Abstract
In this study, the sublimation and vapor pressure characteristics of RuO4 were systematically investigated using ab initio thermodynamic calculations. Structural optimizations and vibrational frequency analyses were performed for gaseous RuO4 and four candidate solid phases (monoclinic Cm, P21/c, C2/c, and cubic P-43n) within the density functional theory (DFT) framework. Gibbs free energies were evaluated by incorporating electronic energies, zero-point corrections, and entropic contributions from translational, rotational, and vibrational modes. The results identify monoclinic C2/c and cubic P-43n as the most stable solid phases across the studied temperature range. Calculated sublimation temperatures of 322 K at 1 atm and 240 K at 1 × 10−3 atm were obtained in good agreement with experimental melting and boiling points. Calculated vapor pressures show reasonable agreement with experimental measurements below the triple point, with deviations at higher temperatures attributable to approximating liquid-gas behavior using solid-gas sublimation data. These findings provide the first theoretical description of RuO4 vapor pressure and offer a computational framework extendable to other transition-metal ALD precursors.
1. Introduction
Ruthenium (Ru) and ruthenium dioxide (RuO2) have attracted significant attention in semiconductor technology due to their favorable electrical properties. Both exhibit low bulk resistivities () (7.1 μΩ·cm for Ru and 35 μΩ·cm for RuO2) and high work functions (4.7 eV and 5.1 eV, respectively) [1,2,3]. These characteristics make Ru a promising candidate to replace copper (Cu) in nanoscale interconnects, as its shorter electron mean free path (6.6 nm compared to 40 nm for Cu) reduces the impact of surface and grain boundary scattering that severely degrades conductivity at nanoscale dimensions [4,5]. RuO2 has likewise been investigated as an alternative electrode to titanium nitride (TiN) in dynamic random-access memory (DRAM) capacitors owing to its superior interfacial stability and reduced degradation [6,7]. Figure 1 compares the resistivity of several candidate metals as a function of thickness (). The metals examined include tungsten (W), platinum (Pt), palladium (Pd), Ru, iridium (Ir), cobalt (Co), and molybdenum (Mo) [8,9,10,11]. The results demonstrate that Ru exhibits a slower increase in resistivity with decreasing thickness than most other metals, highlighting its ability to maintain lower resistance at nanometer dimensions and underscoring its suitability for next-generation interconnect technologies.
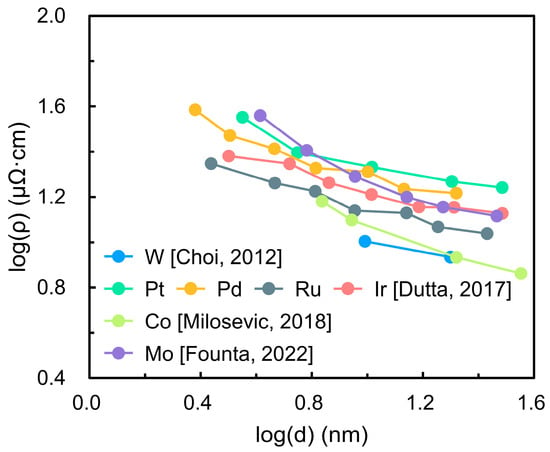
Figure 1.
Thickness ()-dependent resistivity () of candidate interconnect metals (W, Pt, Pd, Ru, Ir, Co, and Mo). The data demonstrate that Ru exhibits a slower resistivity increase with decreasing thickness compared to other metals, highlighting its potential for maintaining lower resistance at nanoscale dimensions [8,9,10,11].
As device scaling advances to atomic dimensions, atomic layer deposition (ALD) has become essential for producing ultrathin films with precise thickness control and excellent conformality [12,13,14]. To date, several Ru precursors have been examined, including bis(cyclopentadienyl)ruthenium(II) (Ru(Cp)2) [15], tris(2,2,6,6-tetramethyl-3,5-heptanedionato)ruthenium(III) (Ru(thd)3) [16], bis(2,4-dimethylpentadienyl)ruthenium(II) (Ru(DMPD)2) [17], and bis(ethylcyclopentadienyl)ruthenium(II) (Ru(EtCp)2) [18]. Despite their widespread use, these precursors exhibit inherent limitations, most notably low vapor pressures (~10−5–10−4 atm at ~330–360 K) [19,20,21,22] and poor conformality in high aspect-ratio structures [23]. Ru(Cp)2 offers stable growth characteristics but suffers from impurity incorporation and limited step coverage [15]. Ru(thd)3 with oxygen (O2) yields high-purity films but exhibits extremely low growth rates (0.035–0.040 nm/cycle) and long nucleation delays [16]. Ru(DMPD)2 offers improved film purity in both thermal and plasma-enhanced ALD (PEALD) yet suffers from notably low PEALD growth rates (~0.025 nm/cycle) that reduce manufacturing throughput and incomplete sidewall coverage (~70%) due to anisotropic radical transport effects [24]. Ru(EtCp)2 with active oxygen (O) radicals enables radical-assisted growth at low temperatures but is susceptible to etching at elevated temperatures, complicating optimization [18]. Together, these results demonstrate the need for alternative Ru precursors with high volatility and improved deposition performance.
Ruthenium tetroxide (RuO4), characterized by lower molecular weight and inherently higher vapor pressure, represents a highly promising candidate. Experimental studies have reported successful ALD of Ru film using RuO4/hydrogen (H2) processes at low temperatures (373 K) with high growth rates (0.1 nm/cycle), minimal nucleation delays, and favorable substrate selectivity [25,26]. Accelerated Ru growth on tantalum pentoxide (Ta2O5) has also been observed, attributed to substrate-mediated effects involving partial reduction and cation incorporation [27]. Processes based on RuO4 have further enabled immediate nucleation of RuO2 and tunable growth rates, as well as area-selective ALD (ASALD) at low temperatures [28,29]. Nevertheless, despite these advances, vapor pressure data for RuO4 remain limited to a few experimental measurements [30,31,32], which are not always consistent.
Computational thermodynamics provides an opportunity to address these limitations. Ab initio methods have been successfully applied to other transition-metal precursors, such as molybdenum oxyhalide (MoO2Cl2) [33] and molybdenum pentachloride (MoCl5) [34], to clarify sublimation energetics and vapor pressure behavior. However, to date, RuO4 has not been systematically studied from this perspective.
This study aims to investigate the sublimation behavior and vapor pressure by performing ab initio thermodynamic calculations. By combining density functional theory (DFT) with vibrational thermodynamics, Gibbs free energies of solid and gaseous RuO4 phases are evaluated, and equilibrium vapor pressures are derived over relevant temperature ranges. The computational results are compared with available experimental data [30,31,32], providing complementary insights that support more effective precursor selection for ALD applications demanding atomic-scale precision.
2. Calculation Details
Density functional theory calculations were carried out using the Vienna Ab initio Simulation Package (VASP version 6.3.2) [35,36,37]. The projector augmented-wave (PAW) method [37,38] was employed in conjunction with the Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation (GGA) for exchange-correlation interactions [39]. Van der Waals interactions were included through the DFT-D3 dispersion correction method [40] to accurately describe the weak intermolecular forces and lattice parameters in RuO4. A plane-wave cutoff energy of 500 eV was selected to balance computational efficiency and accuracy.
For gaseous RuO4 (point group Td), structural optimizations were performed in a cubic box of 15 × 15 × 15 Å3 with Monkhorst-Pack k-point sampling to minimize interactions caused by periodic boundary conditions in DFT calculations [41]. For solid RuO4, four candidate crystal structures—monoclinic Cm, P21/c, C2/c, and cubic P-43n—were considered. The Brillouin zone was sampled with Monkhorst–Pack k-point meshes of 3 × 4 × 3 for Cm, 3 × 3 × 3 for P21/c and C2/c, and 2 × 2 × 2 for P-43n [41]. To ensure adequate system size for phonon calculations, supercell expansions of 1 × 2 × 2, 2 × 1 × 2, and 1 × 2 × 1 were constructed for Cm, P21/c, and C2/c, respectively, while the primitive unit cell was retained for P-43n. The optimized geometries of gaseous RuO4 and the four candidate solids are presented in Figure 2, illustrating the molecular configuration and coordination environment of Ru in each phase.
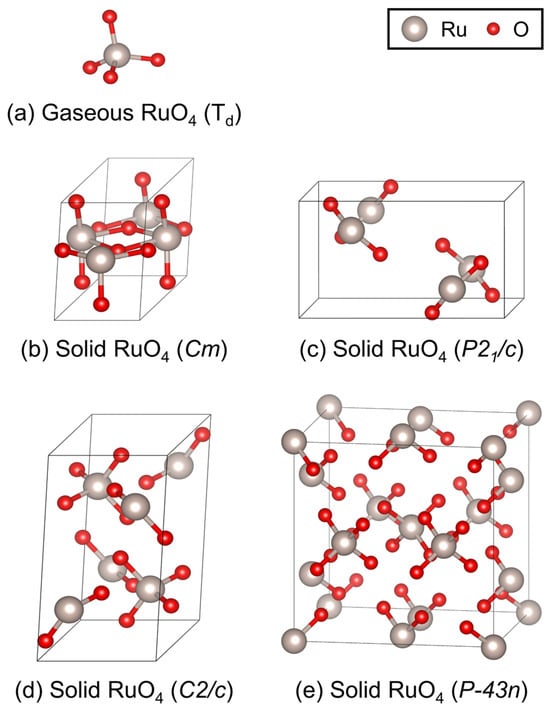
Figure 2.
Optimized geometries of RuO4 in (a) the gaseous phase (point group Td), and four candidate solid structures with space groups: (b) monoclinic Cm, (c) monoclinic P21/c, (d) monoclinic C2/c, and (e) cubic P-43n. Gray and red spheres represent Ru and O atoms, respectively. These optimized structures served as the basis for subsequent vibrational frequency and thermodynamic calculations.
Phonon calculations were performed using the finite-displacement (frozen-phonon) method implemented in the phonopy package (version 2.43.2) [42,43], based on force constants obtained from the DFT supercell calculations. The resulting vibrational properties were used to compute temperature-dependent thermodynamic quantities of each solid phase. Phonon dispersion relations for the four RuO4 polymorphs were also evaluated to examine their dynamic stability. Zero-point energies and temperature-dependent enthalpic corrections were derived from vibrational frequencies. The Gibbs free energy () was evaluated within the framework of ab initio thermodynamics according to
where is the enthalpy, is the temperature, is the entropy, is the DFT total energy, is the pressure, is the volume, is the zero-point energy, is the temperature-dependent enthalpic correction, and , , and are translational, rotational, and vibrational entropies, respectively. Translational entropy was calculated using the Sackur-Tetrode equation, rotational entropy within the rigid-rotor approximation, and vibrational entropy within the harmonic oscillator approximation [44]. For solids, translational and rotational entropy contributions were omitted because of lattice constraints. To prevent overestimation of vibrational entropy, low-frequency modes below 50 cm−1 were adjusted to this cutoff, consistent with established practice [45]. All vibrational modes, excluding imaginary frequencies, were incorporated in the thermodynamic analysis. This methodology ensures reliable Gibbs free energies for both solid and gaseous RuO4, forming the basis for subsequent evaluation of sublimation behavior and vapor pressure.
3. Results and Discussion
The optimized lattice parameters of the four candidate RuO4 structures are summarized in Table 1, showing good agreement with previously reported values [46], which confirms the reliability of the present DFT optimization. Table 2 lists the total energies of the same structures and demonstrates overall consistency with database references [47,48,49]. The relative Gibbs free energies (Figure 3) indicate that the monoclinic Cm and cubic P-43n phases possess the lowest values across the studied temperature range, indicating nearly indistinguishable thermodynamic stability. Given that RuO4-based ALD typically operates near 373 K (below its decomposition temperature of 398 K [25,29]), the C2/c phase was selected for subsequent vapor pressure analysis. Phonon calculations for the four polymorphs yielded a small number of imaginary frequencies (3, 3, 3, and 11 for C2/c, P21/c, P-43n, and Cm, respectively), suggesting localized dynamical instabilities. The phonon band structures for the four RuO4 crystal structures are provided in the Supporting Information (Figure S1). These curves show that the C2/c and P-43n phases exhibit only limited imaginary branches near the Γ-point, whereas the P21/c phase shows broader regions of negative frequencies with smaller magnitudes, and the Cm phase displays pronounced negative branches across most of the Brillouin zone, confirming its dynamic instability.

Table 1.
Calculated lattice parameters of candidate solid RuO4 structures (monoclinic Cm, P21/c, C2/c, and cubic P-43n) compared with experimental data reported by Pley et al. [46].
Table 2.
DFT-calculated total energies of candidate solid RuO4 structures (monoclinic Cm, P21/c, C2/c, and cubic P-43n) compared with literature values from the Materials Project [47] and the Open Quantum Materials Database (OQMD) [48,49]. The results indicate consistent relative stability trends among the polymorphs.
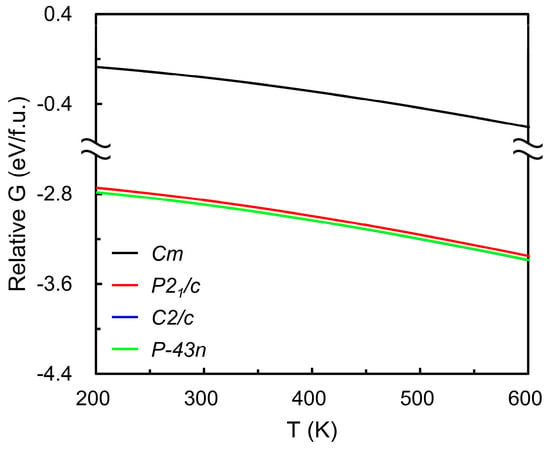
Figure 3.
Temperature-dependent Gibbs free energies of candidate solid RuO4 phases (monoclinic Cm, P21/c, C2/c, and cubic P-43n), referenced to the monoclinic Cm structure at 0 K. Black, red, blue, and green curves denote the Cm, P21/c, C2/c, and cubic P-43n phases, respectively. The blue curve is indistinguishable from the green curve because of their close overlap. The results indicate that the C2/c and P-43n phases exhibit the lowest Gibbs free energies over the studied temperature range, suggesting their thermodynamic stability relative to other polymorphs.
To further elucidate phase stability, Table 3 presents the Gibbs free energy components for solid and gaseous RuO4 at 373 K and 1 atm. For the gas phase, significant contributions arise from translational (0.68 eV) and rotational (0.40 eV) entropy terms, reflecting the molecular degrees of freedom absent in solids. Conversely, solid RuO4 exhibits a much larger vibrational entropy contribution (4.38 eV) due to numerous phonon modes, particularly in the low-frequency region. Zero-point energy (2.92 eV vs. 0.34 eV) and enthalpy contributions (1.97 eV vs. 0.22 eV) are also substantially higher for the solid. These findings highlight the contrasting entropy partitioning between solid and gas phases, which governs their relative Gibbs free energy balance at elevated temperatures.
Table 3.
Thermodynamic contributions to the Gibbs free energy of RuO4 at 373 K and 1 atm for both solid and gaseous phases. Energy contributions include zero-point energy (), temperature-dependent enthalpy change (), and entropy terms including translational (), rotational (), and vibrational () contributions (all values in eV), illustrating the contrasting entropy partitioning between the two phases.
Figure 4 illustrates the temperature-dependent Gibbs free energies of solid and gaseous RuO4 at partial pressures of (a) 1 atm and (b) 1 × 10−3 atm, the latter representing typical ALD operating conditions. The Gibbs free energy of solid RuO4 at 0 K was used as a reference. The intersections of the solid and gaseous free energy curves define the sublimation point (), yielding 322 K at 1 atm and 240 K at 1 × 10−3 atm. Experimentally, RuO4 is known to melt at 298 K and boil at 403 K [32], consistent with the calculated sublimation points falling between these transitions. Direct evaluation of the liquid phase is challenging; therefore, a simple interpolated curve between the experimental melting () and boiling () points is included only for reference. This comparison indicates that the computational framework reasonably captures the thermodynamic behavior of RuO4 under relevant conditions.
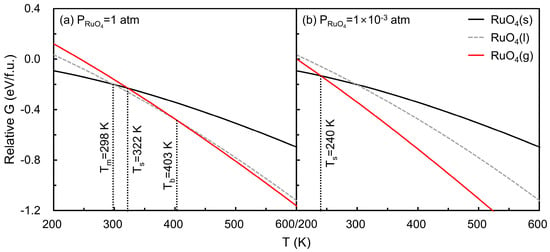
Figure 4.
Gibbs free energies of solid (black curve) and gaseous (red curve) RuO4 as a function of temperature at (a) 1 atm and (b) 1 × 10−3 atm, referenced to solid RuO4 at 0 K. The intersections of the curves define the sublimation point (), yielding 322 K at 1 atm and 240 K at 1 × 10−3 atm. For reference, experimental melting ( = 298 K) and boiling points ( = 403 K) are included, with an illustrative liquid phase curve interpolated between these points.
To extend this analysis, Figure 5 compares the calculated vapor pressure of RuO4 with experimental measurements over a temperature range [30,31,32]. Below the triple point (T < 299 K), the calculated values reproduce the overall temperature dependence and are in reasonable agreement with experimental sublimation data. Nevertheless, the experimental vapor pressures are systematically lower than theoretical predictions, which can be explained by kinetic limitations in the evaporation process. A recent CALPHAD-type thermodynamic assessment of RuO4 reported by Nuta et al. [50] reported a very low evaporation coefficient of ~0.027 which indicates that only a small fraction of surface molecules contributes to vaporization, thereby causing consistent underestimation of experimental measurements. At higher temperatures (T > 299 K), the deviation becomes more pronounced because the experimental data reflect liquid-gas vaporization, while the present calculations employ solid-gas sublimation data as an approximation, inevitably leading to systematic overestimation. For comparison, conventional Ru precursors such as Ru(Cp)2 or Ru(thd)3 typically exhibit vapor pressures on the order of 10−5–10−4 atm near 330–360 K [19,20,21,22], whereas RuO4 already reaches ~10−2 atm at its triple point (299 K). This large difference highlights the exceptional volatility of RuO4. These results confirm that RuO4 possesses a relatively high equilibrium vapor pressure, consistent with prior experimental observations and supporting its suitability as a volatile precursor for atomic layer deposition.
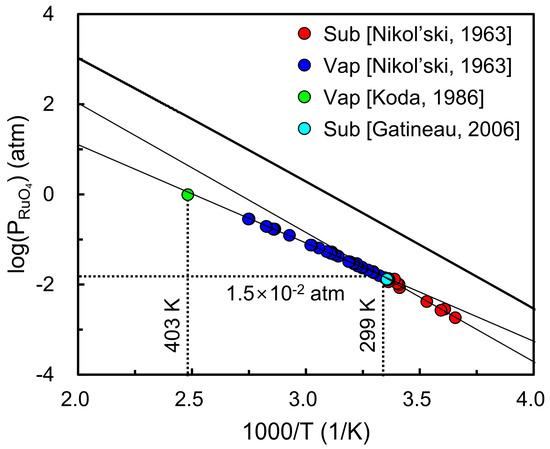
Figure 5.
Calculated vapor pressure of RuO4 as a function of temperature compared with experimental measurements (colored circles) [30,31,32]. The results show reasonable agreement with sublimation data below the triple point, while deviations at higher temperatures arise because liquid-gas vaporization is approximated using solid-gas sublimation data.
4. Conclusions
In this work, the sublimation and vapor pressure behaviors of RuO4 were investigated through ab initio thermodynamic calculations. Structural optimizations and vibrational analyses were performed for gaseous RuO4 and four candidate solid phases, leading to the identification of monoclinic C2/c and cubic P-43n as the most stable crystalline structures. Thermodynamic analysis revealed sublimation temperatures of 322 K at 1 atm and 240 K at 1 × 10−3 atm, values that are consistent with the experimental melting and boiling points of RuO4.
The vapor pressure calculations show reasonable agreement with experimental sublimation data at temperatures below the triple point. Experimental values are consistently lower than theoretical predictions, with modest deviations explained by kinetic limitations in the evaporation process at lower temperatures and more pronounced discrepancies at higher temperatures, where liquid-gas vaporization dominates experimentally while the present calculations employ solid-gas sublimation data as an approximation.
Overall, this work provides the first theoretical description of RuO4 vapor pressure based on ab initio thermodynamics. The results confirm the high volatility of RuO4 and its suitability as an ALD precursor. The computational framework demonstrated here is broadly applicable and can be extended to other transition-metal precursors, thereby contributing to the rational design and evaluation of precursors for future semiconductor technologies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst15110915/s1. Figure S1: Phonon band structures of (a) monoclinic Cm, (b) P21/c, (c) C2/c, and (d) cubic P-43n RuO4 phases, obtained from the frozen-phonon calculations using the phonopy package [42,43].
Author Contributions
Conceptualization, Y.-C.K.; methodology, S.-H.K.; software, S.-H.K.; validation, J.-Y.K., H.-K.K., N.-Y.L., H.-N.K., S.T. and J.-Y.J.; formal analysis, S.-H.K.; investigation, S.-H.K.; resources, J.-Y.J.; data curation, S.-H.K.; writing—original draft preparation, S.-H.K.; writing—review and editing, Y.-C.K.; visualization, S.-H.K. and J.-Y.K.; supervision, Y.-C.K.; project administration, Y.-C.K.; funding acquisition, Y.-C.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Korea Institute for Advancement of Technology (KIAT) grant funded by the Korea Government (MOTIE) (RS-2024-00409639, HRD Program for Industrial Innovation), and by the Education and Research Promotion Program of KOREATECH in 2025.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Acknowledgments
During the preparation of this manuscript, the authors used ChatGPT (OpenAI, GPT-5, 2025) for the purposes of language refinement. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
Author Jun-Yeong Jo was employed by the company Wonik Materials Co., Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors also declare that ChatGPT (OpenAI, GPT-5) was used for language editing assistance in the preparation of this manuscript.
References
- Han, J.H.; Lee, S.W.; Kim, S.K.; Han, S.; Hwang, C.S.; Dussarrat, C.; Gatineau, J. Growth of RuO2 thin films by pulsed-chemical vapor deposition using RuO4 precursor and 5% H2 reduction gas. Chem. Mater. 2010, 22, 5700–5706. [Google Scholar] [CrossRef]
- Han, J.H.; Han, S.; Lee, W.; Lee, S.W.; Kim, S.K.; Gatineau, J.; Dussarrat, C.; Hwang, C.S. Improvement in the leakage current characteristic of metal-insulator-metal capacitor by adopting RuO2 film as bottom electrode. Appl. Phys. Lett. 2011, 99, 022901. [Google Scholar] [CrossRef]
- Lee, H.-J.; Nabeya, S.; Hong, T.E.; Harada, R.; Kim, S.-H. Low temperature atomic layer deposition of Ru thin films using a new carbonyl-based Ru precursor and non-oxidizing reactants; Applications to the seed layer for Cu metallization. In Proceedings of the IEEE International Interconnect Technology Conference (IITC), Hsinchu, Taiwan, 16–18 May 2017. [Google Scholar]
- Kim, Y.-H.; Kim, M.; Kotsugi, Y.; Cheon, T.; Mohapatra, D.; Jang, Y.; Bae, J.-S.; Hong, T.E.; Ramesh, R.; An, K.-S.; et al. Atomic layer deposited RuO2 diffusion barrier for next generation Ru-interconnects. Adv. Funct. Mater. 2022, 32, 2206667. [Google Scholar] [CrossRef]
- Yoon, S.J.; Lee, S.; Lee, T.I.; Yoon, A.; Cho, B.J. Large grain ruthenium for alternative interconnects. IEEE Electron Device Lett. 2018, 40, 91–94. [Google Scholar] [CrossRef]
- Jeon, W.; Yoo, S.; Kim, H.K.; Lee, W.; An, C.H.; Chung, M.J.; Cho, C.J.; Kim, S.K.; Hwang, C.S. Evaluating the top electrode material for achieving an equivalent oxide thickness smaller than 0.4 nm from an Al-doped TiO2 film. ACS Appl. Mater. Interfaces 2014, 6, 21632–21637. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.S.; Kim, T.K.; Lim, J.; Seo, H.; Paik, H.; Hwang, C.S. Enhanced electrical properties of an Al-doped TiO2 dielectric film on a TiN electrode by adopting an atomic layer deposited Ru interlayer. ACS Appl. Electron. Mater. 2022, 4, 2005–2014. [Google Scholar] [CrossRef]
- Choi, D.; Kim, C.S.; Naveh, D.; Chung, S.; Warren, A.P.; Nuhfer, N.T.; Toney, M.F.; Coffey, K.R.; Barmak, K. Electron mean free path of tungsten and the electrical resistivity of epitaxial (110) tungsten films. Phys. Rev. B 2012, 86, 045432. [Google Scholar] [CrossRef]
- Dutta, S.; Sankaran, K.; Moors, K.; Pourtois, G.; Van Elshocht, S.; Bömmels, J.; Vandervorst, W.; Tőkei, Z.; Adelmann, C. Thickness dependence of the resistivity of platinum-group metal thin films. J. Appl. Phys. 2017, 122, 025107. [Google Scholar] [CrossRef]
- Milosevic, E.; Kerdsongpanya, S.; Gall, D. The resistivity size effect in epitaxial Ru(0001) and Co(0001) layers. In Proceedings of the IEEE Nanotechnology Symposium (ANTS), New York, NY, USA, 14–15 November 2018. [Google Scholar]
- Founta, V.; Soulié, J.-P.; Sankaran, K.; Vanstreels, K.; Opsomer, K.; Morin, P.; Lagrain, P.; Franquet, A.; Vanhaeren, D.; Conard, T.; et al. Properties of ultrathin molybdenum films for interconnect applications. Materialia 2022, 24, 101511. [Google Scholar] [CrossRef]
- Suntola, T. Atomic layer epitaxy. Mater. Sci. Rep. 1989, 4, 261–312. [Google Scholar] [CrossRef]
- Ritala, M.; Leskelä, M.; Dekker, J.-P.; Mutsaers, C.; Soininen, P.J.; Skarp, J. Perfectly conformal TiN and Al2O3 films deposited by atomic layer deposition. Chem. Vap. Depos. 1999, 5, 7–9. [Google Scholar] [CrossRef]
- George, S.M. Atomic layer deposition: An overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, T.; Alén, P.; Ritala, M.; Leskelä, M. Ruthenium thin films grown by atomic layer deposition. Chem. Vap. Depos. 2003, 9, 45–49. [Google Scholar] [CrossRef]
- Aaltonen, T.; Ritala, M.; Arstila, K.; Keinonen, J.; Leskelä, M. Atomic layer deposition of ruthenium thin films from Ru(thd)3 and oxygen. Chem. Vap. Depos. 2004, 10, 215–219. [Google Scholar] [CrossRef]
- Methaapanon, R.; Geyer, S.M.; Brennan, S.; Bent, S.F. Size dependent effects in nucleation of Ru and Ru oxide thin films by atomic layer deposition measured by synchrotron radiation X-ray diffraction. Chem. Mater. 2013, 25, 3458–3463. [Google Scholar] [CrossRef]
- Kozodaev, M.G.; Lebedinskii, Y.Y.; Chernikova, A.G.; Korostylev, E.V.; Chouprik, A.A.; Khakimov, R.R.; Markeev, A.M.; Hwang, C.S. Temperature controlled Ru and RuO2 growth via O* radical-enhanced atomic layer deposition with Ru(EtCp)2. J. Chem. Phys. 2019, 151, 204701. [Google Scholar] [CrossRef]
- Green, M.L.; Gross, M.E.; Papa, L.E.; Schnoes, K.J.; Brasen, D. Chemical vapor deposition of ruthenium dioxide films. J. Electrochem. Soc. 1985, 132, 2677–2685. [Google Scholar] [CrossRef]
- Morozova, N.B.; Zherikova, K.V.; Semyannikov, P.P.; Trubin, S.V.; Igumenov, I.K. Study of temperature dependencies of saturated vapor pressure of ruthenium(III) beta-diketonate derivatives. J. Therm. Anal. Calorim. 2009, 98, 395–399. [Google Scholar] [CrossRef]
- Kawano, K.; Kosuge, H.; Oshima, N.; Funakubo, H. Ruthenium and ruthenium oxide film deposition by MOCVD using Ru(DMPD)2. ECS Trans. 2006, 1, 139–144. [Google Scholar] [CrossRef]
- Shibutami, T.; Kawano, K.; Oshima, N.; Yokoyama, S.; Funakubo, H. Ruthenium film with high nuclear density deposited by MOCVD using a novel liquid precursor. Electrochem. Solid-State Lett. 2003, 1, C117–C119. [Google Scholar] [CrossRef]
- Norman, J.A.T.; Perez, M.; Schulz, S.E.; Waechtler, T. New precursors for CVD copper metallization. Microelectron. Eng. 2008, 85, 2159–2163. [Google Scholar] [CrossRef][Green Version]
- Van der Straten, O.; Rossnagel, S.M.; Doyle, J.P.; Rodbell, K.P. Metal-organic atomic layer deposition of metals for applications in interconnect technology. ECS Trans. 2006, 1, 51–56. [Google Scholar] [CrossRef]
- Minjauw, M.M.; Dendooven, J.; Capon, B.; Schaekers, M.; Detavernier, C. Atomic layer deposition of ruthenium at 100 °C using the RuO4-precursor and H2. J. Mater. Chem. C 2015, 3, 132–137. [Google Scholar] [CrossRef]
- Minjauw, M.M.; Rijckaert, H.; Van Driessche, I.; Detavernier, C.; Dendooven, J. Nucleation enhancement and area-selective atomic layer deposition of ruthenium using RuO4 and H2 gas. Chem. Mater. 2019, 31, 1491–1499. [Google Scholar] [CrossRef]
- An, C.H.; Jeon, W.; Kim, S.H.; Cho, C.J.; Kwon, D.S.; Kim, D.G.; Lee, W.; Hwang, C.S. Substrate effects on the growth behavior of atomic-layer-deposited Ru thin films using RuO4 precursor and N2/H2 mixed gas. J. Phys. Chem. 2019, 123, 22539–22549. [Google Scholar] [CrossRef]
- Poonkottil, N.; Minjauw, M.M.; Werbrouck, A.; Checchia, S.; Solano, E.; Nisula, M.; Franquet, A.; Detavernier, C.; Dendooven, J. Atomic layer deposition of ruthenium dioxide based on redox reactions between alcohols and ruthenium tetroxide. Chem. Mater. 2022, 34, 8946–8958. [Google Scholar] [CrossRef]
- Poonkottil, N.; Rijckaert, H.; Rajendran, K.; Petit, R.R.; Martin, L.I.D.J.; Thourhout, D.V.; Driessche, I.V.; Detavernier, C.; Dendooven, J. Low temperature area selective atomic layer deposition of ruthenium dioxide thin films using polymers as inhibition layers. Adv. Mater. Interfaces 2023, 10, 2201934. [Google Scholar] [CrossRef]
- Nikol’skii, A. Saturated vapor pressure of ruthenium tetraoxide. Zh. Neorgan. Khim. 1963, 8, 541–543. [Google Scholar]
- Koda, Y. Boiling points and ideal solutions of ruthenium and osmium tetraoxides. J. Chem. Soc. Chem. Commun. 1986, 17, 1347–1348. [Google Scholar] [CrossRef]
- Gatineau, J.; Yanagita, K.; Dussarrat, C. A new RuO4 solvent solution for pure ruthenium film depositions. Microelectron. Eng. 2006, 83, 2248–2252. [Google Scholar] [CrossRef]
- Kim, H.-K.; Lee, N.-Y.; Kim, Y.-C. Evaluation of vapor pressure of MoO2Cl2 and its initial chemical reaction on a SiO2 surface by ab initio thermodynamics. Curr. Appl. Phys. 2024, 61, 115–120. [Google Scholar] [CrossRef]
- Lee, N.-Y.; Kim, S.-H.; Kim, J.-Y.; Kim, Y.-C. Molecular structure and vapor pressure of molybdenum pentachloride using ab-initio thermodynamics. J. Korean Phys. Soc. 2025, 86, 430–434. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Togo, A.; Chaput, L.; Tadano, T.; Tanaka, I. Implementation strategies in phonopy and phono3py. J. Phys. Condens. Matter 2023, 35, 353001. [Google Scholar] [CrossRef]
- Togo, A. First-principles phonon calculations with phonopy and phono3py. J. Phys. Soc. Jpn. 2023, 92, 012001. [Google Scholar] [CrossRef]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models, 2nd ed.; John Wiley & Sons: Chichester, UK, 2013; pp. 1–596. [Google Scholar]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Pley, M.; Wickleder, M.S. Two crystalln modifications of RuO4. J. Solid State Chem. 2005, 178, 3206–3209. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Saal, J.E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C. Materials design and discovery with high-throughput density functional theory: The Open Quantum Materials Database (OQMD). JOM 2013, 65, 1501–1509. [Google Scholar] [CrossRef]
- Kirklin, S.; Saal, J.E.; Meredig, B.; Thompson, A.; Doak, J.W.; Aykol, M.; Rühl, S.; Wolverton, C. The Open Quantum Materials Database (OQMD): Assessing the accuracy of DFT formation energies. npj Comput. Mater. 2015, 1, 132–137. [Google Scholar] [CrossRef]
- Nuta, I.; Virot, F.; Fischer, E.; Chatillon, C. Thermodynamic assessment of RuO4 oxide. Calphad 2023, 80, 102508. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).