
Estimation of Hardness of Single-Phase Metallic Alloys


Abstract
1. Introduction
2. Results
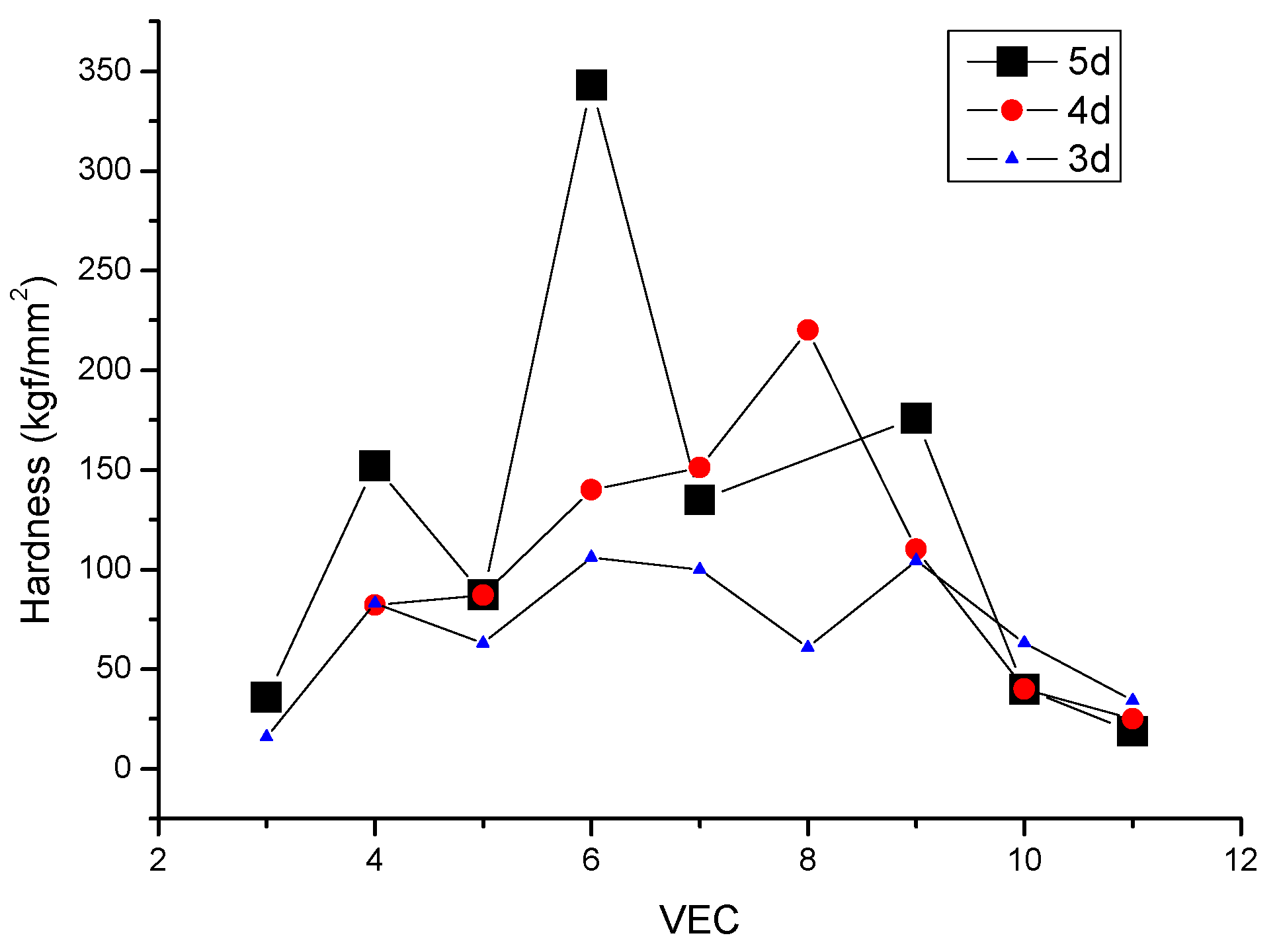
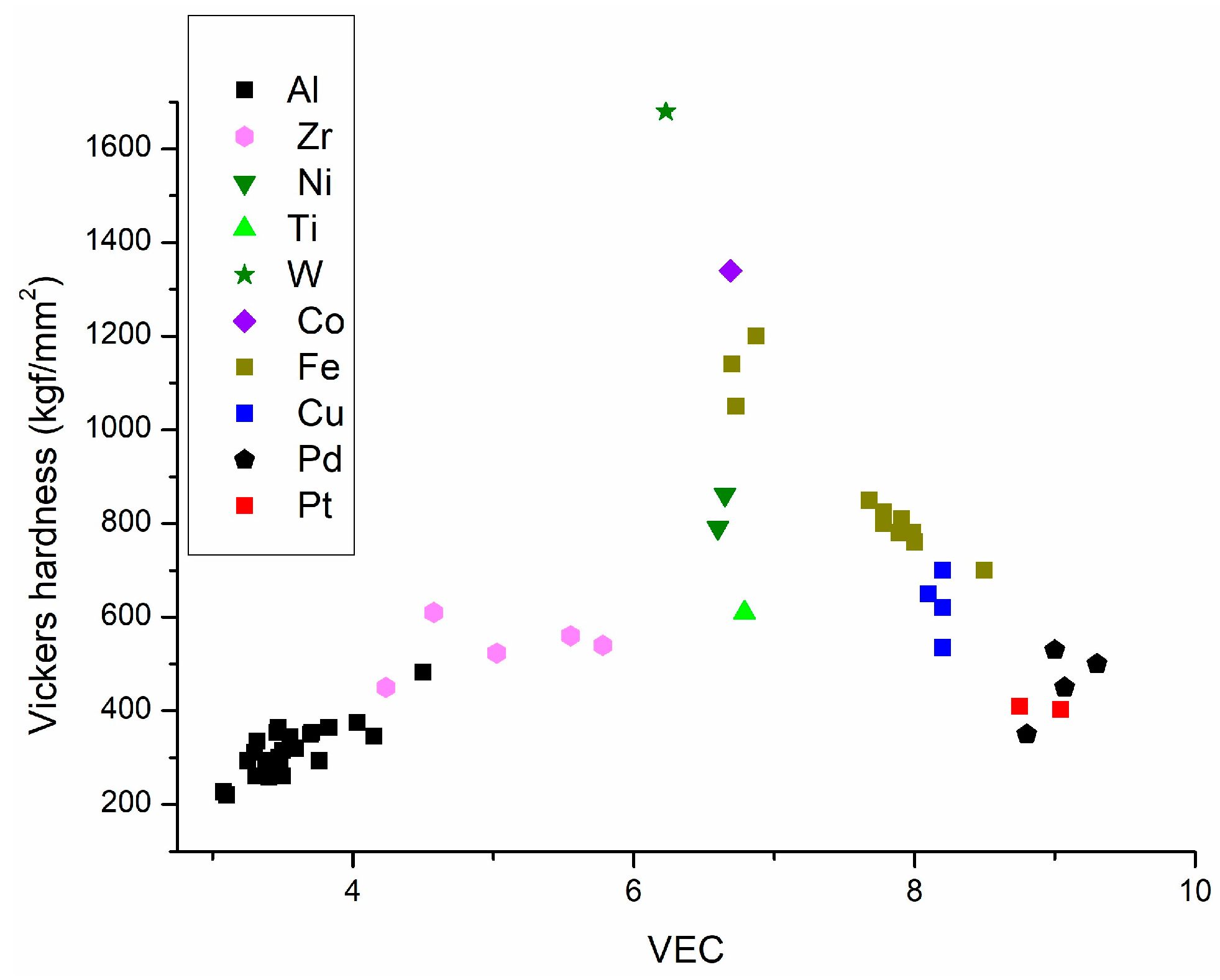
2.1. The Valence Electron Count (VEC) Dependence of Hardness
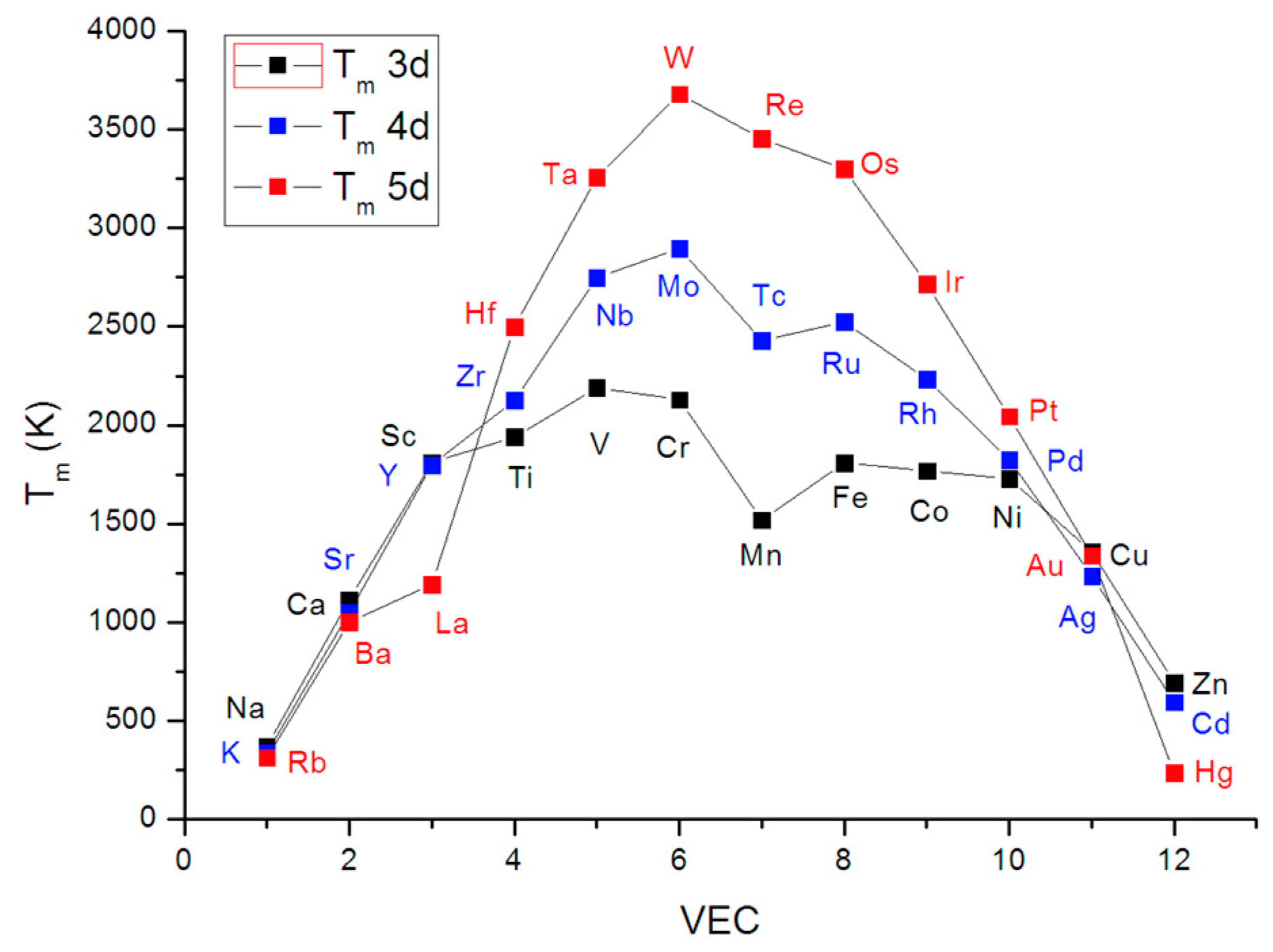
2.2. Estimation of Hardness from Debye Temperature
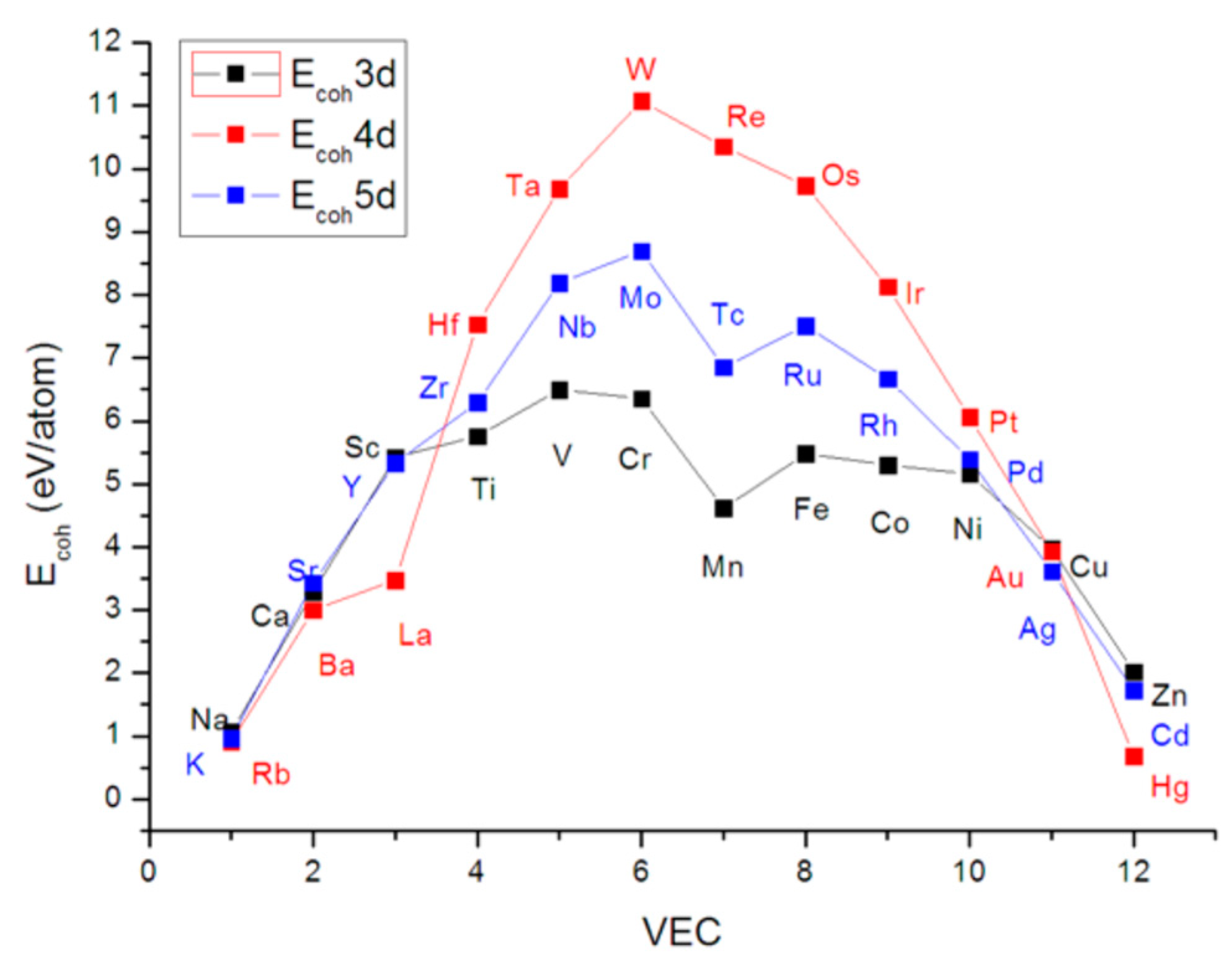
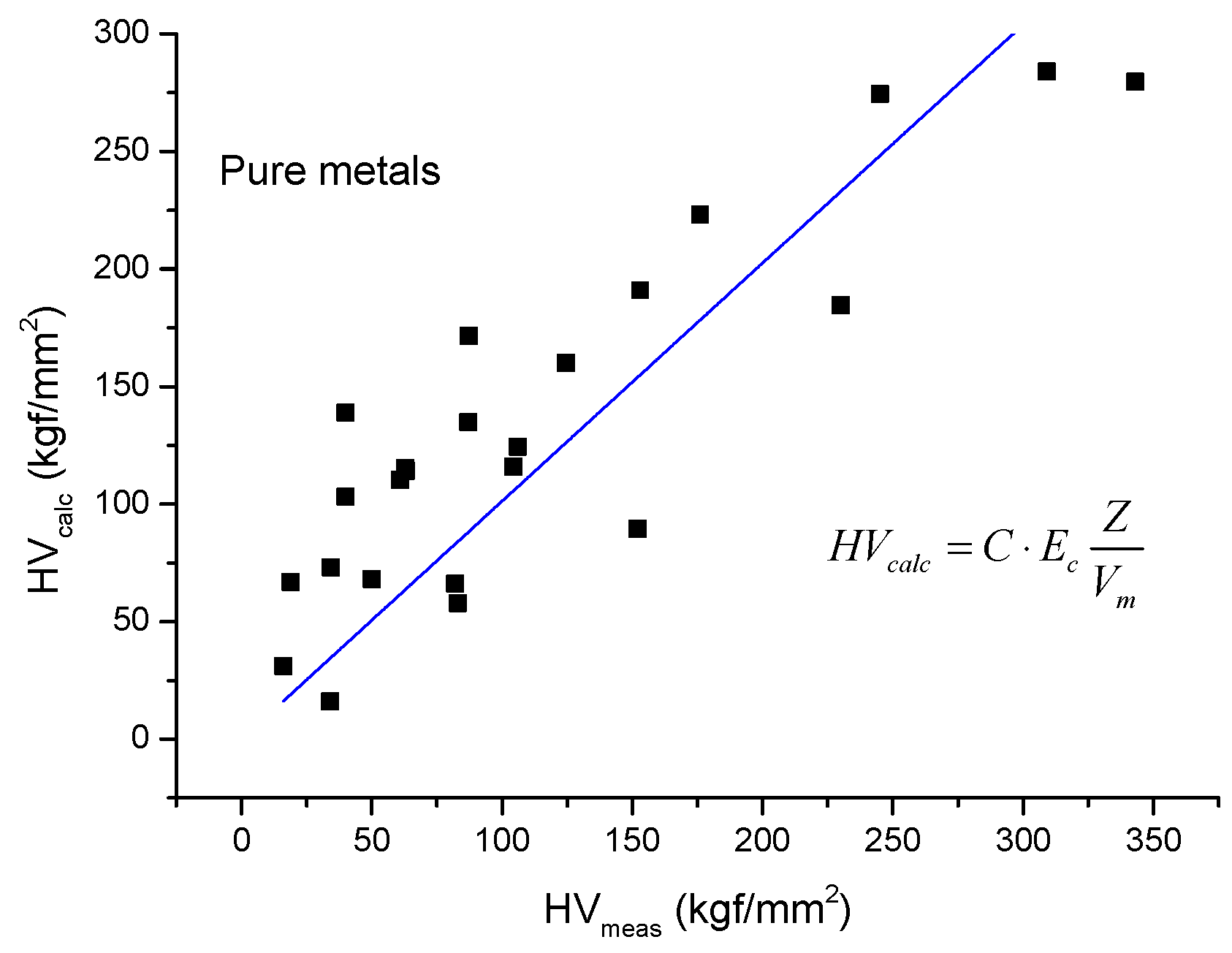
2.3. Estimation of Hardness from Cohesion Energy
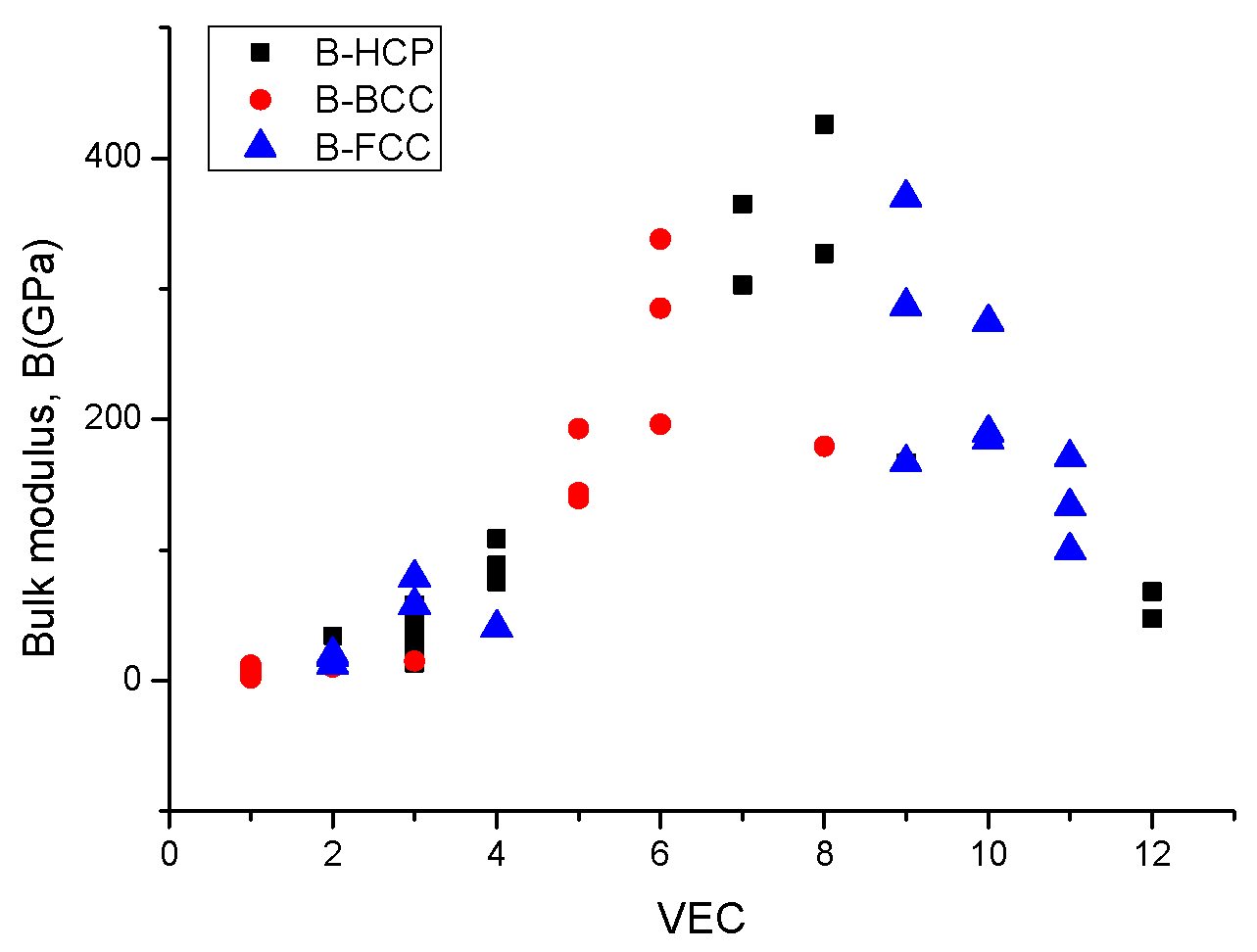
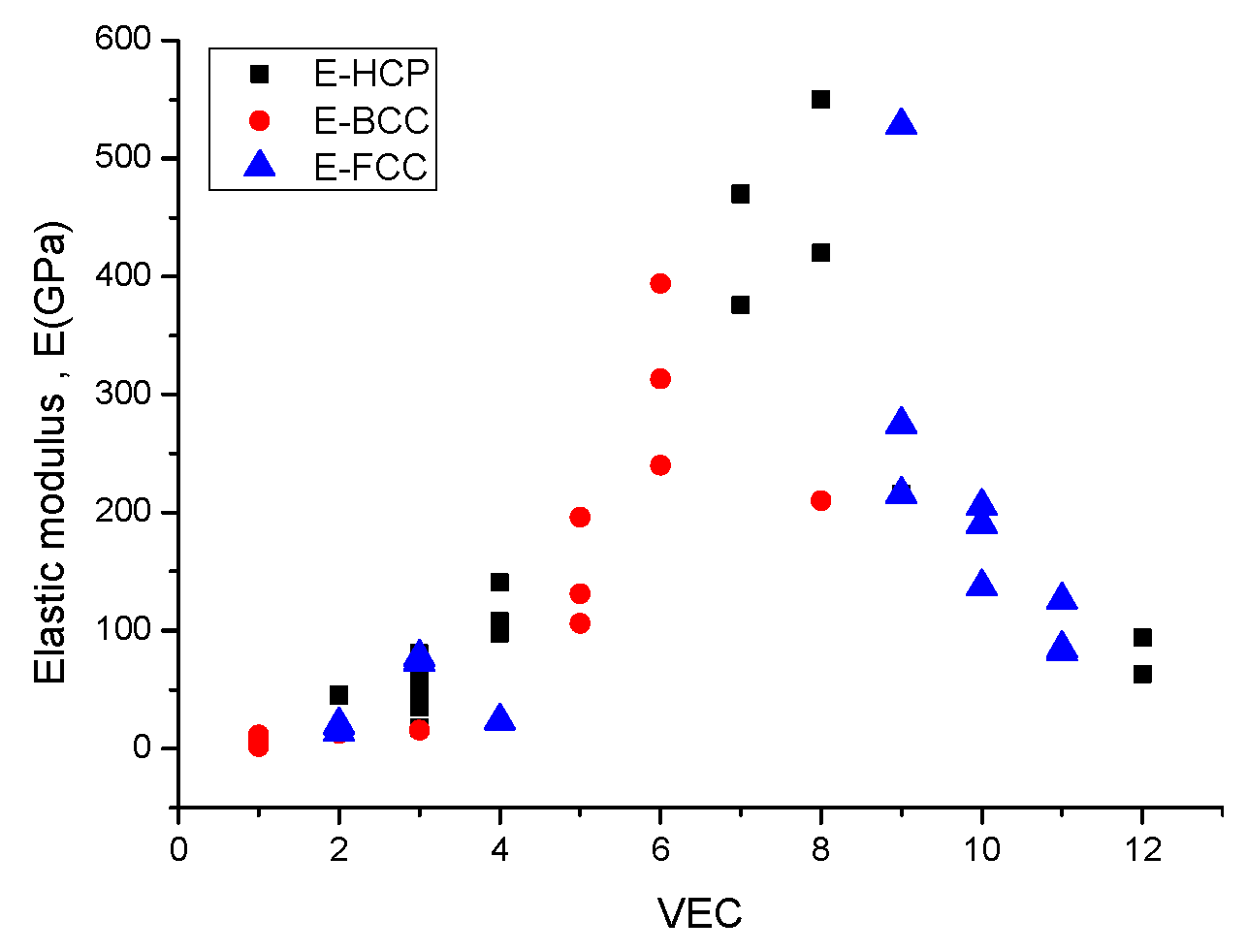
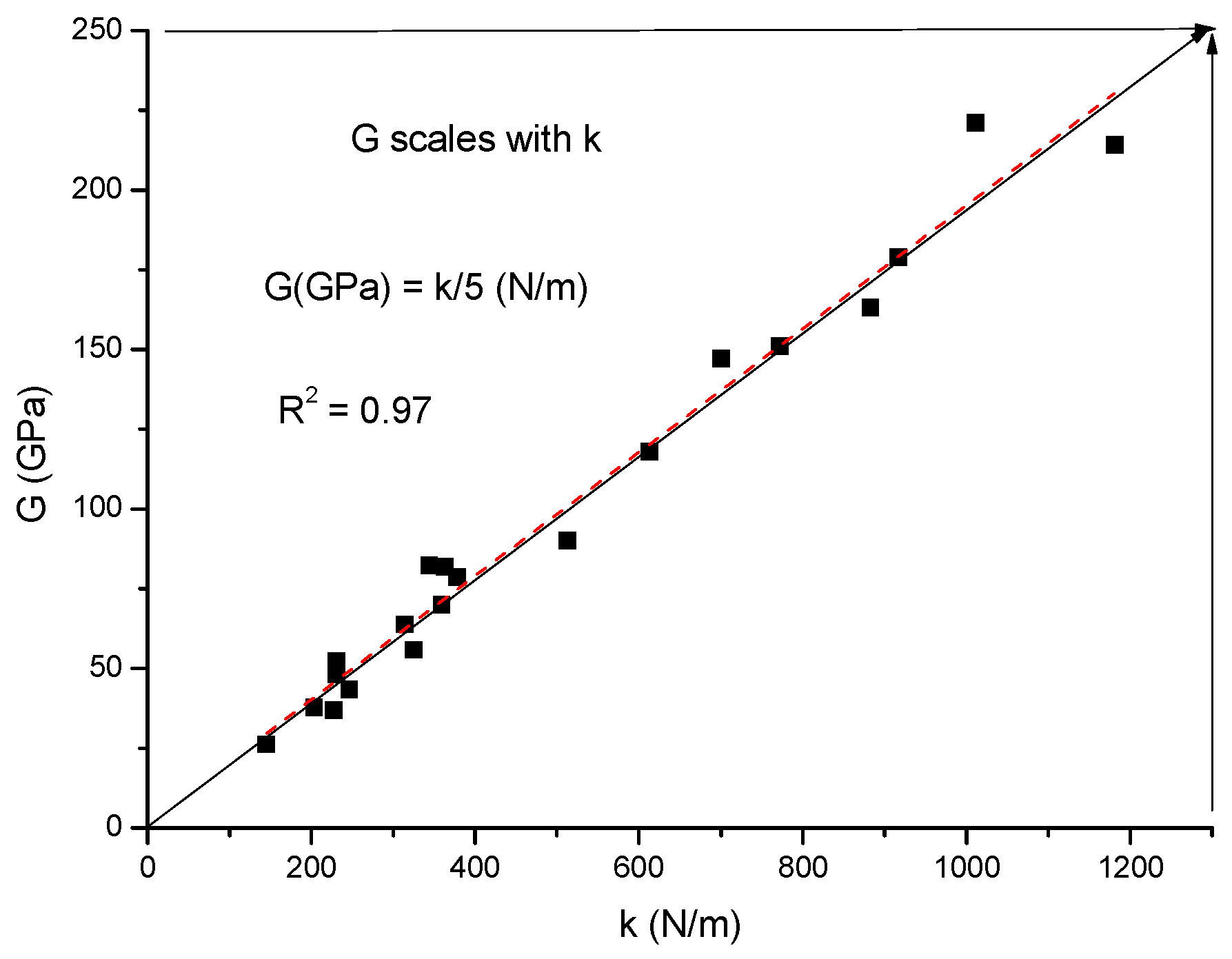
2.4. Estimation of Hardness from Elastic Moduli
E = 16.48 × HV
G = 6.78 × HV
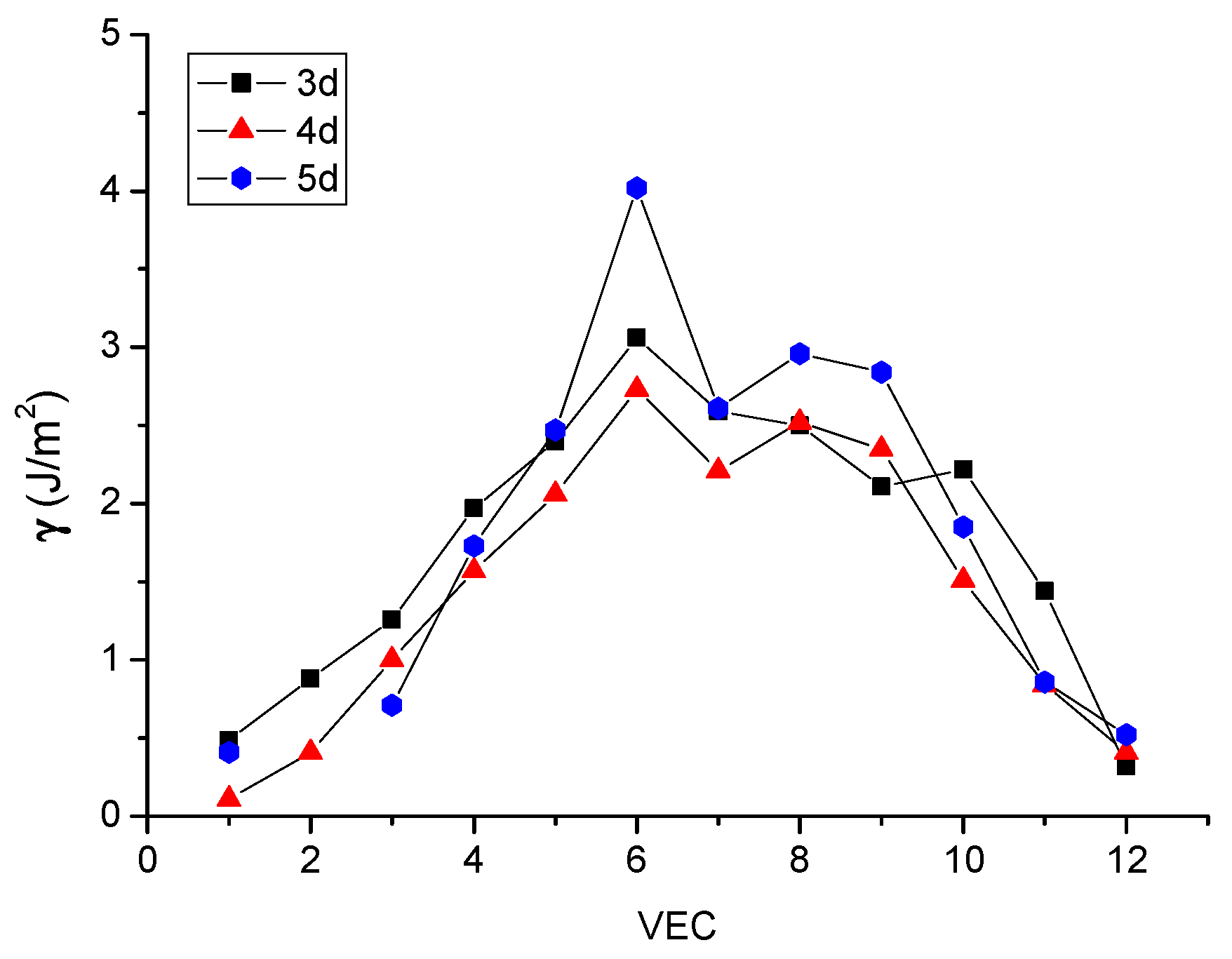
2.5. The Estimation of Hardness Using the Electrostatic Energy Density of Metallic Bonding
b = R(1 − γ/2)
c = R(1 + γ/2)
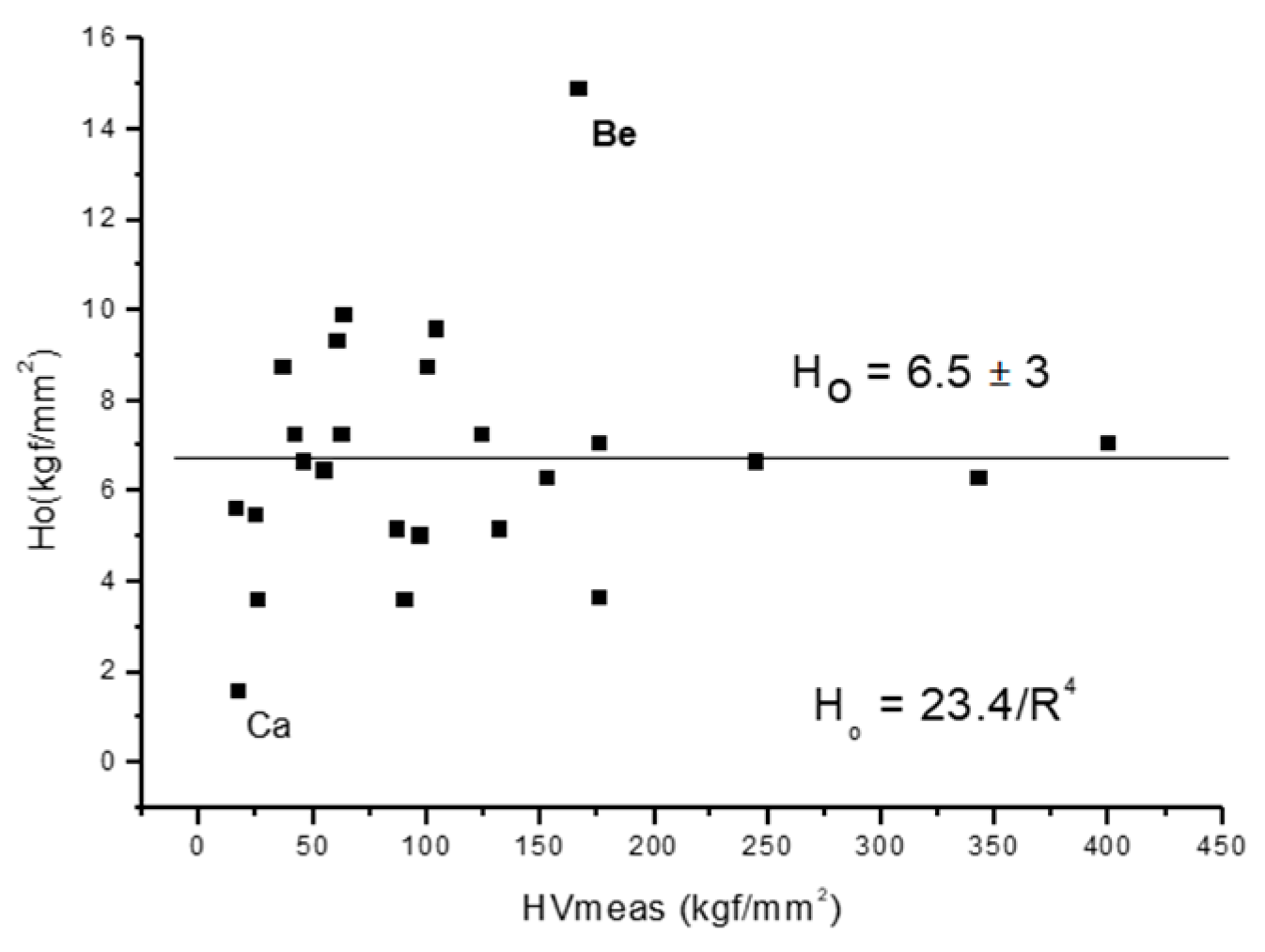
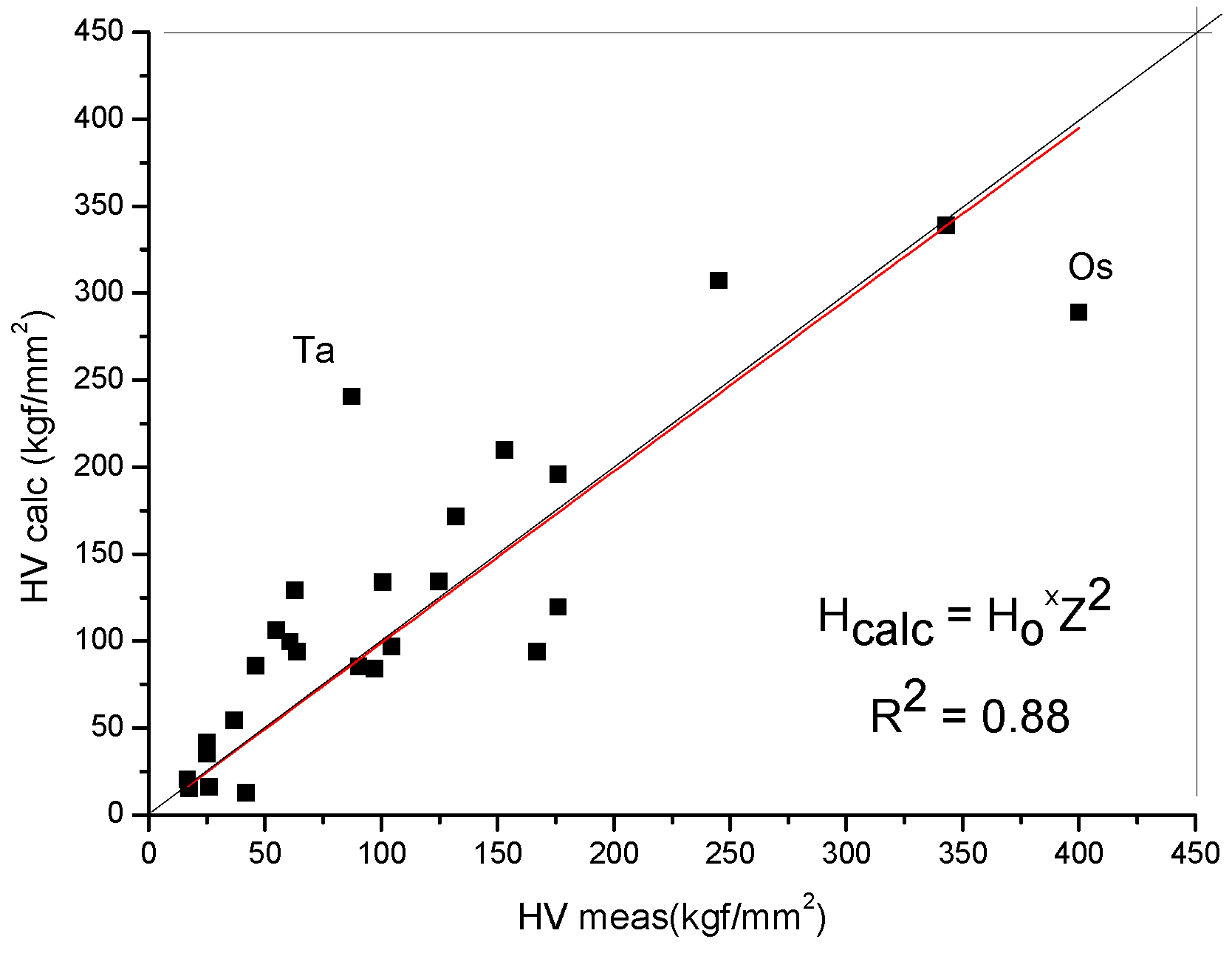
2.6. Estimation of Hardness Based on Selected Atomic Radius and Metallic Valence Data
3. Conclusions
- -
- We have succeeded in interpreting the maximum hardness in the VEC function for single-phase pure metals, amorphous metals, and high-entropy alloys. The maximum is caused by the change in atomic bonds depending on the number of unpaired d electrons. In addition to s-p-type electrons, d-type electrons also participate in the common metallic electron sea and increase the effective valence.
- -
- We have created formulas for calculating hardness from physical properties that directly depend on the strength of atomic bonds: force constant (Equation (7)), cohesion energy (Equation (8)), and shear modulus (Equation (11)).
- -
- We have reduced all these formulas to a common formula that depends only on two parameters: the atomic radius R and the valence Z:H = 0.234 Z2/R4
- -
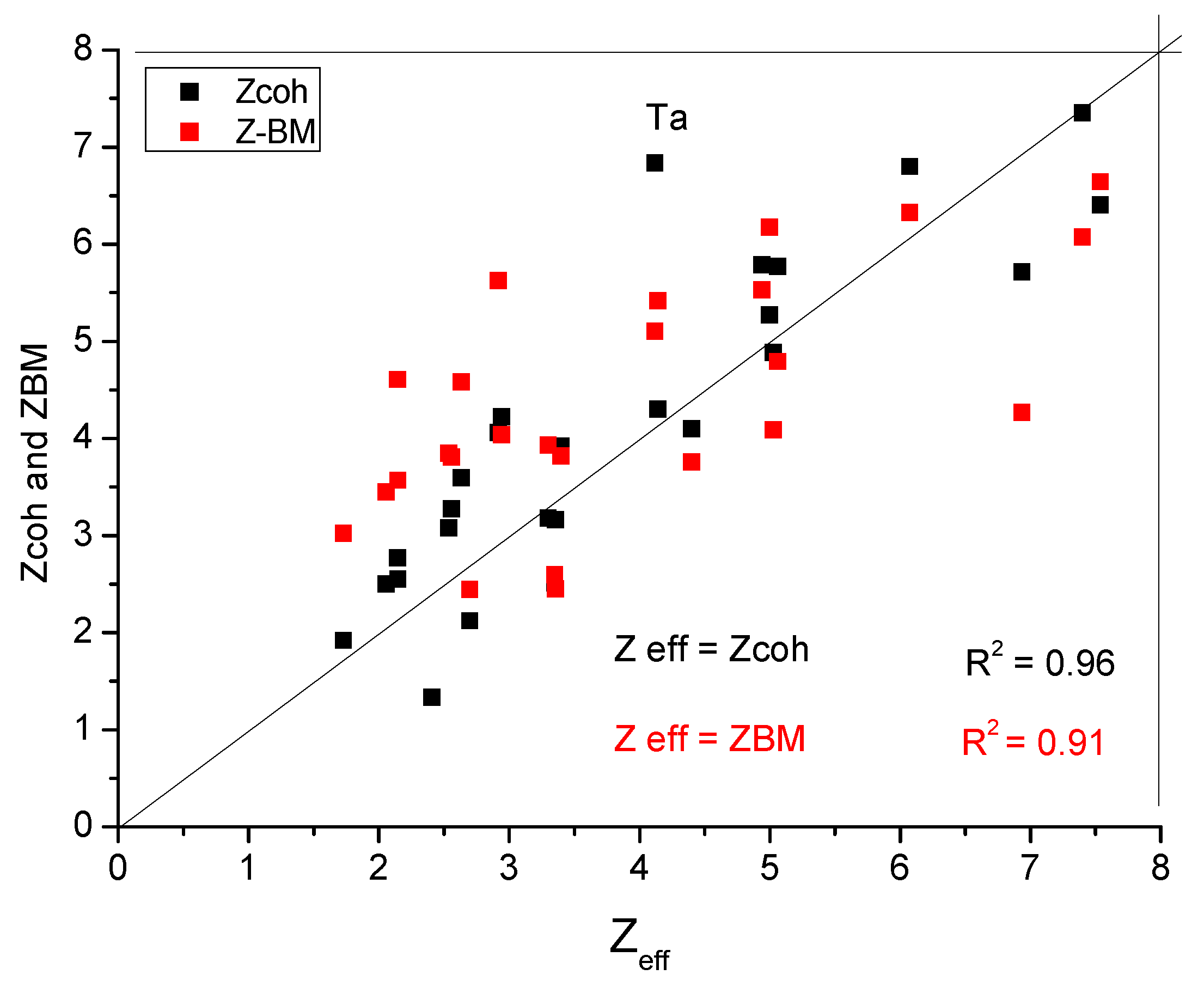
- We have considered the possibilities and decided to use the Goldschmidt radius for R and the valence calculated from the cohesive energy for Zcoh.
- -
- -
- In the case of alloys, our formula gives a lower bound on the hardness only. It is not suitable for calculating the increase of hardness caused by solid solution, grain size, precipitation, and phase mixture hardnesses.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, X.; Wang, H.; Wang, W.; Fu, Z. Simple Method for the Hardness Estimation of Inorganic Crystals by the Bond Valence Model. Inorg. Chem. 2016, 55, 11089–11095. [Google Scholar] [CrossRef]
- Soto, G.; Moreno-Armenta, M.G.; Reyes-Serrato, A. The role of valence electron concentration in the cohesive properties of YBxN1−x, YCxN1−x and YNxO1−x compounds. J. Alloys Compd. 2008, 463, 559–563. [Google Scholar] [CrossRef]
- Lederer, Y.; Toher, C.; Vecchio, K.S.; Curtarolo, S. The search for high entropy alloys: A high-throughput ab-initio approach. Acta Mater. 2018, 159, 364–383. [Google Scholar] [CrossRef]
- Curtarolo, S.; Hart, G.L.W.; Nardelli, M.B.; Mingo, N.; Sanvito, S.; Levy, O. The high-throughput highway to computational materials design. Nat. Mater. 2013, 12, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Varga, L.K.; Chen, N.; Shen, J.; Vitos, L. Empirical design of single phase high-entropy alloys with high hardness. Intermetallics 2015, 58, 1–6. [Google Scholar] [CrossRef]
- Temesi, O.K.; Varga, L.K.; Chinh, N.Q.; Vitos, L. Prediction of the Cohesion Energy, Shear Modulus and Hardness of Single-Phase Metals and High-Entropy Alloys. Materials 2024, 17, 2728. [Google Scholar] [CrossRef]
- Temesi, O.K.; Varga, L.K.; Chinh, N.Q.; Vitos, L. Ductility Index for Refractory High Entropy Alloys. Crystals 2024, 14, 838. [Google Scholar] [CrossRef]
- O’Neill, H. Hardness Measurements of Metals and Alloys; Chapman and Hall: London, UK, 1967; p. 192. [Google Scholar]
- Stubičar, M.; Tonejc, A.; Radić, N. Microhardness characterization of Al–W thin films. Vacuum 2001, 61, 309–316. [Google Scholar] [CrossRef]
- Cho, K.; Schuh, C.A. W-based amorphous phase stable to high temperatures. Acta Mater. 2015, 85, 331–342. [Google Scholar] [CrossRef]
- Fazakas, E.; Varga, L.K. Glass forming ability (GFA) of Cu and Al-based alloys by melt quenching. J. Mater. Sci. Technol. 2007, 15, 211–224. [Google Scholar]
- Han, F.F.; Inoue, A.; Han, Y.; Kong, F.L.; Zhu, S.L.; Shalaan, E.; Al-Marzouki, F.; Greer, A.L. Novel Heating-Induced Reversion during Crystallization of Al-based Glassy Alloys. Sci. Rep. 2017, 7, 46113. [Google Scholar] [CrossRef] [PubMed]
- Fazakas, E.; Russew, K.; Stojanova, L.; Csanady, A.; Varga, L.K. Al85Ni9Ta6, a refractory Al-rich ternary alloy glass and its crystallization kinetics. J. Phys. Conf. Ser. 2009, 144, 012100. [Google Scholar] [CrossRef]
- Fazakas, E.; Varga, L.K. Al-U based amorphous alloys obtained by melt spinning method. Rev. Adv. Mater. Sci. 2008, 18, 494–496. [Google Scholar]
- Inoue, A.; Ohtera, K.; Tsai, A.; Masumoto, T. Aluminum-based amorphous alloys with tensile strength above 980 MPa (100 kg/mm2). Jpn. J. Appl. Phys. 1988, 27, L479. [Google Scholar] [CrossRef]
- Inoue, A. Amorphous, nanocrystalline and nanocrystalline alloys in Al-based systems. Prog. Mater. Sci. 1998, 43, 365–520. [Google Scholar] [CrossRef]
- Inoue, A.; Ohtera, K.; Tsai, A.P.; Masumoto, T. New amorphous alloys with good ductility in Al-Y-M and Al-La-M (M=Fe, Co, Ni or Cu) systems. Jpn. J. Appl. Phys. 1998, 27, L280. [Google Scholar] [CrossRef]
- Inoue, A.; Kawamura, Y.; Kinura, H.M.; Mano, H. Nanocrystalline Al-based bulk alloys with high strength above 1000 MPa. Mater. Sci. Forum 2001, 360–362, 129–136. [Google Scholar] [CrossRef]
- Inoue, A.; Sobu, S.; Louzguine, D.V.; Kimura, H.; Sasamori, K. Ultrahigh strength Al-based amorphous alloys containing Scandium. J. Mater. Res. 2004, 19, 1539–1543. [Google Scholar] [CrossRef]
- Angell, C.A. Formation of Glasses from Liquids and Biopolymers. Science 1995, 267, 1924–1935. [Google Scholar] [CrossRef]
- Yeh, J.-W.; Chen, S.K.; Lin, S.-J.; Gan, J.-Y.; Chin, T.-S.; Shun, T.-T.; Tsau, C.-H.; Chang, S.-Y. Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Lee, J.Y.; Punkkinen, M.P.J.; Schönecker, S.; Nabi, Z.; Kádas, K.; Zólyomi, V.; Koo, Y.M.; Hu, Q.M.; Ahuja, R.; Johansson, B.; et al. The surface energy and stress of metals. Surf. Sci. 2018, 674, 51–68. [Google Scholar] [CrossRef]
- Deus, P.; Schneider, H.A. Estimation of the Debye temperature of diamond-like semiconducting compounds from bulk moduli and microhardness. Cryst. Res. Technol. 1983, 18, 491–500. [Google Scholar] [CrossRef]
- Kaptay, G.; Csicsovszki, G.; Yaghmaee, M.S. An Absolute Scale for the Cohesion Energy of Pure Metals. Mater. Sci. Forum 2003, 414–415, 235–240. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, J.; Jiang, X. Correlation between hardness and elastic moduli of the covalent crystals. Comput. Mater. Sci. 2011, 50, 2287–2290. [Google Scholar] [CrossRef]
- Dovale-Farelo, V.; Tavadze, P.; Lang, L.; Bautista-Hernandez, A.; Romero, A.H. Vickers hardness prediction from machine learning methods. Sci. Rep. 2022, 12, 22475. [Google Scholar] [CrossRef] [PubMed]
- Teter, D.M. Computational alchemy: The search for new superhard materials. MRS Bull. 1998, 23, 22–27. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, J.; Wu, A.; Bai, Y.; Jiang, X. Mechanical and electronic properties of B12-based ternary crystals of orthorhombic phase. J. Phys. Condens. Matter 2010, 22, 315503. [Google Scholar] [CrossRef] [PubMed]
- Miao, N.; Sa, B.; Zhou, J.; Sun, Z. Theoretical investigation on the transition-metal borides with Ta3B4-type structure: A class of hard and refractory materials. Comput. Mater. Sci. 2011, 50, 1559–1566. [Google Scholar] [CrossRef]
- Baiko, D.A. Shear modulus of neutron star crust. Mon. Not. R. Astron. Soc. 2011, 416, 22–31. [Google Scholar] [CrossRef]
- Baiko, D.A. Shear Modulus of a Coulomb Crystal of Ions: Effects of Ion Motion and Electron Background Polarization. Contrib. Plasma Phys. 2012, 52, 157–160. [Google Scholar] [CrossRef]
- Kobyakov, D.; Pethick, C.J. Elastic properties of polycrystalline dense matter. Mon. Not. R. Astron. Soc. Lett. 2015, 449, L110–L112. [Google Scholar] [CrossRef]
- Goldschmidt, V.M. Die Gesetze der Krystallochemie. Sci. Nat. 1926, 14, 477–485. [Google Scholar] [CrossRef]
- Trömel, M.; Hübner, S. Metallradien und Ionenradien. Z. Krist. Cryst. Mater. 2000, 215, 429–432. [Google Scholar] [CrossRef]
- Vitos, L. Computational Quantum Mechanics for Materials Engineers: The EMTO Method and Applications; Engineering Materials and Processes Series; Springer: London, UK, 2007; ISBN 978-1-84628-950-7. [Google Scholar]
- Rose, J.H.; Shore, H.B. Uniform electron gas for transition metals: Input parameters. Phys. Rev. B 1993, 48, 18254–18256. [Google Scholar] [CrossRef]
- Wojciechowski, K.F. Bulk properties of the stabilized uniform ‘metallic’ electron gas in transition metals. Phys. B Condens. Matter 1996, 229, 55–62. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal | HV | R-m | R-G | R-Tak | R-Vit | Zeff | Zcoh | Z-Vit | Z-Trö | Z-Bond | Z-BM |
|---|---|---|---|---|---|---|---|---|---|---|---|
| -- | kgf/mm2 | Å, | Å, | Å, | Å, | -- | -- | ||||
| Li | -- | 1.522 | 1.52 | 1.52 | 1.45 | -- | 0.9913 | 1.118 | 1.000 | 1.090 | 1.439 |
| Be | 167 | 1.143 | 1.12 | 1.13 | 1.05 | 3.351 | 2.512 | 2.041 | 2.000 | 2.080 | 2.593 |
| Na | -- | 1.855 | 1.86 | 1.86 | 1.8 | -- | 0.9919 | 1.104 | 1.000 | 1.110 | 1.478 |
| Mg | 26 | 1.603 | 1.6 | 1.6 | 1.5 | 2.698 | 2.123 | 2.097 | 2.000 | 2.080 | 2.445 |
| Al | 16.7 | 1.431 | 1.43 | 1.43 | 1.25 | 1.728 | 1.919 | 2.928 | 3.000 | 2.760 | 3.022 |
| K | -- | 2.305 | 2.27 | 2.26 | 2.2 | -- | 1.098 | 1.136 | 1.000 | 1.210 | 1.453 |
| Ca | 17.5 | 1.969 | 1.97 | 1.97 | 1.8 | 3.356 | 3.158 | 2.167 | 2.000 | 2.220 | 2.450 |
| Sc | -- | 1.657 | 1.62 | 1.65 | 1.6 | -- | 4.220 | 2.827 | 3.000 | 2.850 | 3.210 |
| Ti | 97 | 1.474 | 1.47 | 1.47 | 1.4 | 4.400 | 4.097 | 3.137 | 3.510 | 3.200 | 3.757 |
| V | 62.8 | 1.311 | 1.34 | 1.32 | 1.35 | 2.942 | 4.218 | 3.401 | 5.000 | 3.450 | 4.034 |
| Cr | 100.6 | 1.252 | 1.28 | 1.25 | 1.4 | 3.397 | 3.919 | 3.501 | 6.000 | 3.530 | 3.821 |
| Mn | -- | 1.258 | 1.27 | 1.12 | 1.4 | -- | 2.773 | 4.413 | 3.000 | 3.410 | 2.929 |
| Fe | 60.8 | 1.241 | 1.26 | 1.24 | 1.4 | 2.559 | 3.277 | 3.216 | 4.000 | 3.330 | 3.808 |
| Co | 104.3 | 1.251 | 1.25 | 1.25 | 1.35 | 3.299 | 3.177 | 3.073 | 4.000 | 3.030 | 3.931 |
| Ni | 63.8 | 1.245 | 1.24 | 1.25 | 1.35 | 2.539 | 3.080 | 2.888 | 4.000 | 2.830 | 3.847 |
| Cu | 36.9 | 1.278 | 1.28 | 1.28 | 1.35 | 2.057 | 2.499 | 2.558 | 3.000 | 2.570 | 3.447 |
| Zn | 42 | 1.332 | 1.34 | 1.33 | 1.35 | 2.406 | 1.334 | 2.675 | 2.000 | 2.400 | -- |
| Y | -- | 1.823 | 1.8 | 1.82 | 1.8 | -- | 4.655 | 3.042 | 3.000 | 3.210 | 3.199 |
| Zr | 90.3 | 1.615 | 1.6 | 1.62 | 1.55 | 5.029 | 4.887 | 3.635 | 3.590 | 3.750 | 4.086 |
| Nb | 132 | 1.429 | 1.46 | 1.43 | 1.45 | 5.063 | 5.772 | 4.087 | 5.000 | 4.140 | 4.793 |
| Mo | 153 | 1.36 | 1.39 | 1.36 | 1.45 | 4.940 | 5.787 | 4.389 | 6.000 | 4.420 | 5.527 |
| Tc | -- | 1.371 | 1.36 | 1.35 | 1.35 | -- | -- | 4.332 | 4.900 | 4.240 | 5.663 |
| Ru | -- | 1.344 | 1.34 | 1.33 | 1.3 | -- | 4.860 | 4.177 | 6.000 | 4.050 | 5.555 |
| Rh | 124.6 | 1.345 | 1.34 | 1.34 | 1.35 | 4.143 | 4.301 | 3.804 | 4.850 | 3.670 | 5.414 |
| Pd | 46.1 | 1.374 | 1.37 | 1.37 | 1.4 | 2.634 | 3.594 | 3.214 | 4.000 | 3.150 | 4.581 |
| Ag | 25.1 | 1.445 | 1.44 | 1.44 | 1.6 | 2.148 | 2.554 | 2.665 | 3.000 | 2.700 | 3.570 |
| Hf | 176 | 1.595 | 1.59 | 1.6 | 1.55 | 6.933 | 5.714 | 3.986 | 3.750 | 3.970 | 4.263 |
| Ta | 87.3 | 1.43 | 1.46 | 1.43 | 1.45 | 4.117 | 6.838 | 4.399 | 5.000 | 4.510 | 5.100 |
| W | 343 | 1.37 | 1.39 | 1.37 | 1.35 | 7.397 | 7.353 | 4.853 | 6.000 | 4.790 | 6.074 |
| Re | 245 | 1.384 | 1.37 | 1.37 | 1.35 | 6.073 | 6.800 | 4.939 | 6.000 | 4.790 | 6.322 |
| Os | 414 | 1.36 | 1.35 | 1.35 | 1.3 | 7.535 | 6.404 | 4.907 | 6.000 | 4.720 | 6.645 |
| Ir | 176 | 1.317 | 1.35 | 1.35 | 1.35 | 4.998 | 5.271 | 4.598 | 5.000 | 4.360 | 6.174 |
| Pt | 54.9 | 1.387 | 1.38 | 1.39 | 1.35 | 2.917 | 4.057 | 3.989 | 4.000 | 3.900 | 5.622 |
| Au | 25 | 1.441 | 1.44 | 1.44 | 1.35 | 2.143 | 2.770 | 3.289 | 3.000 | 3.260 | 4.606 |
| Pb | -- | 1.75 | 1.8 | 1.76 | 1.8 | -- | 1.554 | 2.639 | 4.000 | 2.350 | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Temesi, O.K.; Chinh, N.Q.; Vitos, L.; Varga, L.K. Estimation of Hardness of Single-Phase Metallic Alloys. Crystals 2025, 15, 156. https://doi.org/10.3390/cryst15020156
Temesi OK, Chinh NQ, Vitos L, Varga LK. Estimation of Hardness of Single-Phase Metallic Alloys. Crystals. 2025; 15(2):156. https://doi.org/10.3390/cryst15020156
Chicago/Turabian StyleTemesi, Ottó K., Nguyen Q. Chinh, Levente Vitos, and Lajos K. Varga. 2025. "Estimation of Hardness of Single-Phase Metallic Alloys" Crystals 15, no. 2: 156. https://doi.org/10.3390/cryst15020156
APA StyleTemesi, O. K., Chinh, N. Q., Vitos, L., & Varga, L. K. (2025). Estimation of Hardness of Single-Phase Metallic Alloys. Crystals, 15(2), 156. https://doi.org/10.3390/cryst15020156