
Exploring the Premelting Transition through Molecular Simulations Powered by Neural Network Potentials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Neural Network Potential
2.2. Simulation Settings
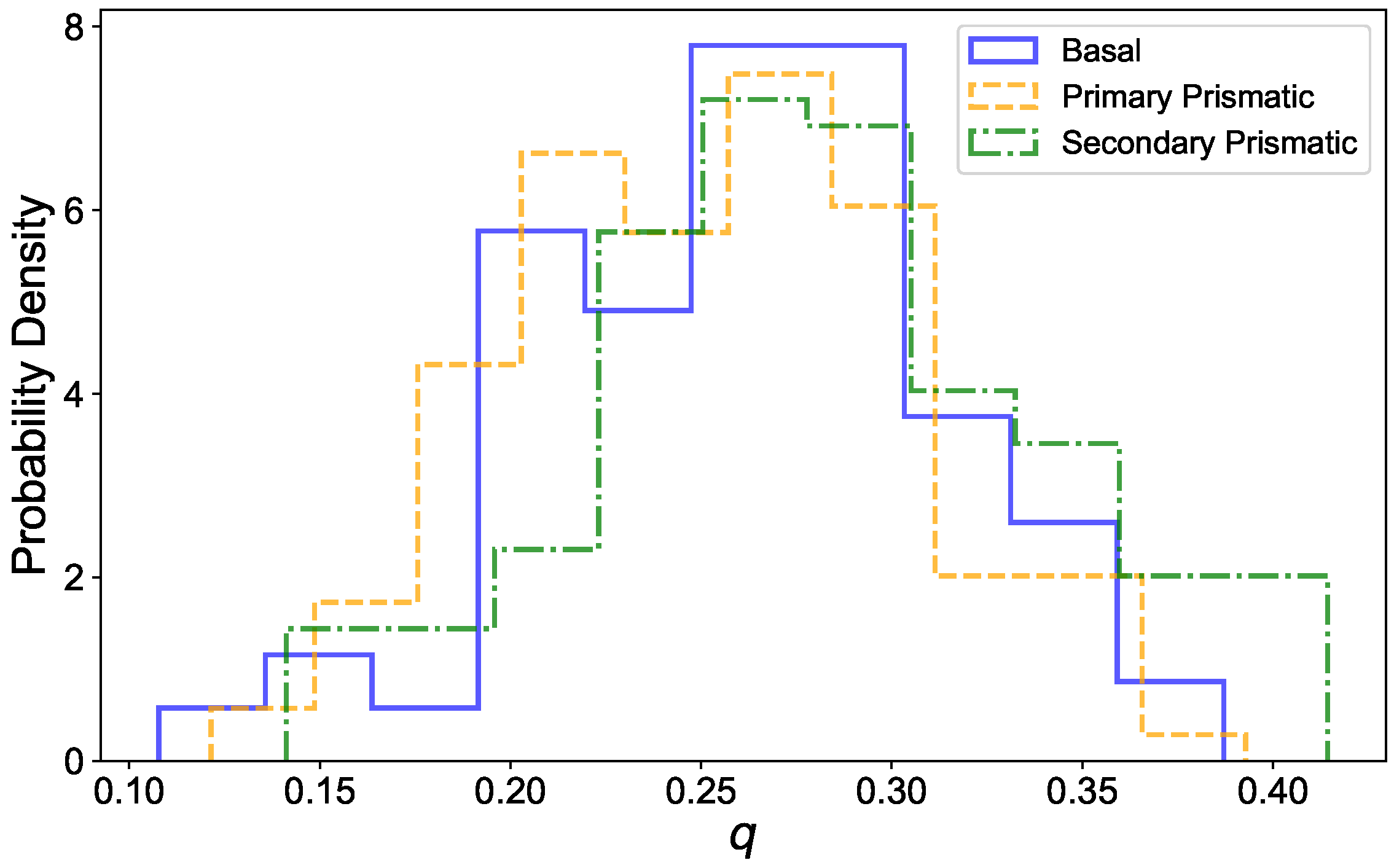
2.3. Local Structural Order Parameter
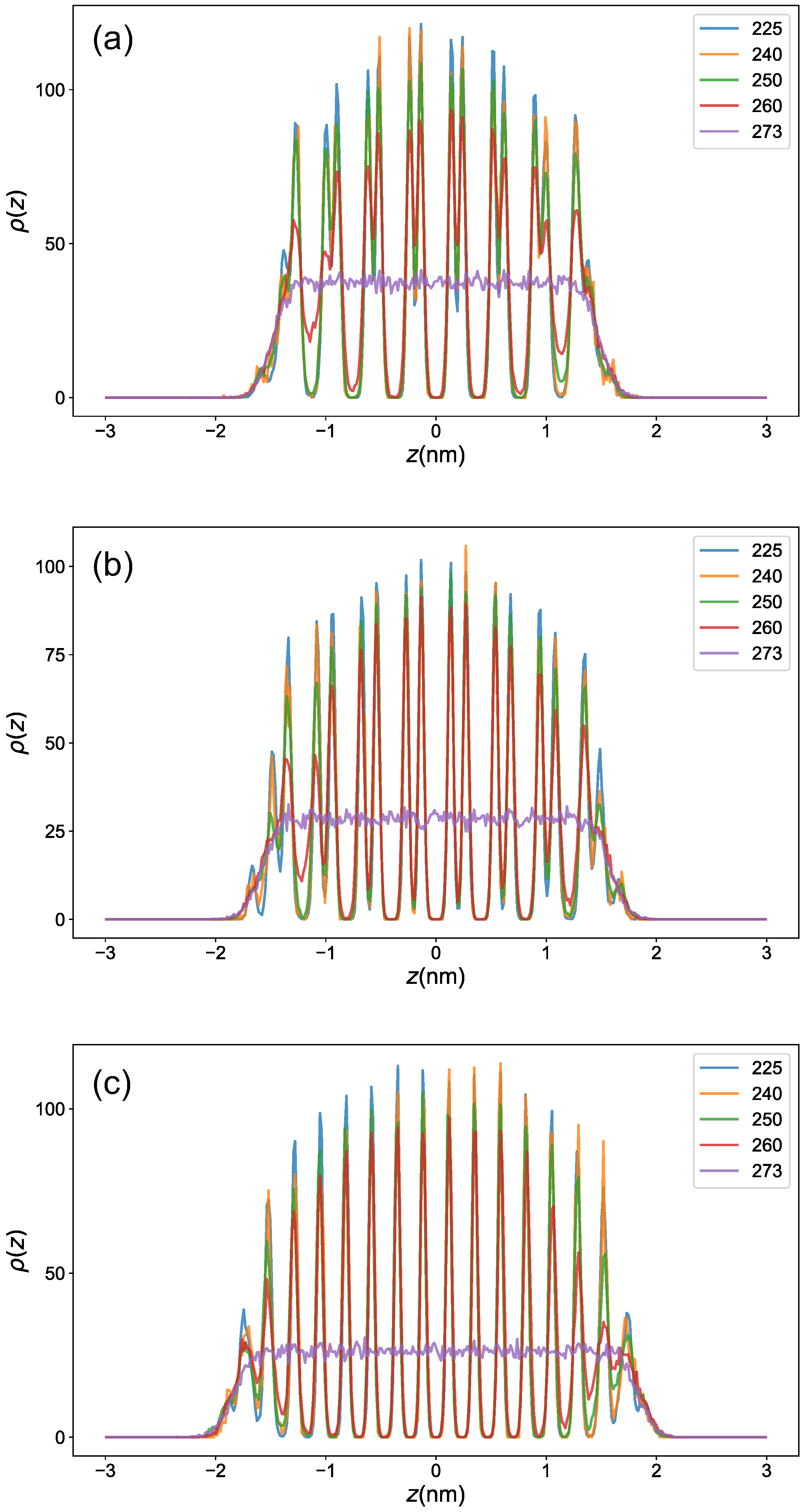
2.4. Density Profile across the Interface
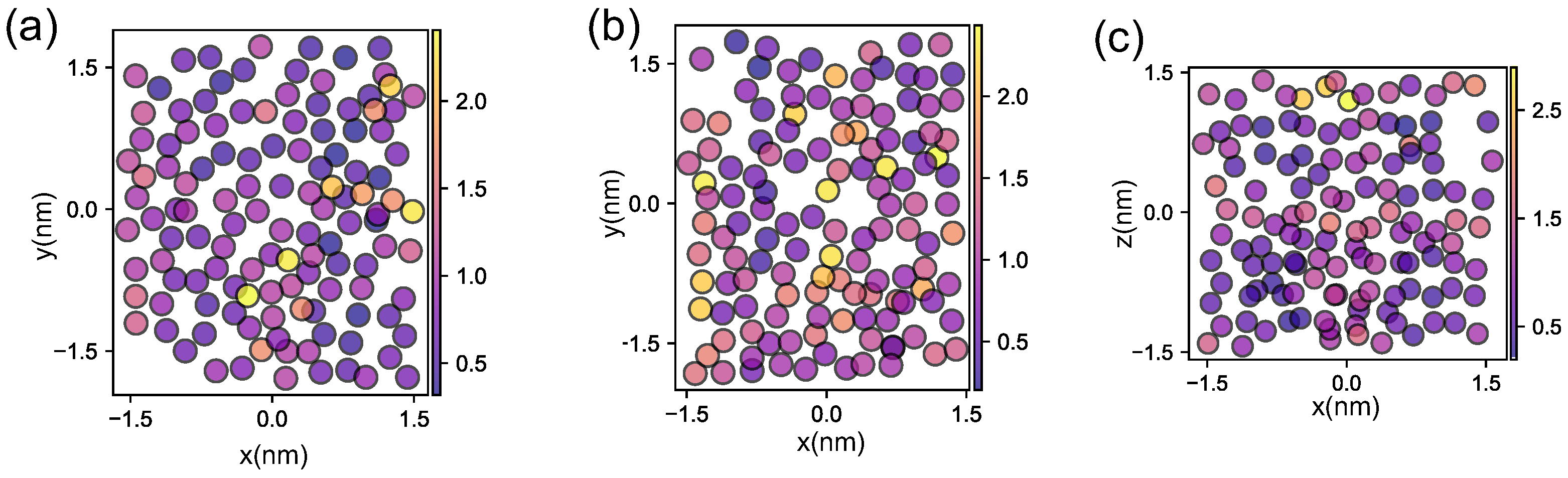
2.5. Propensity
3. Results
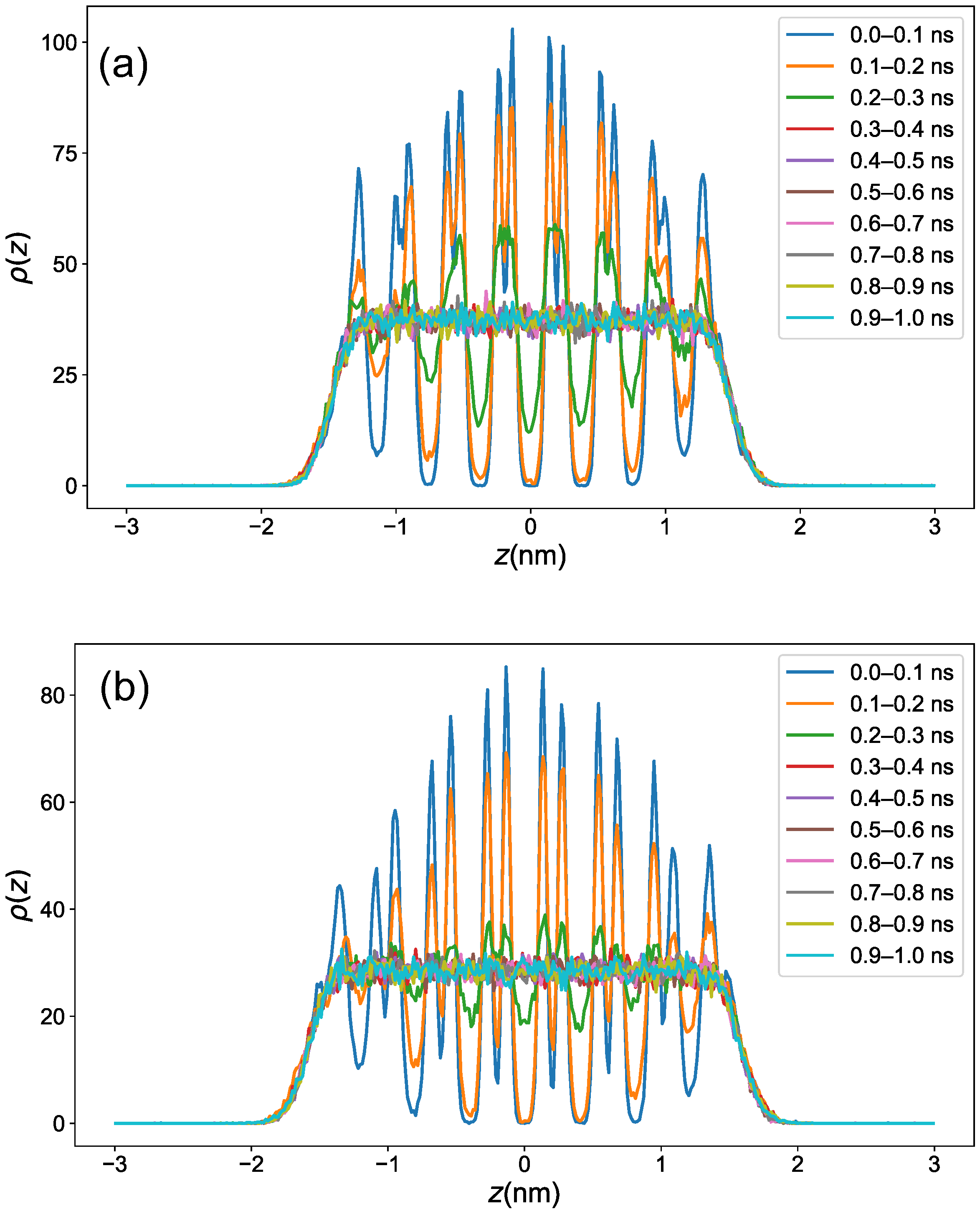
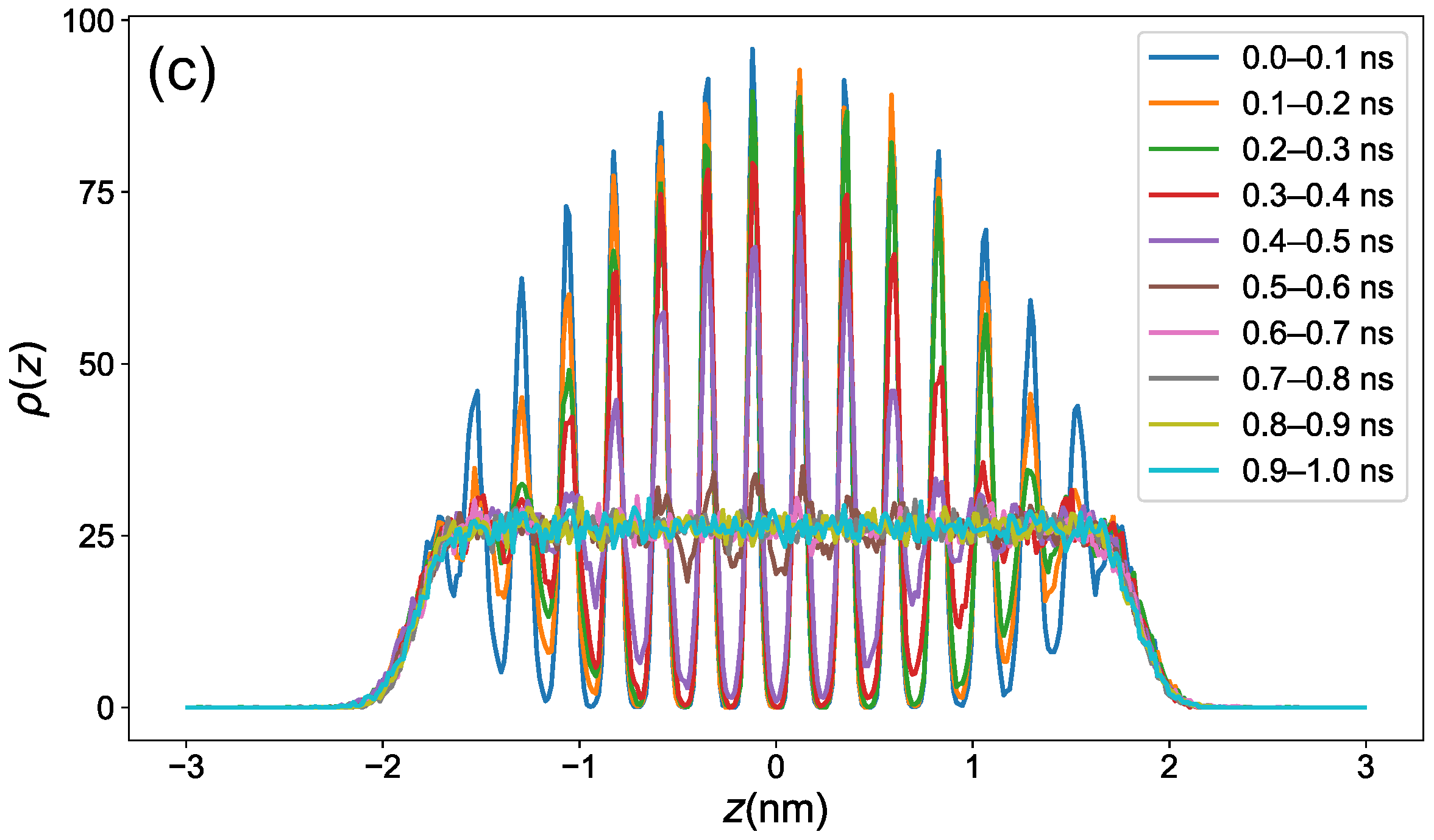
3.1. Equilibration
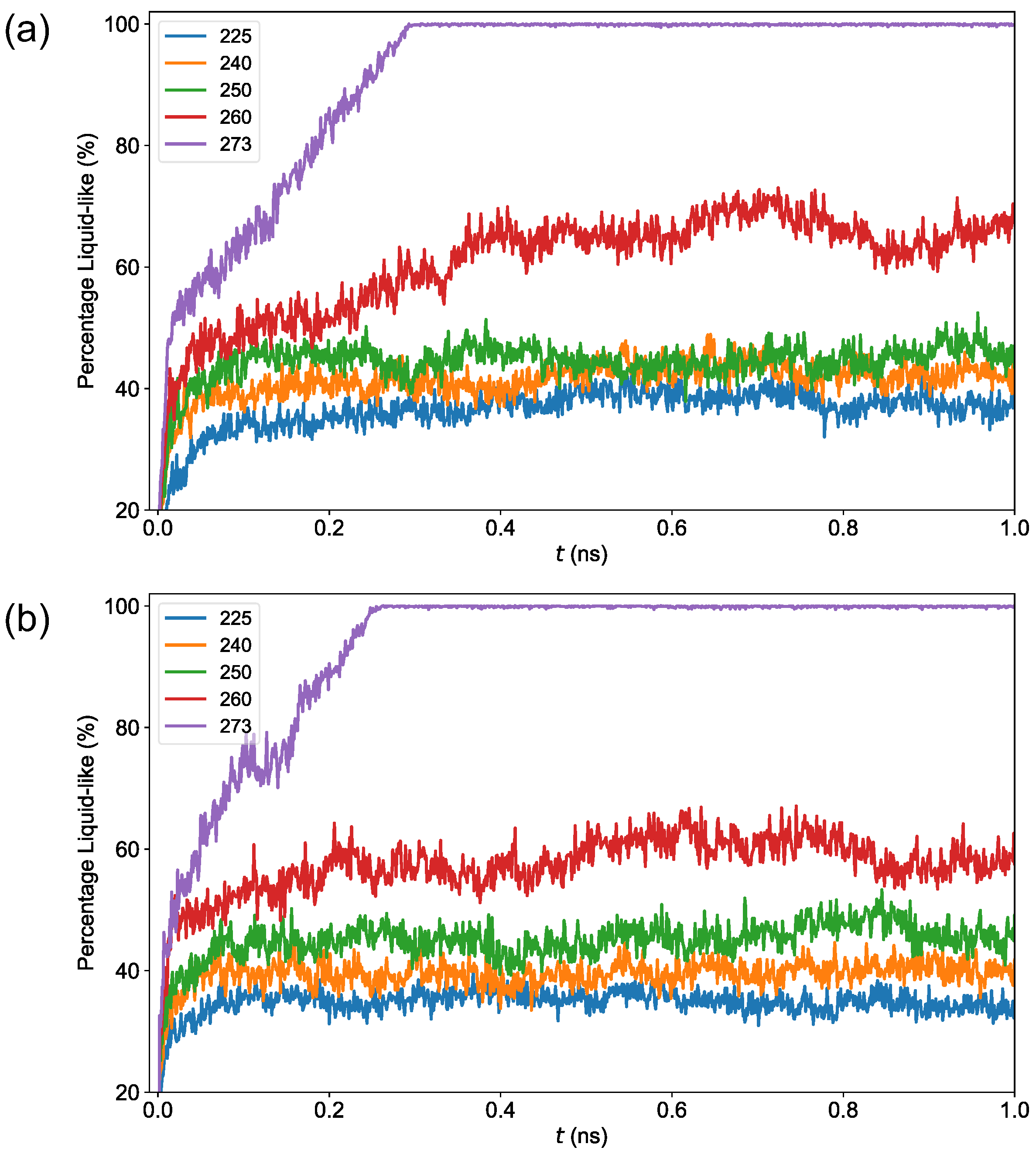
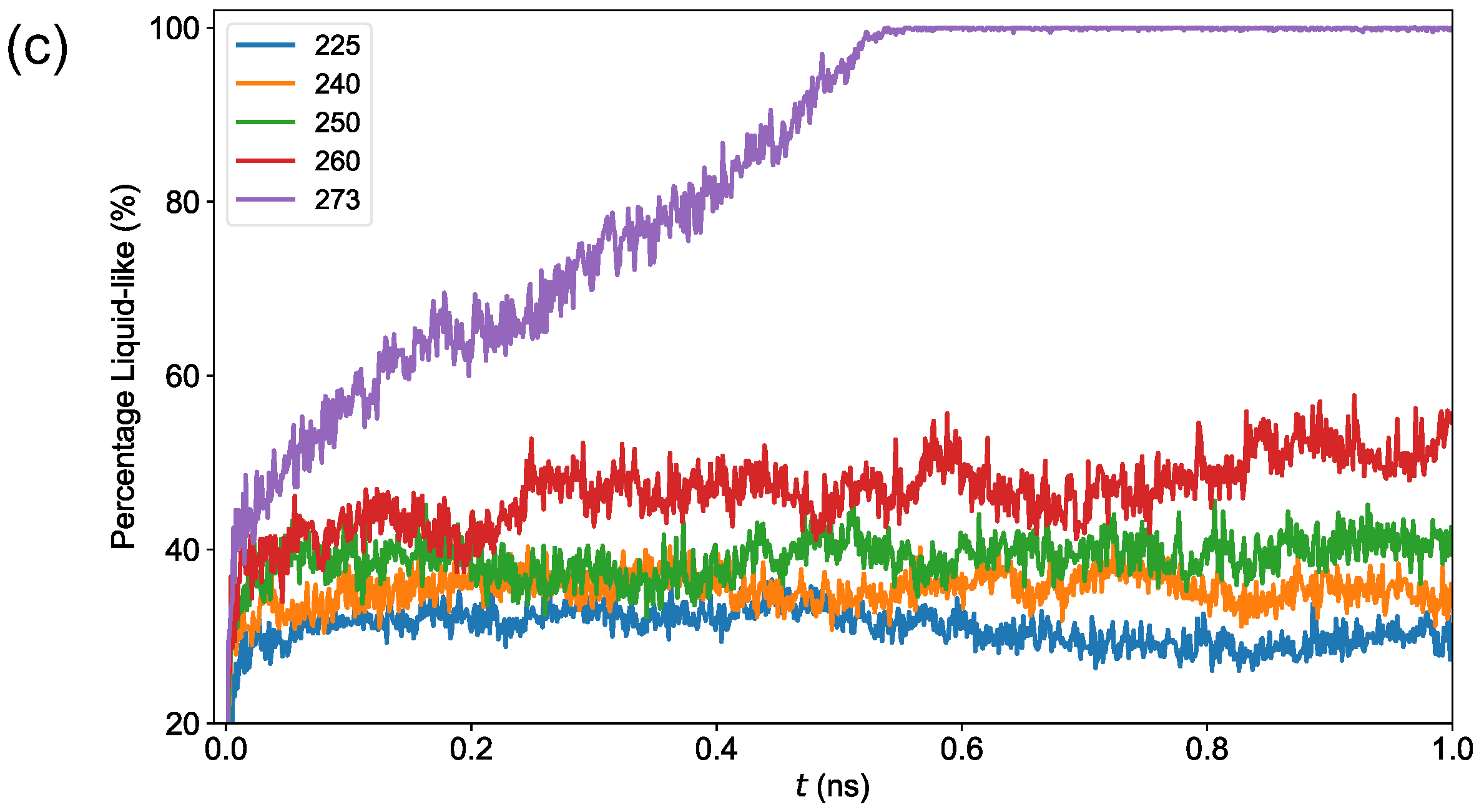
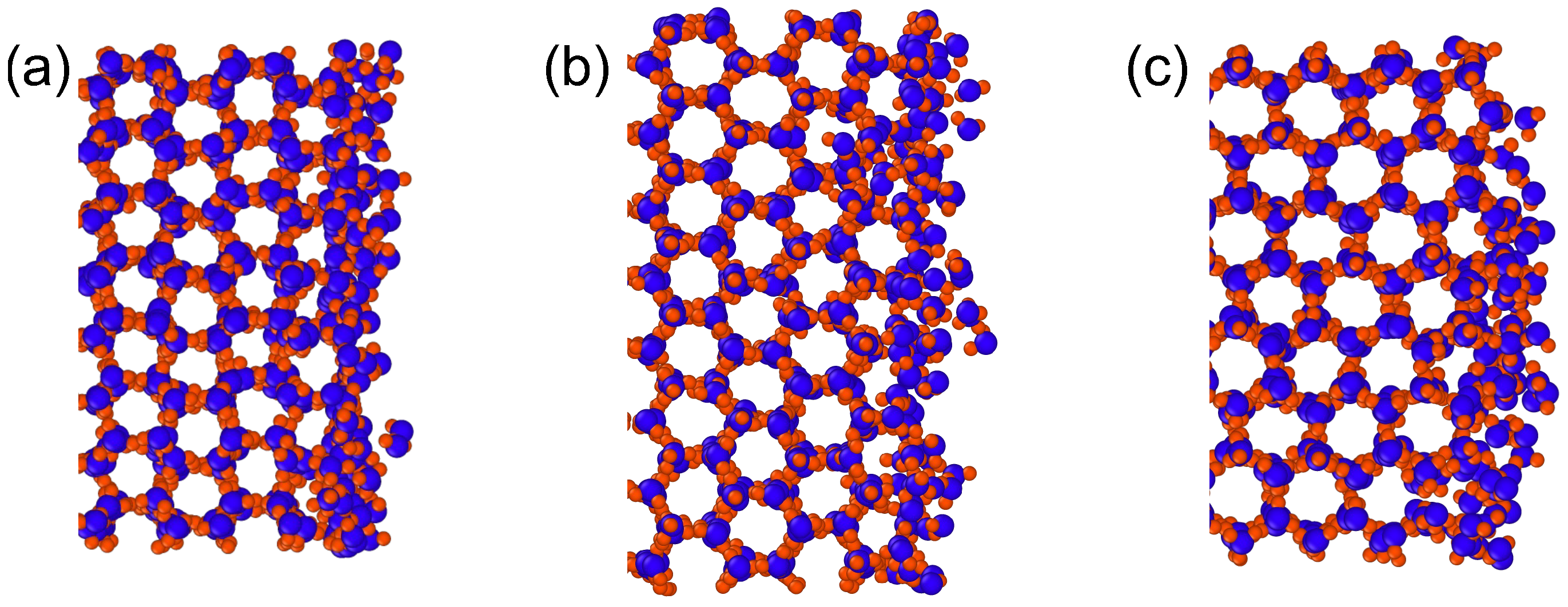
3.2. Premelting Transition
3.3. Dynamic Heterogeneity
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Faraday, M. Experimental Researches in Chemistry and Physics; Taylor and Francis: London, UK, 1859. [Google Scholar]
- Kouchi, A.; Furukawa, Y.; Kuroda, T. X-ray diffraction pattern of quasi-liquid layer on ice crystal surface. J. Phys. Colloq. 1987, 48, C1-675–C1-677. [Google Scholar] [CrossRef]
- Pluis, B.; Taylor, T.N.; Frenkel, D.; Van Der Veen, J.F. Role of long-range interactions in the melting of a metallic surface. Phys. Rev. B 1989, 40, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Dash, J.G.; Mason, B.L.; Wettlaufer, J.S. Theory of charge and mass transfer in ice-ice collisions. J. Geophys. Res. Atmos. 2001, 106, 20395–20402. [Google Scholar] [CrossRef]
- Bianco, R.; Hynes, J.T. Heterogeneous Reactions Important in Atmospheric Ozone Depletion: A Theoretical Perspective. Accounts Chem. Res. 2006, 39, 159–165. [Google Scholar] [CrossRef]
- Abbatt, J.P.D. Interactions of Atmospheric Trace Gases with Ice Surfaces: Adsorption and Reaction. Chem. Rev. 2003, 103, 4783–4800. [Google Scholar] [CrossRef]
- Conklin, M.H.; Sommerfeld, R.A.; Kay Laird, S.; Villinski, J.E. Sulfur dioxide reactions on ice surfaces: Implications for dry deposition to snow. Atmos. Environment. Part A. Gen. Top. 1993, 27, 159–166. [Google Scholar] [CrossRef]
- Hudait, A.; Allen, M.T.; Molinero, V. Sink or Swim: Ions and Organics at the Ice-Air Interface. J. Am. Chem. Soc. 2017, 139, 10095–10103. [Google Scholar] [CrossRef]
- Pickering, I.; Paleico, M.; Sirkin, Y.A.; Scherlis, D.A.; Factorovich, M.H. Grand Canonical Investigation of the Quasi Liquid Layer of Ice: Is It Liquid? J. Phys. Chem. B 2018, 122, 4880–4890. [Google Scholar] [CrossRef]
- Sazaki, G.; Zepeda, S.; Nakatsubo, S.; Yokomine, M.; Furukawa, Y. Quasi-liquid layers on ice crystal surfaces are made up of two different phases. Proc. Natl. Acad. Sci. USA 2012, 109, 1052–1055. [Google Scholar] [CrossRef] [PubMed]
- Sazaki, G.; Asakawa, H.; Nagashima, K.; Nakatsubo, S.; Furukawa, Y. How do Quasi-Liquid Layers Emerge from Ice Crystal Surfaces? Cryst. Growth Des. 2013, 13, 1761–1766. [Google Scholar] [CrossRef]
- Asakawa, H.; Sazaki, G.; Nagashima, K.; Nakatsubo, S.; Furukawa, Y. Prism and Other High-Index Faces of Ice Crystals Exhibit Two Types of Quasi-Liquid Layers. Cryst. Growth Des. 2015, 15, 3339–3344. [Google Scholar] [CrossRef]
- Murata, K.I.; Asakawa, H.; Nagashima, K.; Furukawa, Y.; Sazaki, G. Thermodynamic origin of surface melting on ice crystals. Proc. Natl. Acad. Sci. USA 2016, 113, E6741–E6748. [Google Scholar] [CrossRef]
- Dosch, H.; Lied, A.; Bilgram, J. Glancing-angle X-ray scattering studies of the premelting of ice surfaces. Surf. Sci. 1995, 327, 145–164. [Google Scholar] [CrossRef]
- Bluhm, H.; Ogletree, D.F.; Fadley, C.S.; Hussain, Z.; Salmeron, M. The premelting of ice studied with photoelectron spectroscopy. J. Physics: Condens. Matter 2002, 14, L227–L233. [Google Scholar] [CrossRef]
- Dash, J.G.; Rempel, A.W.; Wettlaufer, J.S. The physics of premelted ice and its geophysical consequences. Rev. Mod. Phys. 2006, 78, 695–741. [Google Scholar] [CrossRef]
- Dzyaloshinskii, I.; Lifshitz, E.; Pitaevskii, L. The general theory of van der Waals forces. Adv. Phys. 1961, 10, 165–209. [Google Scholar] [CrossRef]
- Elbaum, M.; Schick, M. Application of the theory of dispersion forces to the surface melting of ice. Phys. Rev. Lett. 1991, 66, 1713–1716. [Google Scholar] [CrossRef]
- Limmer, D.T.; Chandler, D. Premelting, fluctuations, and coarse-graining of water-ice interfaces. J. Chem. Phys. 2014, 141, 18C505. [Google Scholar] [CrossRef]
- Luengo-Márquez, J.; Izquierdo-Ruiz, F.; MacDowell, L.G. Intermolecular forces at ice and water interfaces: Premelting, surface freezing, and regelation. J. Chem. Phys. 2022, 157, 044704. [Google Scholar] [CrossRef] [PubMed]
- Sibley, D.N.; Llombart, P.; Noya, E.G.; Archer, A.J.; MacDowell, L.G. How ice grows from premelting films and water droplets. Nat. Commun. 2021, 12, 239. [Google Scholar] [CrossRef] [PubMed]
- Niblett, S.P.; Limmer, D.T. Ion Dissociation Dynamics in an Aqueous Premelting Layer. J. Phys. Chem. B 2021, 125, 2174–2181. [Google Scholar] [CrossRef]
- Paesani, F.; Voth, G.A. Quantum effects strongly influence the surface premelting of ice. J. Phys. Chem. C 2008, 112, 324–327. [Google Scholar] [CrossRef]
- Watkins, M.; Pan, D.; Wang, E.G.; Michaelides, A.; Vandevondele, J.; Slater, B. Large variation of vacancy formation energies in the surface of crystalline ice. Nat. Mater. 2011, 10, 794–798. [Google Scholar] [CrossRef]
- Behler, J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 2011, 134. [Google Scholar] [CrossRef]
- Behler, J. Four Generations of High-Dimensional Neural Network Potentials. Chem. Rev. 2021, 121, 10037–10072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H.; Car, R.; Weinan, E. Phase Diagram of a Deep Potential Water Model. Phys. Rev. Lett. 2021, 126, 236001. [Google Scholar] [CrossRef]
- Gao, A.; Remsing, R.C. Self-consistent determination of long-range electrostatics in neural network potentials. Nat. Commun. 2022, 13, 1572. [Google Scholar] [CrossRef]
- Ko, T.W.; Finkler, J.A.; Goedecker, S.; Behler, J. A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer. Nat. Commun. 2021, 12, 398. [Google Scholar] [CrossRef]
- Ko, T.W.; Finkler, J.A.; Goedecker, S.; Behler, J. Accurate Fourth-Generation Machine Learning Potentials by Electrostatic Embedding. J. Chem. Theory Comput. 2023, 19, 3567–3579. [Google Scholar] [CrossRef]
- Deringer, V.L.; Bartók, A.P.; Bernstein, N.; Wilkins, D.M.; Ceriotti, M.; Csányi, G. Gaussian Process Regression for Materials and Molecules. Chem. Rev. 2021, 121, 10073–10141. [Google Scholar] [CrossRef] [PubMed]
- Mailoa, J.P.; Kornbluth, M.; Batzner, S.; Samsonidze, G.; Lam, S.T.; Vandermause, J.; Ablitt, C.; Molinari, N.; Kozinsky, B. A fast neural network approach for direct covariant forces prediction in complex multi-element extended systems. Nat. Mach. Intell. 2019, 1, 471–479. [Google Scholar] [CrossRef]
- Bapst, V.; Keck, T.; Grabska-Barwińska, A.; Donner, C.; Cubuk, E.D.; Schoenholz, S.S.; Obika, A.; Nelson, A.W.R.; Back, T.; Hassabis, D.; et al. Unveiling the predictive power of static structure in glassy systems. Nat. Phys. 2020, 16, 448–454. [Google Scholar] [CrossRef]
- Wohlfahrt, O.; Dellago, C.; Sega, M. Ab initio structure and thermodynamics of the RPBE-D3 water/vapor interface by neural-network molecular dynamics. J. Chem. Phys. 2020, 153, 144710. [Google Scholar] [CrossRef]
- Morawietz, T.; Singraber, A.; Dellago, C.; Behler, J. How van der waals interactions determine the unique properties of water. Proc. Natl. Acad. Sci. United States Am. 2016, 113, 8368–8373. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Singraber, A.; Behler, J.; Dellago, C. Library-Based LAMMPS Implementation of High-Dimensional Neural Network Potentials. J. Chem. Theory Comput. 2019, 15, 1827–1840. [Google Scholar] [CrossRef]
- Singraber, A.; Morawietz, T.; Behler, J.; Dellago, C. Parallel Multistream Training of High-Dimensional Neural Network Potentials. J. Chem. Theory Comput. 2019, 15, 3075–3092. [Google Scholar] [CrossRef]
- Matsumoto, M.; Yagasaki, T.; Tanaka, H. GenIce: Hydrogen-Disordered Ice Generator. J. Comput. Chem. 2018, 39, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Steinhardt, P.J.; Nelson, D.R.; Ronchetti, M. Bond-orientational order in liquids and glasses. Phys. Rev. B 1983, 28, 784–805. [Google Scholar] [CrossRef]
- Lechner, W.; Dellago, C. Accurate determination of crystal structures based on averaged local bond order parameters. J. Chem. Phys. 2008, 129, 114707. [Google Scholar] [CrossRef] [PubMed]
- Widmer-Cooper, A.; Harrowell, P. On the study of collective dynamics in supercooled liquids through the statistics of the isoconfigurational ensemble. J. Chem. Phys. 2007, 126, 154503. [Google Scholar] [CrossRef] [PubMed]
- Berthier, L.; Jack, R.L. Structure and dynamics of glass formers: Predictability at large length scales. Phys. Rev. E 2007, 76, 041509. [Google Scholar] [CrossRef]
- Slater, B.; Michaelides, A. Surface premelting of water ice. Nat. Rev. Chem. 2019, 3, 172–188. [Google Scholar] [CrossRef]
- Li, Y.; Somorjai, G.A. Surface premelting of ice. J. Phys. Chem. C 2007, 111, 9631–9637. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Muniz, M.C.; Panagiotopoulos, A.Z.; Car, R.; E, W. A deep potential model with long-range electrostatic interactions. J. Chem. Phys. 2022, 156, 124107. [Google Scholar] [CrossRef]
- Grisafi, A.; Ceriotti, M. Incorporating long-range physics in atomic-scale machine learning. J. Chem. Phys. 2019, 151, 204105. [Google Scholar] [CrossRef]
- Debenedetti, P.G.; Sciortino, F.; Zerze, G.H. Second critical point in two realistic models of water. Science 2020, 369, 289–292. [Google Scholar] [CrossRef]
- Kim, K.H.; Amann-Winkel, K.; Giovambattista, N.; Späh, A.; Perakis, F.; Pathak, H.; Parada, M.L.; Yang, C.; Mariedahl, D.; Eklund, T.; et al. Experimental observation of the liquid–liquid transition in bulk supercooled water under pressure. Science 2020, 370, 978–982. [Google Scholar] [CrossRef]
- Henry, L.; Mezouar, M.; Garbarino, G.; Sifré, D.; Weck, G.; Datchi, F. Liquid–liquid transition and critical point in sulfur. Nature 2020, 584, 382–386. [Google Scholar] [CrossRef]
- Brovchenko, I.; Geiger, A.; Oleinikova, A. Liquid-liquid phase transitions in supercooled water studied by computer simulations of various water models. J. Chem. Phys. 2005, 123, 044515. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.C.; Martelli, F.; Liu, Y.; Car, R.; Panagiotopoulos, A.Z.; Debenedetti, P.G. Metastable liquid–liquid transition in a molecular model of water. Nature 2014, 510, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Sastry, S.; Austen Angell, C. Liquid–liquid phase transition in supercooled silicon. Nat. Mater. 2003, 2, 739–743. [Google Scholar] [CrossRef] [PubMed]
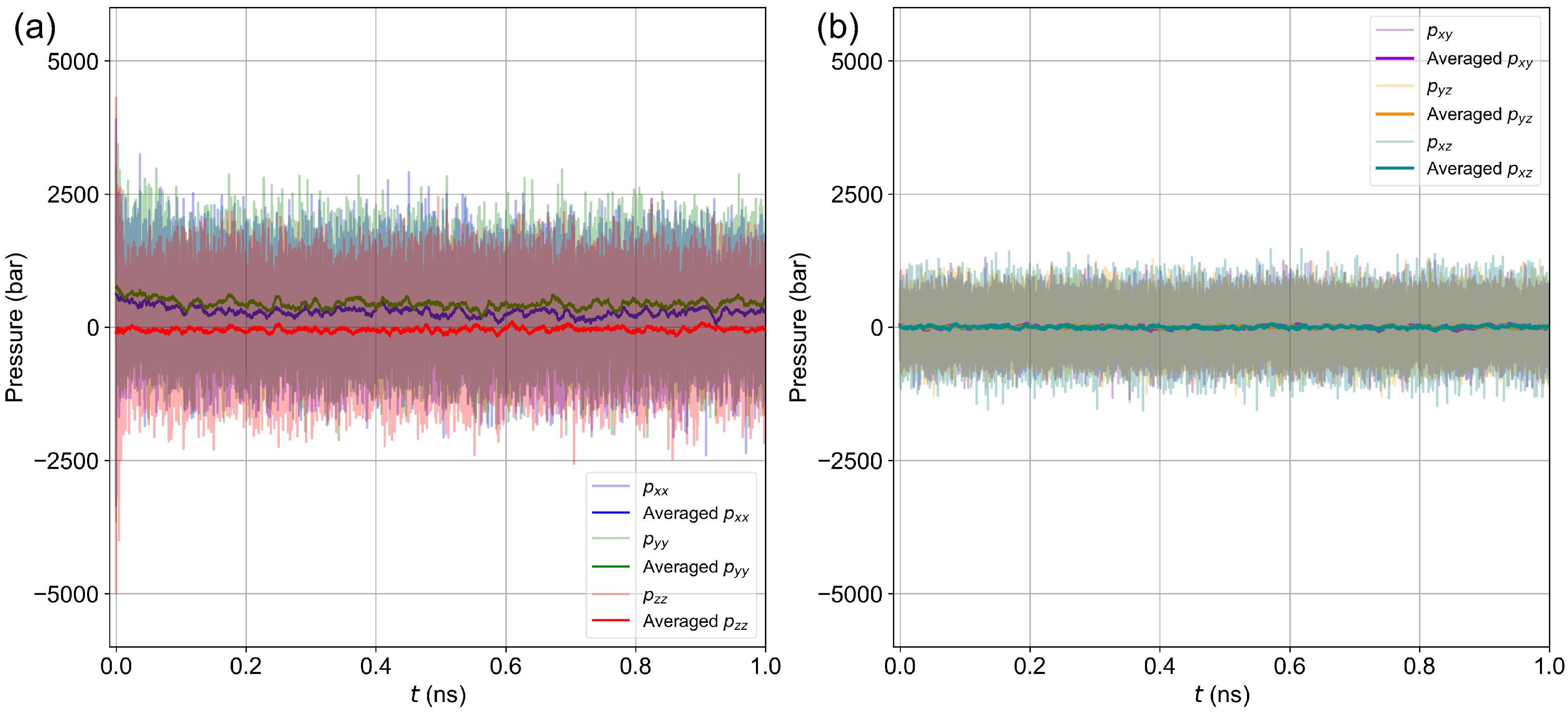
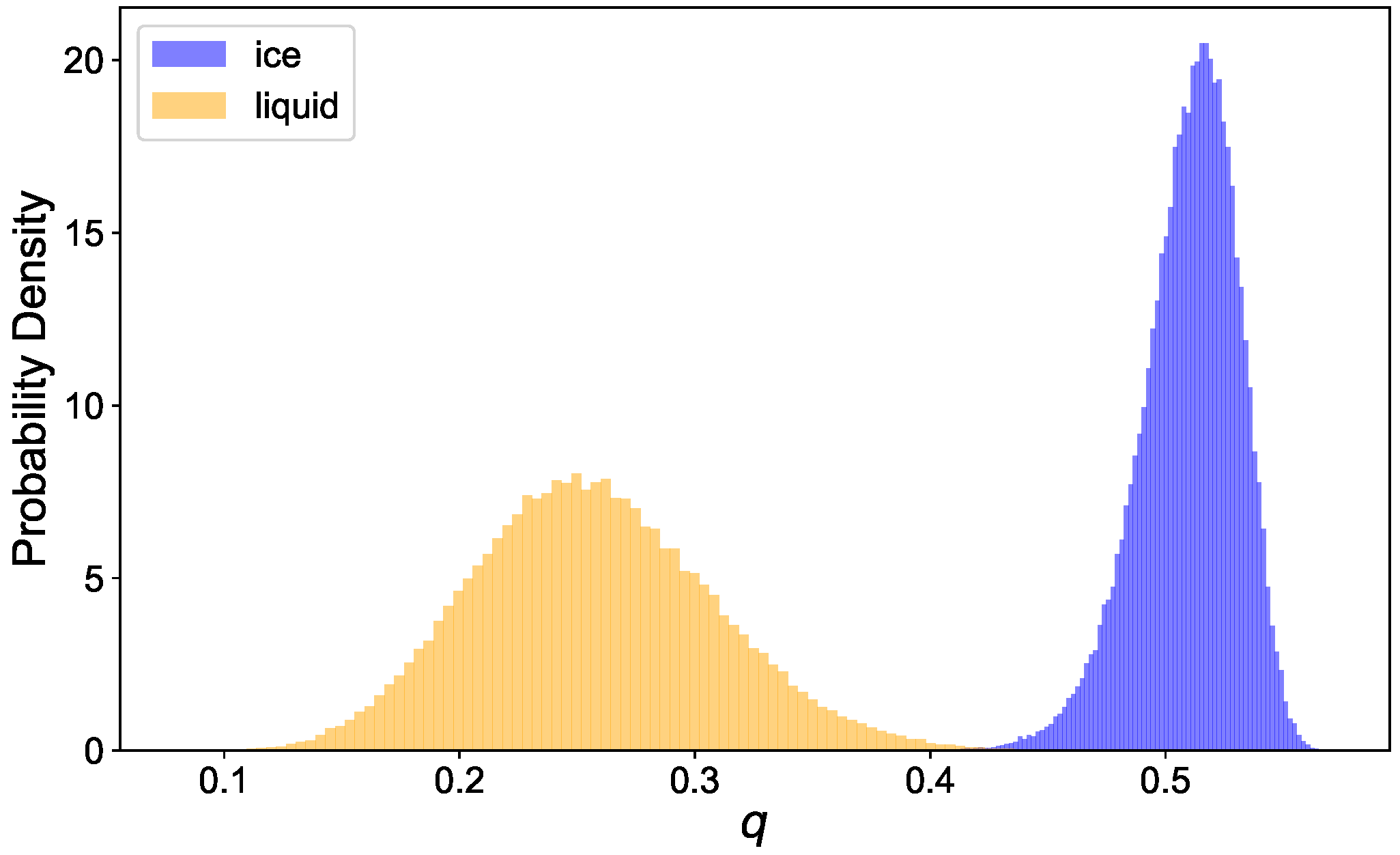

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, L.; Gao, A. Exploring the Premelting Transition through Molecular Simulations Powered by Neural Network Potentials. Crystals 2024, 14, 737. https://doi.org/10.3390/cryst14080737
Zeng L, Gao A. Exploring the Premelting Transition through Molecular Simulations Powered by Neural Network Potentials. Crystals. 2024; 14(8):737. https://doi.org/10.3390/cryst14080737
Chicago/Turabian StyleZeng, Limin, and Ang Gao. 2024. "Exploring the Premelting Transition through Molecular Simulations Powered by Neural Network Potentials" Crystals 14, no. 8: 737. https://doi.org/10.3390/cryst14080737
APA StyleZeng, L., & Gao, A. (2024). Exploring the Premelting Transition through Molecular Simulations Powered by Neural Network Potentials. Crystals, 14(8), 737. https://doi.org/10.3390/cryst14080737