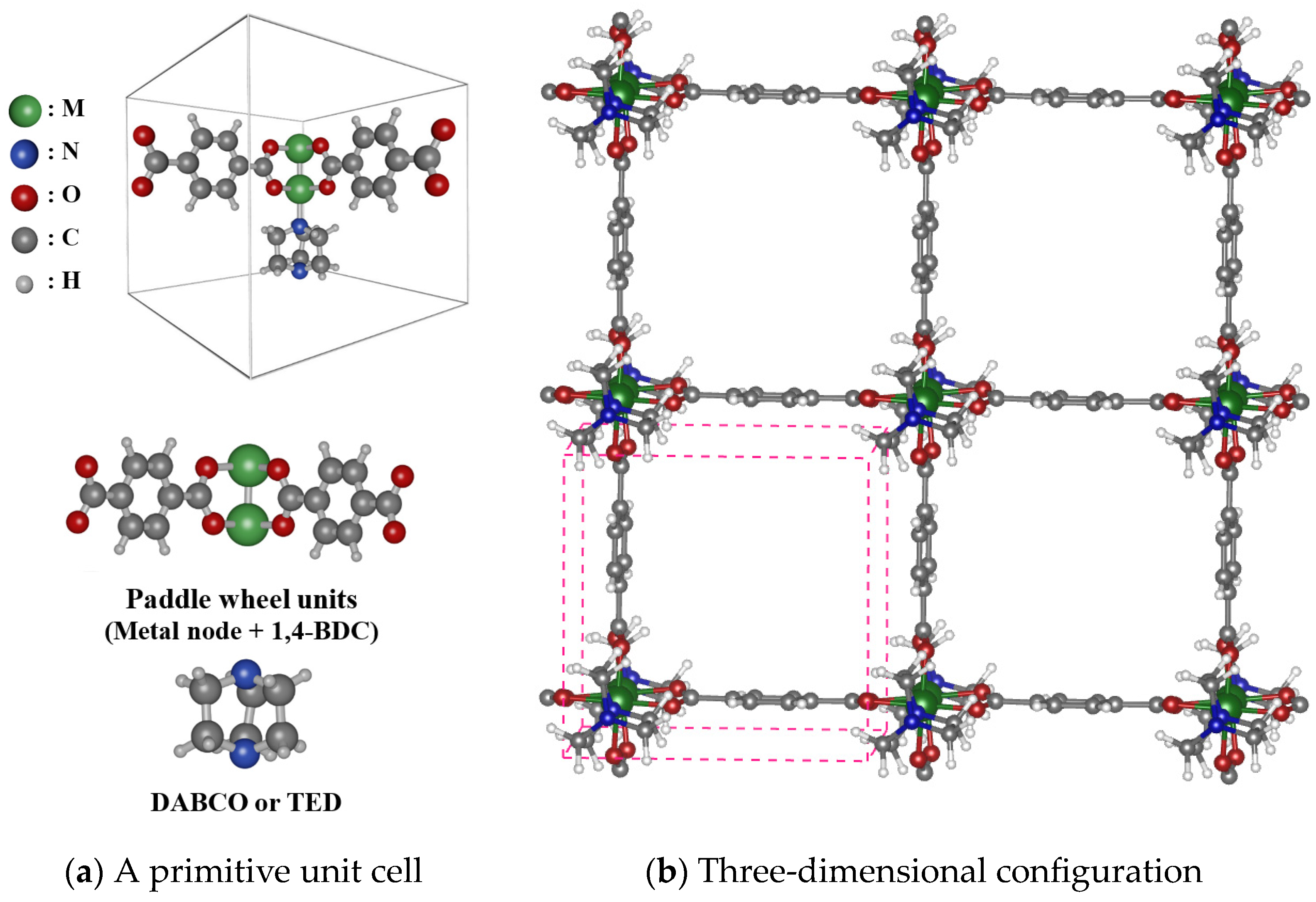
2.1. Modeling of M2(BDC)2(DABCO) MOFs
2.2. Grand Canonical Monte Carlo Simulation
2.3. Density Functional Theory Calculation
3. Results and Discussion
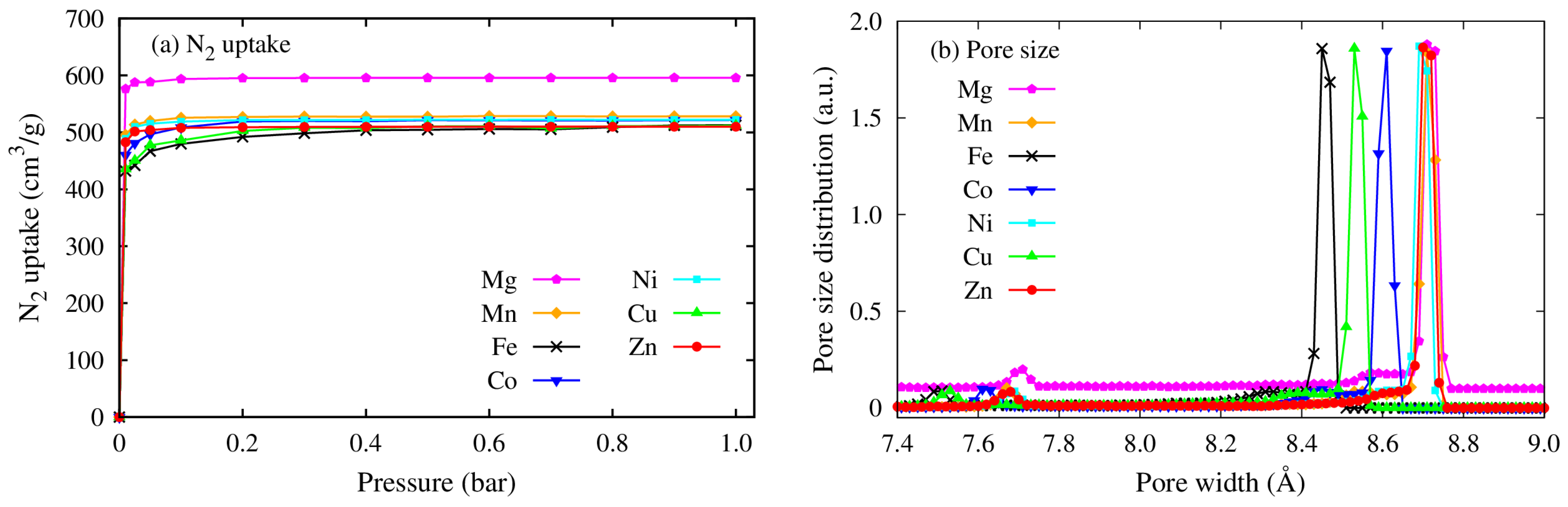
3.1. Porosity and Surface Analysis of M(DABCO)
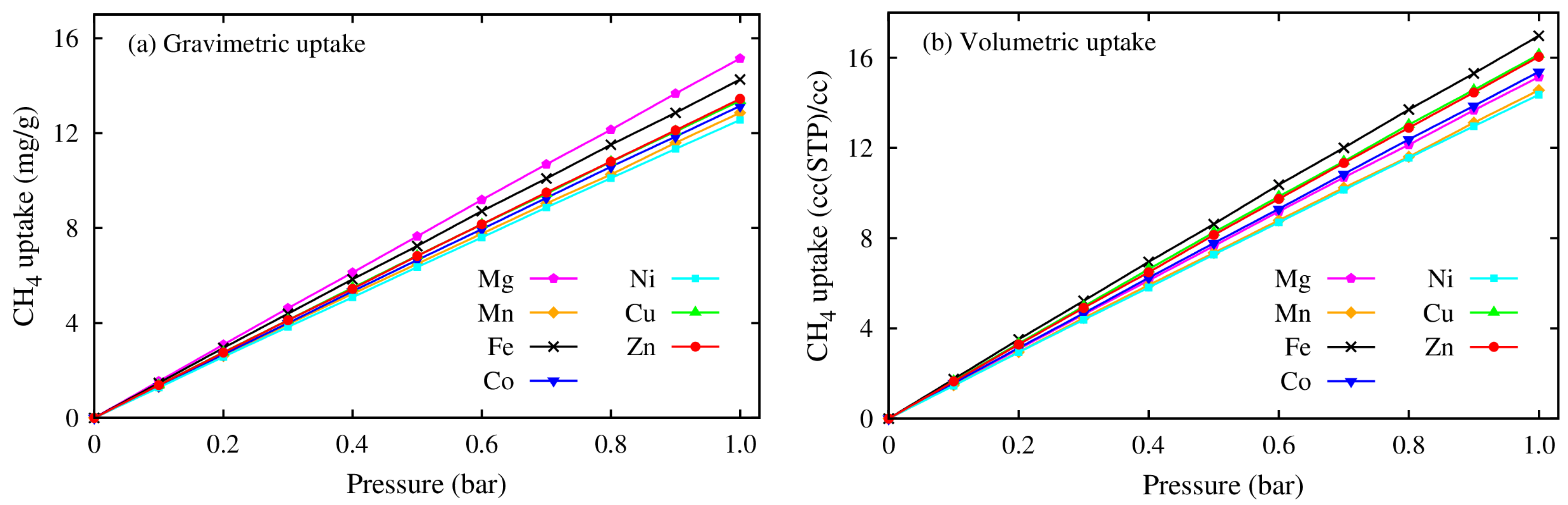
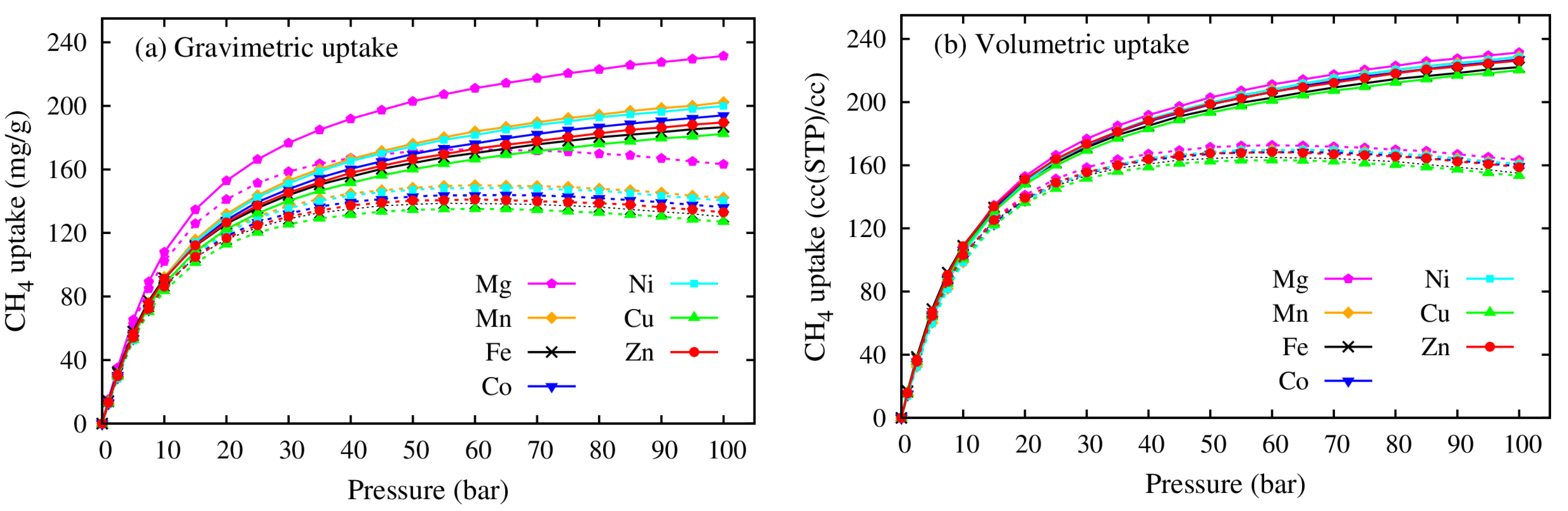
3.2. Methane Adsorption on M(DABCO)
3.3. Adsorption Heat of Methane on M(DABCO)
3.4. Stable CH4 Adsorption Sites on M(DABCO)
4. Conclusions
- (i)
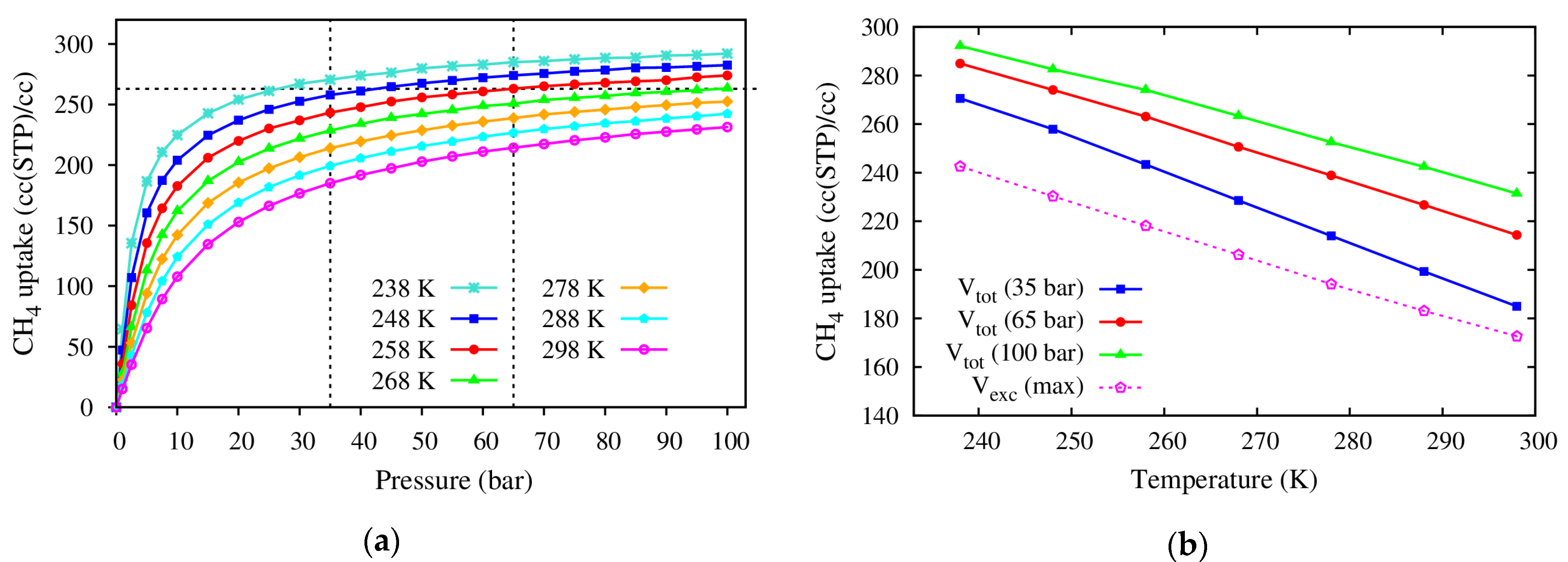
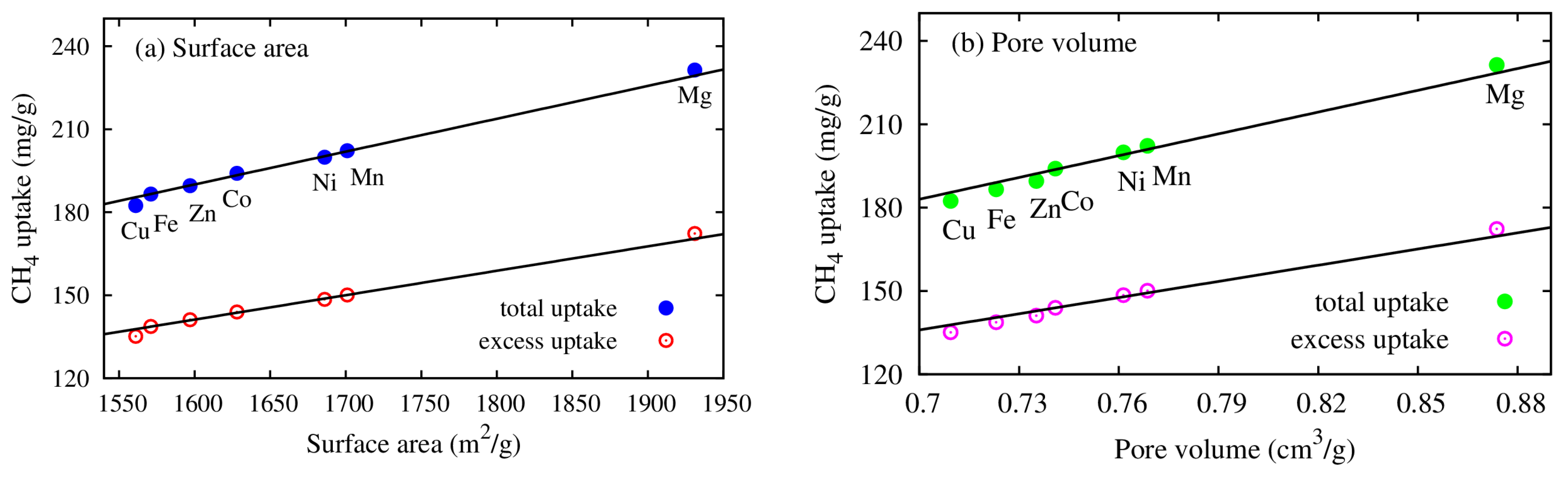
- The CH4 storage capacities in regards to both the gravimetric and volumetric uptakes of the M(DABCO) series are evaluated, and the volumetric uptakes of CH4 in M(DABCO) are remarkable. Both uptakes are in the increasing order of M(DABCO), as: Cu < Fe < Zn < Co < Ni < Mn < Mg. Among these MOFs, Mg (DABCO) shows the highest CH4 adsorption, with the maximum total and excess uptakes of 231.39 mg/g (at 100 bar) and 172.30 mg/g (at 65 bar), for gravimetric uptakes, and 231.43 cc(STP)/cc (at 100 bar) and 172.33 cc(STP)/cc (at 65 bar), for volumetric uptakes. Moreover, the methane adsorption capacities increase almost linearly with temperature, and the gravimetric methane adsorption strongly depends on the geometrical features (specific surface area and pore volume) of the M(DABCO) MOFs. Our simulations predicted that the volumetric CH4 storage capacity could meet the DOE target at lower temperatures, ca. 238 K–268 K in the 35–100 bar pressure range.
- (ii)
- The interaction between CH4 and M(DABCO) is mainly governed by the vdW interaction, and electrostatic interactions play a minor role, as indicated by the DFT calculations.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, Y.; Zhou, W.; Qian, G.; Chen, B. Methane Storage in Metal-Organic Frameworks. Chem. Soc. Rev. 2014, 43, 5657. [Google Scholar] [CrossRef] [PubMed]
- Huong, T.T.T.; Thanh, P.N.; Huynh, N.T.X.; Son, D.N. Metal—Organic Frameworks: State-of-the-Art Material for Gas Capture and Storage. VNU J. Sci. Math.—Phys. 2016, 32, 67–85. [Google Scholar]
- Ribeiro, R.P.P.L.; Mota, J.P.B. Surface Area and Porosity of Co3(ndc)3(dabco) Metal-Organic Framework and Its Methane Storage Capacity: A Combined Experimental and Simulation Study. J. Phys. Chem. C 2021, 125, 2411–2423. [Google Scholar] [CrossRef]
- Arutyunov, V.; Savchenko, V.; Sedov, I.; Arutyunov, A.; Nikitin, A. The Fuel of Our Future: Hydrogen or Methane? Methane 2022, 1, 96–106. [Google Scholar] [CrossRef]
- Koh, H.S.; Rana, M.K.; Wong-Foy, A.G.; Siegel, D.J. Predicting Methane Storage in Open-Metal-Site Metal-Organic Frameworks. J. Phys. Chem. C 2015, 119, 13451–13458. [Google Scholar] [CrossRef]
- Peng, Y.; Krungleviciute, V.; Eryazici, I.; Hupp, J.T.; Farha, O.K.; Yildirim, T. Methane Storage in Metal-Organic Frameworks: Current Records, Surprise Findings, and Challenges. J. Am. Chem. Soc. 2013, 135, 11887–11894. [Google Scholar] [CrossRef] [PubMed]
- Granja-DelRío, A.; Cabria, I. Exploring the Hydrogen and Methane Storage Capacities of Novel DUT MOFs at Room Temperature: A Grand Canonical Monte Carlo Simulation Study. Int. J. Hydrogen Energy 2024, 54, 665–677. [Google Scholar] [CrossRef]
- Li, H.; Li, L.; Lin, R.-B.B.; Zhou, W.; Zhang, Z.; Xiang, S.; Chen, B. Porous Metal-Organic Frameworks for Gas Storage and Separation: Status and Challenges. EnergyChem 2019, 1, 100006. [Google Scholar] [CrossRef] [PubMed]
- Safaei, M.; Foroughi, M.M.; Ebrahimpoor, N.; Jahani, S.; Omidi, A.; Khatami, M. A Review on Metal-Organic Frameworks: Synthesis and Applications. TrAC—Trends Anal. Chem. 2019, 118, 401–425. [Google Scholar] [CrossRef]
- Li, D.; Yadav, A.; Zhou, H.; Roy, K.; Thanasekaran, P.; Lee, C. Advances and Applications of Metal-Organic Frameworks (MOFs) in Emerging Technologies: A Comprehensive Review. Glob. Chall. 2024, 8, 2300244. [Google Scholar] [CrossRef]
- Dutt, S.; Kumar, A.; Singh, S. Synthesis of Metal Organic Frameworks (MOFs) and Their Derived Materials for Energy Storage Applications. Clean Technol. 2023, 5, 140–166. [Google Scholar] [CrossRef]
- Sandhu, Z.A.; Raza, M.A.; Awwad, N.S.; Ibrahium, H.A.; Farwa, U.; Ashraf, S.; Dildar, A.; Fatima, E.; Ashraf, S.; Ali, F. Metal-Organic Frameworks for next-Generation Energy Storage Devices; a Systematic Review. Mater. Adv. 2024, 5, 30–50. [Google Scholar] [CrossRef]
- Ursueguía, D.; Díaz, E.; Ordóñez, S. Metal-Organic Frameworks (MOFs) as Methane Adsorbents: From Storage to Diluted Coal Mining Streams Concentration. Sci. Total Environ. 2021, 790, 148211. [Google Scholar] [CrossRef] [PubMed]
- Getman, R.; Bae, Y.; Wilmer, C.; Snurr, R. Review and Analysis of Molecular Simulations of Methane, Hydrogen, and Acetylene Storage in Metal–Organic Frameworks. Chem. Rev. 2012, 112, 703–723. [Google Scholar] [CrossRef] [PubMed]
- Guo, K. Applications of Metal-Organic Frameworks in Methane Storage. Highlights Sci. Eng. Technol. 2023, 58, 338–343. [Google Scholar] [CrossRef]
- Forrest, K.A.; Verma, G.; Ye, Y.; Ren, J.; Ma, S.; Pham, T.; Space, B. Methane Storage in Flexible and Dynamical Metal–Organic Frameworks. Chem. Phys. Rev. 2022, 3, 021308. [Google Scholar] [CrossRef]
- Kondo, M.; Yoshitomi, T.; Seki, K.; Matsuzaka, H.; Kitagawa, S. Three-Dimensional Framework with Channeling Cavities for Small Molecules: {[M2(4, 4′-Bpy)3(NO3)4]·xH2O}n (M=Co, Ni, Zn). Angew. Chemie Int. Ed. English 1997, 36, 1725–1727. [Google Scholar] [CrossRef]
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic Design of Pore Size and Functionality in Isoreticular MOFs and Their Application in Methane Storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Alezi, D.; Belmabkhout, Y.; Suyetin, M.; Bhatt, P.M.; Weseliński, L.J.; Solovyeva, V.; Adil, K.; Spanopoulos, I.; Trikalitis, P.N.; Emwas, A.H.; et al. MOF Crystal Chemistry Paving the Way to Gas Storage Needs: Aluminum-Based Soc -MOF for CH4, O2, and CO2 Storage. J. Am. Chem. Soc. 2015, 137, 13308–13318. [Google Scholar] [CrossRef]
- Spanopoulos, I.; Tsangarakis, C.; Klontzas, E.; Tylianakis, E.; Froudakis, G.; Adil, K.; Belmabkhout, Y.; Eddaoudi, M.; Trikalitis, P.N. Reticular Synthesis of HKUST-like Tbo-MOFs with Enhanced CH4 Storage. J. Am. Chem. Soc. 2016, 138, 1568–1574. [Google Scholar] [CrossRef]
- Moreau, F.; Kolokolov, D.I.; Stepanov, A.G.; Easun, T.L.; Dailly, A.; Lewis, W.; Blake, A.J.; Nowell, H.; Lennox, M.J.; Besley, E.; et al. Tailoring Porosity and Rotational Dynamics in a Series of Octacarboxylate Metal-Organic Frameworks. Proc. Natl. Acad. Sci. USA 2017, 114, 3056–3061. [Google Scholar] [CrossRef]
- Mason, J.A.; Veenstra, M.; Long, J.R. Evaluating Metal-Organic Frameworks for Natural Gas Storage. Chem. Sci. 2014, 5, 32–51. [Google Scholar] [CrossRef]
- Stoeck, U.; Krause, S.; Bon, V.; Senkovska, I.; Kaskel, S. A Highly Porous Metal–Organic Framework, Constructed from a Cuboctahedral Super-Molecular Building Block, with Exceptionally High Methane Uptake. Chem. Commun. 2012, 48, 10841–10843. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, Z.; Li, Y.; Yao, K.; Zhu, Y.; Deng, Z.; Yang, F.; Zhou, X.; Li, G.; Wu, H.; et al. Enhanced Binding Affinity, Remarkable Selectivity, and High Capacity of CO2 by Dual Functionalization of a Rht-Type Metal–Organic Framework. Angew. Chemie 2012, 124, 1441–1444. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, R.B.; Alothman, Z.A.; Alduhaish, O.; Yildirim, T.; Zhou, W.; Li, J.R.; Chen, B. Promotion of Methane Storage Capacity with Metal-Organic Frameworks of High Porosity. Inorg. Chem. Front. 2022, 10, 454–459. [Google Scholar] [CrossRef]
- Zhang, M.; Zhou, W.; Pham, T.; Forrest, K.A.; Liu, W.; He, Y.; Wu, H.; Yildirim, T.; Chen, B.; Space, B.; et al. Fine Tuning of MOF-505 Analogues To Reduce Low-Pressure Methane Uptake and Enhance Methane Working Capacity. Angew. Chemie—Int. Ed. 2017, 56, 11426–11430. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wen, H.M.; Wang, H.; Wu, H.; Tyagi, M.; Yildirim, T.; Zhou, W.; Chen, B. A Porous Metal-Organic Framework with Dynamic Pyrimidine Groups Exhibiting Record High Methane Storage Working Capacity. J. Am. Chem. Soc. 2014, 136, 6207–6210. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.M.; Li, B.; Li, L.; Lin, R.B.; Zhou, W.; Qian, G.; Chen, B. A Metal–Organic Framework with Optimized Porosity and Functional Sites for High Gravimetric and Volumetric Methane Storage Working Capacities. Adv. Mater. 2018, 30, 1704792. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Xu, F.; Zhou, X.; Xiao, J.; Xia, Q.; Li, Y.; Li, Z. Ethane Selective Adsorbent Ni(bdc)(ted)0.5 with High Uptake and Its Significance in Adsorption Separation of Ethane and Ethylene. Chem. Eng. Sci. 2016, 148, 275–281. [Google Scholar] [CrossRef]
- Xiang, H.; Ameen, A.; Gorgojo, P.; Siperstein, F.R.; Holmes, S.M.; Fan, X. Selective Adsorption of Ethane over Ethylene on M(bdc)(ted)0.5 (M = Co, Cu, Ni, Zn) Metal-Organic Frameworks (MOFs). Microporous Mesoporous Mater. 2020, 292, 109724. [Google Scholar] [CrossRef]
- Peng, L.; Wu, S.; Yang, X.; Hu, J.; Fu, X.; Huo, Q.; Guan, J. Application of Metal Organic Frameworks M(bdc)(ted)0.5 (M = Co, Zn, Ni, Cu) in the Oxidation of Benzyl Alcohol. RSC Adv. 2016, 6, 72433–72438. [Google Scholar] [CrossRef]
- Tan, K.; Canepa, P.; Gong, Q.; Liu, J.; Johnson, D.H.; Dyevoich, A.; Thallapally, P.K.; Thonhauser, T.; Li, J.; Chabal, Y.J. Mechanism of Preferential Adsorption of SO2 into Two Microporous Paddle Wheel Frameworks M(bdc)(ted)0.5. Chem. Mater. 2013, 25, 4653–4662. [Google Scholar] [CrossRef]
- Son, D.N.; Thuy Huong, T.T.; Chihaia, V. Simultaneous Adsorption of SO2 and CO2 in an Ni(bdc)(ted)0.5 Metal-Organic Framework. RSC Adv. 2018, 8, 38648–38655. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lee, J.Y.; Pan, L.; Obermyer, R.T.; Simizu, S.; Zande, B.; Li, J.; Sankar, S.G.; Johnson, J.K. Adsorption and Diffusion of Hydrogen in a New Metal-Organic Framework Material: [Zn(bdc)(ted)0.5]. J. Phys. Chem. C 2008, 112, 2911–2917. [Google Scholar] [CrossRef]
- Li, R.; Han, X.; Liu, Q.; Qian, A.; Zhu, F.; Hu, J.; Fan, J.; Shen, H.; Liu, J.; Pu, X.; et al. Enhancing Hydrogen Adsorption Capacity of Metal Organic Frameworks M(BDC)TED0.5 through Constructing a Bimetallic Structure. ACS Omega 2022, 7, 20081–20091. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Li, Y.; He, B.; Yang, J.; Zheng, J.; Li, X. Hydrogen Storage Properties of Two Pillared-Layer Ni(II) Metal-Organic Frameworks. Microporous Mesoporous Mater. 2011, 142, 208–213. [Google Scholar] [CrossRef]
- Kozlova, S.G.; Mirzaeva, I.V.; Ryzhikov, M.R. DABCO Molecule in the M2(C8H4O4)2·C6H12N2 (M = Co, Ni, Cu, Zn) Metal-Organic Frameworks. Coord. Chem. Rev. 2018, 376, 62–74. [Google Scholar] [CrossRef]
- Seki, K.; Mori, W. Syntheses and Characterization of Microporous Coordination Polymers with Open Frameworks. J. Phys. Chem. B 2002, 106, 1380–1385. [Google Scholar] [CrossRef]
- Wang, H.; Getzschmann, J.; Senkovska, I.; Kaskel, S. Structural Transformation and High Pressure Methane Adsorption of Co2(1,4-bdc)2dabco. Microporous Mesoporous Mater. 2008, 116, 653–657. [Google Scholar] [CrossRef]
- Senkovska, I.; Kaskel, S. High Pressure Methane Adsorption in the Metal-Organic Frameworks Cu3(btc)2, Zn2(bdc)2dabco, and Cr3F(H2O)2O(bdc)3. Microporous Mesoporous Mater. 2008, 112, 108–115. [Google Scholar] [CrossRef]
- Lee, J.Y.; Pan, L.; Huang, X.; Emge, T.J.; Li, J. A Systematic Approach to Building Highly Porous, Noninterpenetrating Metal-Organic Frameworks with a Large Capacity for Adsorbing H2 and CH4. Adv. Funct. Mater. 2011, 21, 993–998. [Google Scholar] [CrossRef]
- Senthil Raja, D.; Pan, C.C.; Chen, C.W.; Kang, Y.H.; Chen, J.J.; Lin, C.H. Synthesis of Mixed Ligand and Pillared Paddlewheel MOFs Using Waste Polyethylene Terephthalate Material as Sustainable Ligand Source. Microporous Mesoporous Mater. 2016, 231, 186–191. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, W.; Yildirim, T. High-Capacity Methane Storage in Metal—Organic Frameworks M2(dhtp): The Important Role of Open Metal Sites. J. Am. Chem. Soc. 2009, 131, 4995–5000. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Z.; Chen, L.; Liu, G.; Yuan, Z.Y.; Li, B.F.; Zhang, X.; Wei, J.Q. Porous Metal-Organic Frameworks for Methane Storage and Capture: Status and Challenges. New Carbon Mater. 2021, 36, 468–496. [Google Scholar] [CrossRef]
- Hu, T.D.; Jiang, Y.; Ding, Y.H. Computational Screening of Metal-Substituted HKUST-1 Catalysts for Chemical Fixation of Carbon Dioxide into Epoxides. J. Mater. Chem. A 2019, 7, 14825–14834. [Google Scholar] [CrossRef]
- Jiang, Y.; Hu, T.D.; Yu, L.Y.; Ding, Y.H. A More Effective Catalysis of the CO2 Fixation with Aziridines: Computational Screening of Metal-Substituted HKUST-1. Nanoscale Adv. 2021, 3, 4079–4088. [Google Scholar] [CrossRef] [PubMed]
- Zong, S.; Zhang, Y.; Lu, N.; Ma, P.; Wang, J.; Shi, X.R. A DFT Screening of M-HKUST-1 MOFs for Nitrogen-Containing Compounds Adsorption. Nanomaterials 2018, 8, 958. [Google Scholar] [CrossRef] [PubMed]
- Goings, J.J.; Ohlsen, S.M.; Blaisdell, K.M.; Schofield, D.P. Sorption of H2 to Open Metal Sites in a Metal-Organic Framework: A Symmetry-Adapted Perturbation Theory Analysis. J. Phys. Chem. A 2014, 118, 7411–7417. [Google Scholar] [CrossRef] [PubMed]
- Avci, G.; Velioglu, S.; Keskin, S. In Silico Design of Metal Organic Frameworks with Enhanced CO2 Separation Performances: Role of Metal Sites. J. Phys. Chem. C 2019, 123, 28255–28265. [Google Scholar] [CrossRef]
- Fu, Z.; Yi, J.; Chen, Y.; Liao, S.; Guo, N.; Dai, J.; Yang, G.; Lian, Y.; Wu, X. From Interwoven to Noninterpenetration: Crystal Structural Motifs of Two New Manganese-Organic Frameworks Mediated by the Substituted Group of the Bridging Ligand. Eur. J. Inorg. Chem. 2008, 2008, 628–634. [Google Scholar] [CrossRef]
- Lestari, W.W.; Ni’Maturrohmah, D.; Putra, R.; Suwarno, H.; Arrozi, U.S.F. Enhanced Hydrogen Sorption Properties over Mg2+ Modified Solvothermal Synthesized HKUST-1 (Mg2+/HKUST-1). IOP Conf. Ser. Mater. Sci. Eng. 2019, 509, 012152. [Google Scholar] [CrossRef]
- Świrk, K.; Delahay, G.; Zaki, A.; Adil, K.; Cadiau, A. Facile Modifications of HKUST-1 by V, Nb and Mn for Low-Temperature Selective Catalytic Reduction of Nitrogen Oxides by NH3. Catal. Today 2022, 384–386, 25–32. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Wojtas, L.; Eddaoudi, M.; Zaworotko, M.J. Template-Directed Synthesis of Nets Based upon Octahemioctahedral Cages That Encapsulate Catalytically Active Metalloporphyrins. J. Am. Chem. Soc. 2012, 134, 928–933. [Google Scholar] [CrossRef]
- Huynh, N.T.X.; Na, O.M.; Chihaia, V.; Son, D.N. A Computational Approach towards Understanding Hydrogen Gas Adsorption in Co-MIL-88A. RSC Adv. 2017, 7, 39583–39593. [Google Scholar] [CrossRef]
- Vinh, N.Q.; Duyen, N.T.M.; Tran, N.L.B.; Van Nghia, N.; Vien, L.T.T.V.; Thanh, H.T.M.; Huynh, N.T.X. Computational Study on Enhancing SO2 Capture Capacity of M2(BDC)2TED (M = Mg, V, Co, or Ni). Quy Nhon Univ. J. Sci. 2024, 18, 91–100. [Google Scholar]
- Huynh, N.T.X.; Chihaia, V.; Son, D.N. Enhancing Hydrogen Storage by Metal Substitution in MIL-88A Metal-Organic Framework. Adsorption 2020, 26, 509–519. [Google Scholar] [CrossRef]
- Dubbeldam, D.; Calero, S.; Ellis, D.E.; Snurr, R.Q. RASPA: Molecular Simulation Software for Adsorption and Diffusion in Flexible Nanoporous Materials. Mol. Simul. 2016, 42, 81–101. [Google Scholar] [CrossRef]
- Lorentz, H.A. Ueber Die Anwendung Des Satzes Vom Virial in Der Kinetischen Theorie Der Gase. Ann. Phys. 1881, 248, 127–136. [Google Scholar] [CrossRef]
- Berthelot, D. Sur Le Mélange Des Gaz. Acad. Sci. 1898, 126, 1703–1855. [Google Scholar]
- Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 1. United-Atom Description of n-Alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Lyubchyk, A.; Esteves, I.A.A.C.; Cruz, F.J.A.L.; Mota, J.P.B. Experimental and Theoretical Studies of Supercritical Methane Adsorption in the Mil-53(Al) Metal Organic Framework. J. Phys. Chem. C 2011, 115, 20628–20638. [Google Scholar] [CrossRef]
- Dubbeldam, D.; Calero, S.; Vlugt, T.J.H.; Krishna, R.; Maesen, T.L.M.; Smit, B. United Atom Force Field for Alkanes in Nanoporous Materials. J. Phys. Chem. B 2004, 108, 12301–12313. [Google Scholar] [CrossRef]
- Dubbeldam, D.; Calero, S.; Vlugt, T.J.H.; Krishna, R.; Maesen, T.L.M.; Beerdsen, E.; Smit, B. Force Field Parametrization through Fitting on Inflection Points in Isotherms. Phys. Rev. Lett. 2004, 93, 088302. [Google Scholar] [CrossRef]
- Klimeš, J.; Bowler, D.R.; Michaelides, A. Chemical Accuracy for the Van der Waals Density Functional. J. Phys. Condens. Matter 2010, 22, 022201. [Google Scholar] [CrossRef] [PubMed]
- Klimeš, J.; Bowler, D.R.; Michaelides, A. Van der Waals Density Functionals Applied to Solids. Phys. Rev. B—Condens. Matter Mater. Phys. 2011, 83, 195131. [Google Scholar] [CrossRef]
- Dion, M.; Rydberg, H.; Schröder, E.; Langreth, D.C.; Lundqvist, B.I. Van der Waals Density Functional for General Geometries. Phys. Rev. Lett. 2004, 92, 246401. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B—Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Pack, J.D.; Monkhorst, H.J. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A Fast and Robust Algorithm for Bader Decomposition of Charge Density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved Grid-Based Algorithm for Bader Charge Allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wu, Y.; Wu, J.; Lv, D.; Yan, J.; Wang, X.; Chen, X.; Liu, Z.; Peng, J. A Microporous Zn(bdc)(ted)0.5 with Super High Ethane Uptake for Efficient Selective Adsorption and Separation of Light Hydrocarbons. Molecules 2023, 28, 6000. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Medina, J.; Mass-González, G.; Pacheco-Londoño, L.; Hernández-Rivera, S.P.; Fu, R.; Hernández-Maldonado, A.J.; Hern, A.J.; Hern, S.P. Long and Local Range Structural Changes in M[(bdc)(ted)0.5] (M = Zn, Ni or Cu) Metal Organic Frameworks upon Spontaneous Thermal Dispersion of LiCl and Adsorption of Carbon Dioxide. Microporous Mesoporous Mater. 2015, 212, 8–17. [Google Scholar] [CrossRef]
- Mason, J.A.; Oktawiec, J.; Taylor, M.K.; Hudson, M.R.; Rodriguez, J.; Bachman, J.E.; Gonzalez, M.I.; Cervellino, A.; Guagliardi, A.; Brown, C.M.; et al. Methane Storage in Flexible Metal–Organic Frameworks with Intrinsic Thermal Management. Nature 2015, 527, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Howe, J.D.; Lin, L.C.; Smit, B.; Neaton, J.B. Small-Molecule Adsorption in Open-Site Metal-Organic Frameworks: A Systematic Density Functional Theory Study for Rational Design. Chem. Mater. 2015, 27, 668–678. [Google Scholar] [CrossRef]
- Vlaisavljevich, B.; Huck, J.; Hulvey, Z.; Lee, K.; Mason, J.A.; Neaton, J.B.; Long, J.R.; Brown, C.M.; Alfè, D.; Michaelides, A.; et al. Performance of van Der Waals Corrected Functionals for Guest Adsorption in the M2(dobdc) Metal-Organic Frameworks. J. Phys. Chem. A 2017, 121, 4139–4151. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoms | (K) | (Å) |
|---|---|---|
| H a | 7.65 | 2.85 |
| C a | 47.86 | 3.47 |
| N a | 38.95 | 3.26 |
| O a | 48.16 | 3.03 |
| Mg b | 55.86 | 2.69 |
| Mn b | 6.54 | 2.64 |
| Fe b | 6.54 | 2.59 |
| Co b | 7.05 | 2.56 |
| Ni b | 7.55 | 2.52 |
| Cu b | 2.52 | 3.11 |
| Zn b | 62.40 | 2.46 |
| CH4 (Dubbeldam et al.) [62,63] | 158.50 | 3.72 |
| CH4 (TraPPE-UA) (Martin et al.) [60] | 148.00 | 3.73 |
| MOFs | (m2/g) | Vp (cm3/g) | Pore Size (Å) |
|---|---|---|---|
| Cu(DABCO) | 1561 | 0.71 | 8.53 |
| Fe(DABCO) | 1571 | 0.72 | 8.45 |
| Zn(DABCO) | 1597 | 0.73 | 8.72 |
| Co(DABCO) | 1628 | 0.74 | 8.61 |
| Ni(DABCO) | 1686 | 0.76 | 8.69 |
| Mn(DABCO) | 1701 | 0.77 | 8.71 |
| Mg(DABCO) | 1931 | 0.87 | 8.72 |
| Cu(DABCO) | 1631 [30], 1572 [76] | 0.63 [30], 0.65 [76] | 7.98 [76] |
| Zn(DABCO) | 1781 [30], 1523 [76], 1904 [75] | 0.65 [30], 0.56 [76], 0.73 [75] | 8.00 [75] |
| Co(DABCO) | 1708 [30], 1600 [39] | 0.62 [30], 0.82 [39] | |
| Ni(DABCO) | 1905 [30], 1698 [76] | 0.76 [30], 0.71 [76] | 7.88 [76] |
| HKUST-1 [6] | 1850 | 0.78 |
| MOFs | CH4 Adsorption | (kJ/mol) | |||||
|---|---|---|---|---|---|---|---|
| Gravimetric Uptake | Volumetric Uptake cc(STP)/cc | ||||||
| mg/g | cm3/g | ||||||
| Cu(DABCO) | 182.39 | 135.12 | 254.83 | 188.79 | 220.26 | 163.17 | 17.14 |
| Fe(DABCO) | 186.58 | 138.71 | 260.69 | 193.80 | 222.14 | 165.14 | 17.19 |
| Zn(DABCO) | 189.59 | 141.12 | 264.89 | 197.17 | 226.30 | 168.44 | 17.21 |
| Co(DABCO) | 194.05 | 143.93 | 271.12 | 201.09 | 227.03 | 168.39 | 17.17 |
| Ni(DABCO) | 199.90 | 148.48 | 279.30 | 207.46 | 228.68 | 169.86 | 17.10 |
| Mn(DABCO) | 202.22 | 150.06 | 282.53 | 209.65 | 228.94 | 169.89 | 17.13 |
| Mg(DABCO) | 231.39 | 172.30 | 323.29 | 240.73 | 231.43 | 172.33 | 17.27 |
| Co(DABCO) [39] | 14 wt.% (75 bar) a | ||||||
| Cu(DABCO) [38] | ca. 185 (35 bar) | 188 (35 bar) a | 16.25 | ||||
| HKUST-1 [6] | 216 (184) b | 178 (165) c | 267 (227) b | 220 (204) b | 17 | ||
| PCN-14 [6] | 197 (169) b | 157 (146) c | 230 (195) b | 183 (171) b | 18.7 | ||
| MIL-101(Cr) | 215 (150) b | ||||||
| Al-soc-MOF-1 [13,19] | 420 (263) b | - | 579 (362) b | 197 (123) b | 10.5 | ||
| NJU-Bai 43 [26] | 396 (315) b | 254 (202) b | 14.45 | ||||
| UTSA-76 [27] | 263 (216) b | 302 (257) b | 257 (211) b | 15.44 | |||
| UTSA-110a [8] | 402 (312) b | 241 (187) b | 15.49 | ||||
| DOE | 500 | 263 | |||||
| Temperature (K) | (Max) | |||
|---|---|---|---|---|
| 35 bar | 65 bar | 100 bar (Max) | ||
| 238 | 270.61 | 284.95 | 292.15 | 242.58 (30.0 bar) |
| 248 | 257.89 | 274.09 | 282.54 | 230.32 (35.0 bar) |
| 258 | 243.37 | 263.09 | 274.05 | 218.13 (37.5 bar) |
| 268 | 228.56 | 250.63 | 263.34 | 206.24 (42.5 bar) |
| 278 | 214.01 | 238.87 | 252.54 | 194.15 (47.5 bar) |
| 288 | 199.34 | 226.72 | 242.40 | 183.14 (52.5 bar) |
| 298 | 184.99 | 214.38 | 231.43 | 172.71 (60.0 bar) |
| Adsorption Configurations | Site 1 (Metal Cluster) | Site 2 (M-O-C Cluster—TED Interface) | ||
|---|---|---|---|---|
| Mg | −76.58 | 2.92 | −76.23 | 2.46 |
| Mn | −77.73 | 3.08 | −76.15 | 2.58 |
| Fe | −76.97 | 3.00 | −75.78 | 2.50 |
| Co | −76.56 | 2.97 | −76.55 | 2.59 |
| Ni | −73.35 | 3.01 | −75.40 | 2.46 |
| Cu | −20.31 | 2.87 | −45.33 | 2.74 |
| Zn | −61.46 | 2.89 | −62.13 | 2.49 |
| CH4@M(DABCO) at Site 1 | M = Mg | M = Mn | M = Fe | M = Co | M = Ni | M = Cu | M = Zn | |
|---|---|---|---|---|---|---|---|---|
| CH4 molecule | 4 H | +0.011 | −0.011 | +0.037 | −0.028 | −0.011 | −0.007 | −0.001 |
| 1 C | −0.018 | +0.005 | −0.030 | +0.016 | +0.001 | +0.012 | −0.006 | |
| Total | −0.007 | −0.006 | +0.007 | −0.012 | −0.010 | −0.011 | −0.007 | |
| M(DABCO) | 20 H | −0.008 | +0.053 | −0.043 | +0.020 | −0.053 | −0.007 | +0.013 |
| 12 C1 | −0.052 | +0.021 | +0.017 | +0.035 | +0.151 | +0.165 | −0.091 | |
| 4 C2 | +0.081 | +0.024 | +0.023 | −0.029 | −0.160 | −0.178 | +0.108 | |
| 6 C3 | −0.016 | −0.056 | +0.016 | −0.021 | −0.003 | −0.034 | +0.033 | |
| 2 N | +0.014 | +0.011 | −0.0004 | −0.010 | +0.006 | −0.002 | −0.019 | |
| 8 O | −0.013 | −0.036 | −0.010 | +0.059 | +0.066 | +0.099 | −0.043 | |
| 2 M | +0.001 | −0.012 | −0.009 | −0.042 | +0.003 | −0.035 | +0.004 | |
| Total | +0.007 | +0.006 | −0.006 | +0.012 | +0.010 | +0.011 | +0.007 | |
| CH4@M(DABCO) at Site 2 | M = Mg | M = Mn | M = Fe | M = Co | M = Ni | M = Cu | M = Zn | |
|---|---|---|---|---|---|---|---|---|
| CH4 molecule | 4 H | +0.032 | +0.022 | +0.033 | −0.004 | +0.028 | +0.022 | −0.043 |
| 1 C | −0.038 | −0.027 | −0.024 | −0.001 | −0.036 | −0.031 | +0.036 | |
| Total | −0.006 | −0.005 | +0.009 | −0.005 | −0.008 | −0.009 | −0.007 | |
| M(DABCO) | 20 H | +0.060 | +0.008 | −0.077 | +0.061 | +0.009 | +0.034 | +0.020 |
| 12 C1 | −0.044 | +0.037 | +0.069 | −0.016 | +0.130 | +0.096 | −0.089 | |
| 4 C2 | −0.011 | +0.007 | −0.015 | −0.023 | −0.170 | −0.162 | +0.111 | |
| 6 C3 | −0.032 | −0.020 | +0.0003 | −0.042 | −0.022 | −0.001 | +0.023 | |
| 2 N | +0.016 | +0.015 | +0.006 | +0.002 | +0.006 | +0.032 | −0.034 | |
| 8 O | +0.016 | −0.034 | +0.012 | +0.047 | +0.054 | +0.042 | −0.054 | |
| 2 M | +0.001 | −0.007 | −0.001 | −0.025 | +0.001 | −0.032 | +0.031 | |
| Total | +0.006 | +0.005 | −0.006 | +0.005 | +0.008 | +0.009 | +0.007 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huynh, N.T.X.; Nguyen-Van, T.; Tran, N.L.B.; Nghia, N.V.; Thanh, P.N. Exploring Methane Storage Capacities of M2(BDC)2(DABCO) Sorbents: A Multiscale Computational Study. Crystals 2024, 14, 596. https://doi.org/10.3390/cryst14070596
Huynh NTX, Nguyen-Van T, Tran NLB, Nghia NV, Thanh PN. Exploring Methane Storage Capacities of M2(BDC)2(DABCO) Sorbents: A Multiscale Computational Study. Crystals. 2024; 14(7):596. https://doi.org/10.3390/cryst14070596
Chicago/Turabian StyleHuynh, Nguyen Thi Xuan, Tue Nguyen-Van, Nguyen Le Bao Tran, Nguyen Van Nghia, and Pham Ngoc Thanh. 2024. "Exploring Methane Storage Capacities of M2(BDC)2(DABCO) Sorbents: A Multiscale Computational Study" Crystals 14, no. 7: 596. https://doi.org/10.3390/cryst14070596
APA StyleHuynh, N. T. X., Nguyen-Van, T., Tran, N. L. B., Nghia, N. V., & Thanh, P. N. (2024). Exploring Methane Storage Capacities of M2(BDC)2(DABCO) Sorbents: A Multiscale Computational Study. Crystals, 14(7), 596. https://doi.org/10.3390/cryst14070596