
What Is More Important When Calculating the Thermodynamic Properties of Organic Crystals, Density Functional, Supercell, or Energy Second-Order Derivative Method Choice?


Abstract
:1. Introduction
2. Materials and Methods
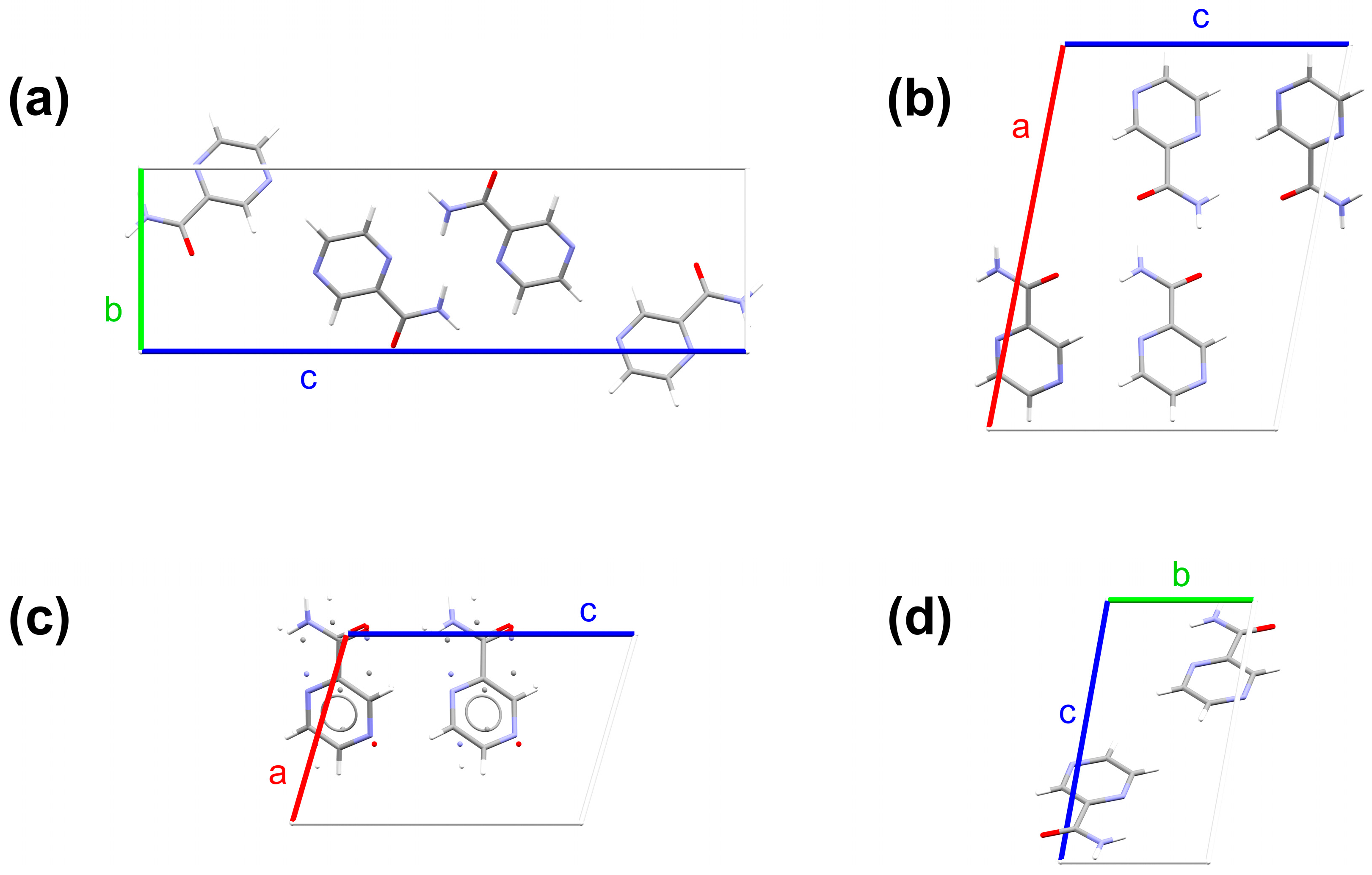
2.1. Crystallographic Structures
2.2. Periodic DFT Calculations
2.3. Phonon Calculations
3. Results
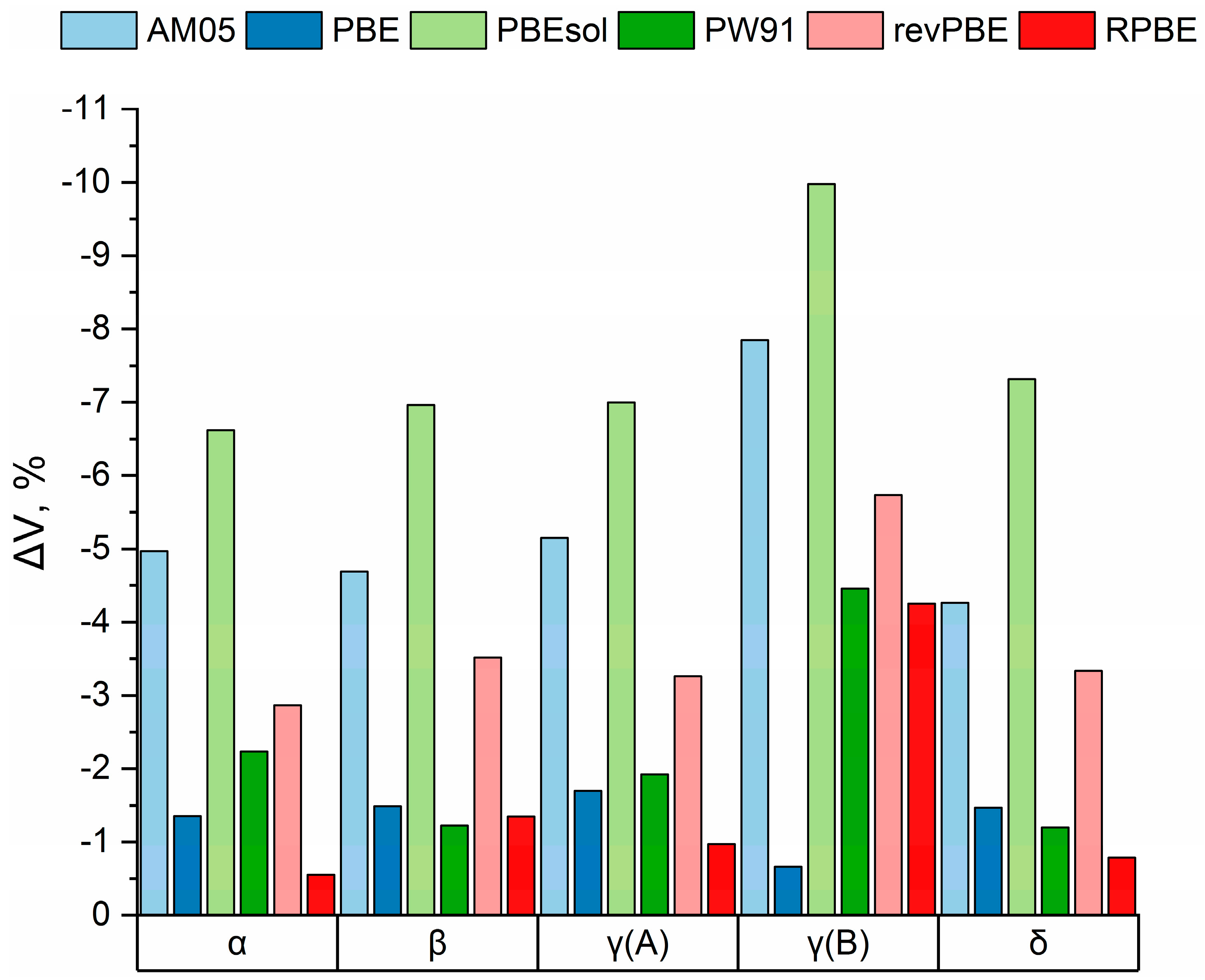
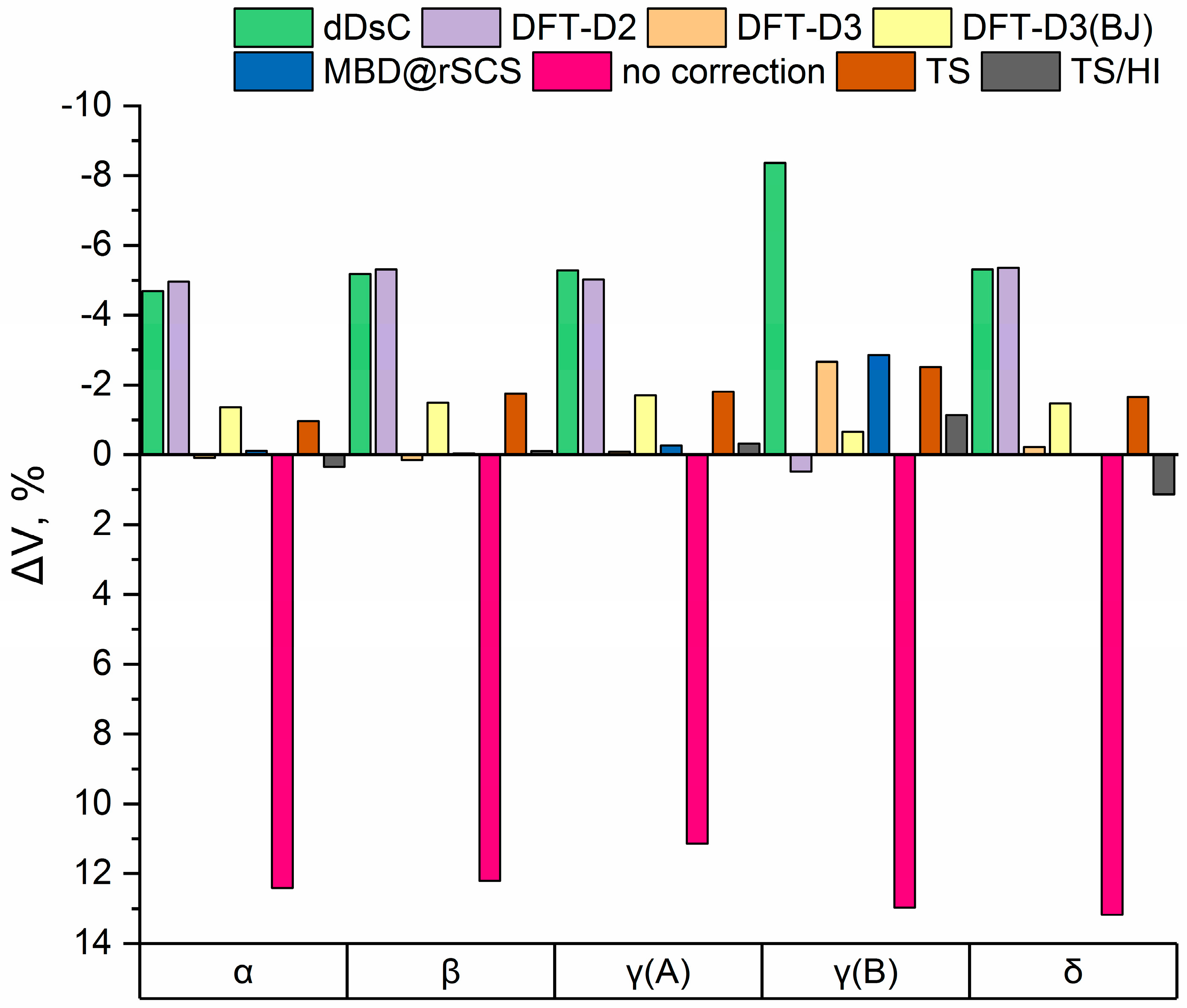
3.1. Electronic Structure Energies and Unit Cell Parameters
3.2. Gibbs Energy Calculations at 0 K
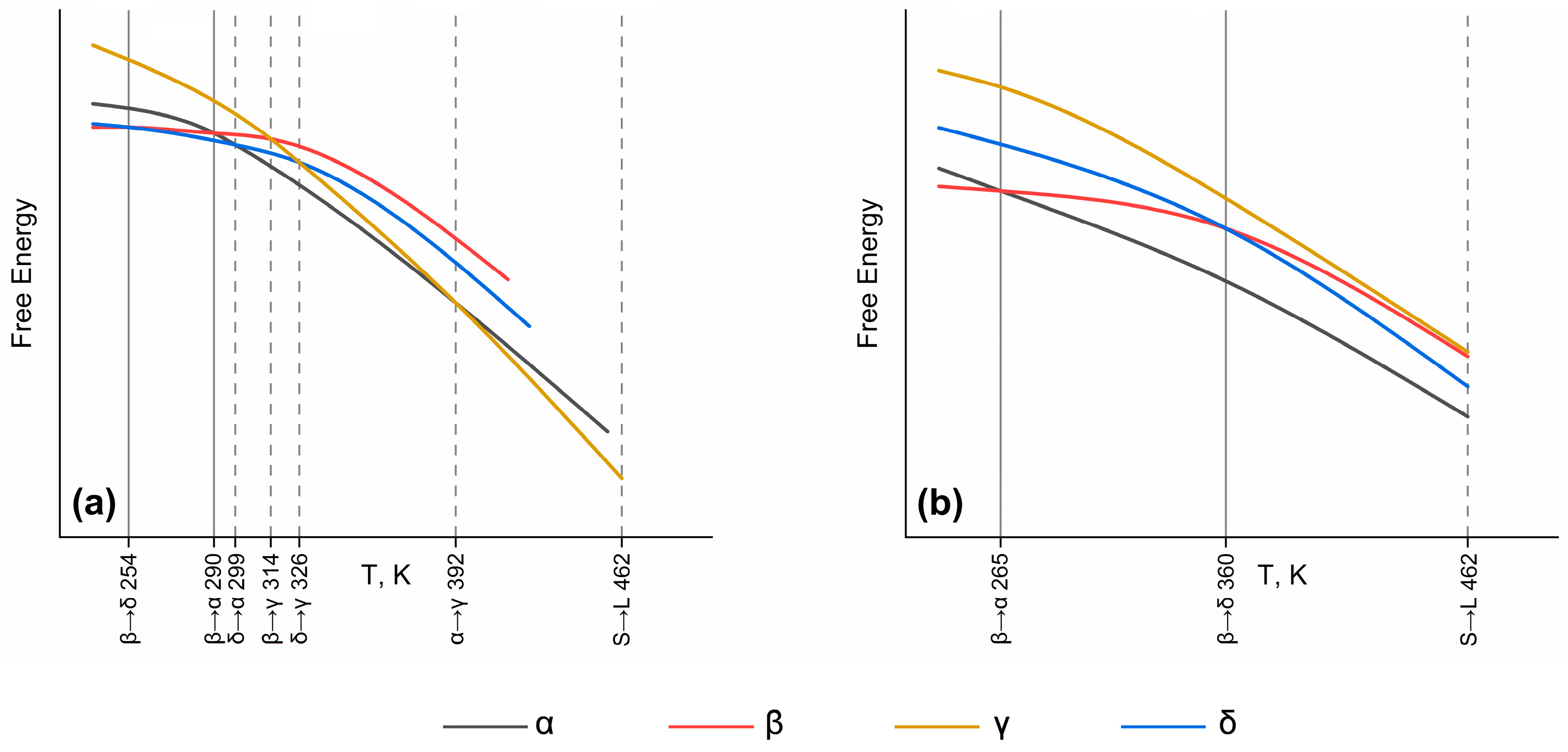
3.3. Thermodynamic Potential Calculations in the 0 K–470 K Temperature Range
4. Conclusions and Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FD | Finite difference |
| DFPT | Density functional perturbation theory |
| DFT | Density functional theory |
| PAW | Projector augmented wave (atomic pseudopotentials) |
| D3BJ | Grimme D3 with Becke–Johnson damping (function) |
| PBE | Perdew–Burke–Ernzerhof (functional) |
| rev-vdW-DF2 | Revised van der Waals density (functional) |
| AM05 | Armiento–Mattsson (functional) |
| PBEsol | Revised PBE for solids (functional) |
| PW91 | Perdew–Wang (functional) |
| revPBE | Revised PBE from Zhang and Yang (functional) |
| RPBE | Revised PBE from Hammer et al. (functional) |
| dDsC | dDsC dispersion correction method (dDsC) |
| DFT-D2 | DFT-D2 method of Grimme (dispersion correction) |
| DFT-D3 | DFT-D3 method of Grimme with zero-damping function (dispersion correction) |
| DFT-D3BJ | DFT-D2 method of Grimme with Becke–Johnson damping (dispersion correction) |
| TS | Tkatchenko–Scheffler method (dispersion correction) |
| TS/HI | Tkatchenko–Scheffler method with iterative Hirshfeld partitioning (dispersion correction) |
| MBD@rsSCS | Many-body dispersion energy method (dispersion correction) |
References
- Brog, J.-P.; Chanez, C.-L.; Crochet, A.; Fromm, K.M. Polymorphism, What It Is and How to Identify It: A Systematic Review. RSC Adv. 2013, 3, 16905. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and Fictions about Polymorphism. Chem. Soc. Rev. 2015, 44, 8619–8635. [Google Scholar] [CrossRef] [PubMed]
- Kersten, K.; Kaur, R.; Matzger, A. Survey and Analysis of Crystal Polymorphism in Organic Structures. IUCrJ 2018, 5, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J. Polymorphism of Pharmaceuticals. In Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2020; pp. 342–375. [Google Scholar]
- Tandon, R.; Tandon, N.; Gupta, N.; Gupta, R. Art of Synthesis of Desired Polymorphs: A Review. Asian J. Chem. 2018, 30, 5–14. [Google Scholar] [CrossRef]
- Ainurofiq, A.; Dinda, K.E.; Pangestika, M.W.; Himawati, U.; Wardhani, W.D.; Sipahutar, Y.T. The Effect of Polymorphism on Active Pharmaceutical Ingredients: A Review. Int. J. Res. Pharm. Sci. 2020, 11, 1621–1630. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Feeder, N.; Davey, R.J. Open Questions in Organic Crystal Polymorphism. Commun. Chem. 2020, 3, 142. [Google Scholar] [CrossRef]
- Braga, D.; Casali, L.; Grepioni, F. The Relevance of Crystal Forms in the Pharmaceutical Field: Sword of Damocles or Innovation Tools? Int. J. Mol. Sci. 2022, 23, 9013. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Ito, S.; Itai, S.; Yamamoto, K. Physicochemical Properties and Bioavailability of Carbamazepine Polymorphs and Dihydrate. Int. J. Pharm. 2000, 193, 137–146. [Google Scholar] [CrossRef]
- Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Xiao, Y.; Wang, T.; Huang, X. The Effects of Polymorphism on Physicochemical Properties and Pharmacodynamics of Solid Drugs. Curr. Pharm. Des. 2018, 24, 2375–2382. [Google Scholar] [CrossRef]
- Llinàs, A.; Box, K.J.; Burley, J.C.; Glen, R.C.; Goodman, J.M. A New Method for the Reproducible Generation of Polymorphs: Two Forms of Sulindac with Very Different Solubilities. J. Appl. Crystallogr. 2007, 40, 379–381. [Google Scholar] [CrossRef]
- Nicoud, L.; Licordari, F.; Myerson, A.S. Estimation of the Solubility of Metastable Polymorphs: A Critical Review. Cryst. Growth Des. 2018, 18, 7228–7237. [Google Scholar] [CrossRef]
- McGregor, L.; Rychkov, D.A.D.A.; Coster, P.L.P.L.; Day, S.; Drebushchak, V.A.V.A.; Achkasov, A.F.A.F.; Nichol, G.S.G.S.; Pulham, C.R.C.R.; Boldyreva, E.V.E.V. A New Polymorph of Metacetamol. CrystEngComm 2015, 17, 6183–6192. [Google Scholar] [CrossRef]
- Bonilha Dezena, R.M. Ritonavir Polymorphism: Analytical Chemistry Approach to Problem Solving in the Pharmaceutical Industry. Braz. J. Anal. Chem. 2020, 7, 12–17. [Google Scholar] [CrossRef]
- Anwar, J.; Zahn, D. Polymorphic Phase Transitions: Macroscopic Theory and Molecular Simulation. Adv. Drug Deliv. Rev. 2017, 117, 47–70. [Google Scholar] [CrossRef] [PubMed]
- Belenguer, A.M.; Lampronti, G.I.; Cruz-Cabeza, A.J.; Hunter, C.A.; Sanders, J.K.M. Solvation and Surface Effects on Polymorph Stabilities at the Nanoscale. Chem. Sci. 2016, 7, 6617–6627. [Google Scholar] [CrossRef]
- Kras, W.; Carletta, A.; Montis, R.; Sullivan, R.A.; Cruz-Cabeza, A.J. Switching Polymorph Stabilities with Impurities Provides a Thermodynamic Route to Benzamide Form III. Commun. Chem. 2021, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Brits, M.; Liebenberg, W.; de Villiers, M.M. Characterization of Polymorph Transformations That Decrease the Stability of Tablets Containing the WHO Essential Drug Mebendazole. J. Pharm. Sci. 2010, 99, 1138–1151. [Google Scholar] [CrossRef]
- Ho, R.; Shin, Y.; Chen, Y.; Poloni, L.; Chen, S.; Sheikh, A.Y. Multiscale Assessment of Api Physical Properties in the Context of Materials Science Tetrahedron Concept. In Chemical Engineering in the Pharmaceutical Industry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2019; pp. 689–712. ISBN 9781119600800. [Google Scholar]
- Drebushchak, V.A.; McGregor, L.; Rychkov, D.A. Cooling Rate “Window” in the Crystallization of Metacetamol Form II. J. Therm. Anal. Calorim. 2017, 127, 1807–1814. [Google Scholar] [CrossRef]
- Mishra, M.K.; Ramamurty, U.; Desiraju, G.R. Solid Solution Hardening of Molecular Crystals: Tautomeric Polymorphs of Omeprazole. J. Am. Chem. Soc. 2015, 137, 1794–1797. [Google Scholar] [CrossRef]
- Bag, P.P.; Chen, M.; Sun, C.C.; Reddy, C.M. Direct Correlation among Crystal Structure, Mechanical Behaviour and Tabletability in a Trimorphic Molecular Compound. CrystEngComm 2012, 14, 3865. [Google Scholar] [CrossRef]
- Ghosh, S.; Reddy, C.M. Elastic and Bendable Caffeine Cocrystals: Implications for the Design of Flexible Organic Materials. Angew. Chem. Int. Ed. 2012, 51, 10319–10323. [Google Scholar] [CrossRef]
- Raju, K.B.; Ranjan, S.; Vishnu, V.S.; Bhattacharya, M.; Bhattacharya, B.; Mukhopadhyay, A.K.; Reddy, C.M. Rationalizing Distinct Mechanical Properties of Three Polymorphs of a Drug Adduct by Nanoindentation and Energy Frameworks Analysis: Role of Slip Layer Topology and Weak Interactions. Cryst. Growth Des. 2018, 18, 3927–3937. [Google Scholar] [CrossRef]
- Masunov, A.E.; Wiratmo, M.; Dyakov, A.A.; Matveychuk, Y.V.; Bartashevich, E.V. Virtual Tensile Test for Brittle, Plastic, and Elastic Polymorphs of 4-Bromophenyl 4-Bromobenzoate. Cryst. Growth Des. 2020, 20, 6093–6100. [Google Scholar] [CrossRef]
- Domalski, E.S.; Hearing, E.D. Heat Capacities and Entropies of Organic Compounds in the Condensed Phase. Volume III. J. Phys. Chem. Ref. Data 1996, 25, 1. [Google Scholar] [CrossRef]
- De Wit, H.G.; Van Miltenburg, J.; De Kruif, C. Thermodynamic Properties of Molecular Organic Crystals Containing Nitrogen, Oxygen, and Sulphur 1. Vapour Pressures and Enthalpies of Sublimation. J. Chem. Thermodyn. 1983, 15, 651–663. [Google Scholar] [CrossRef]
- De Wit, H.G.; De Kruif, C.; Van Miltenburg, J. Thermodynamic Properties of Molecular Organic Crystals Containing Nitrogen, Oxygen, and Sulfur II. Molar Heat Capacities of Eight Compounds by Adiabatic Calorimetry. J. Chem. Thermodyn. 1983, 15, 891–902. [Google Scholar] [CrossRef]
- De Wit, H.G.M.; Offringa, J.C.A.; De Kruif, C.G.; Van Miltenburg, J.C. Thermodynamic Properties of Molecular Organic Crystals Containing Nitrogen, Oxygen and Sulfur. III. Molar Heat Capacities Measured by Differential Scanning Calorimetry. Thermochim. Acta 1983, 65, 43–51. [Google Scholar] [CrossRef]
- Schnieders, M.J.; Baltrusaitis, J.; Shi, Y.; Chattree, G.; Zheng, L.; Yang, W.; Ren, P. The Structure, Thermodynamics, and Solubility of Organic Crystals from Simulation with a Polarizable Force Field. J. Chem. Theory Comput. 2012, 8, 1721–1736. [Google Scholar] [CrossRef]
- Palmer, D.S.; McDonagh, J.L.; Mitchell, J.B.O.; van Mourik, T.; Fedorov, M.V. First-Principles Calculation of the Intrinsic Aqueous Solubility of Crystalline Druglike Molecules. J. Chem. Theory Comput. 2012, 8, 3322–3337. [Google Scholar] [CrossRef]
- Dybeck, E.C.; Schieber, N.P.; Shirts, M.R. Effects of a More Accurate Polarizable Hamiltonian on Polymorph Free Energies Computed Efficiently by Reweighting Point-Charge Potentials. J. Chem. Theory Comput. 2016, 12, 3491–3505. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, J.G.; Grimme, S. Accurate Modeling of Organic Molecular Crystals by Dispersion-Corrected Density Functional Tight Binding (DFTB). J. Phys. Chem. Lett. 2014, 5, 1785–1789. [Google Scholar] [CrossRef] [PubMed]
- Červinka, C.; Fulem, M.; Stoffel, R.P.; Dronskowski, R. Thermodynamic Properties of Molecular Crystals Calculated within the Quasi-Harmonic Approximation. J. Phys. Chem. A 2016, 120, 2022–2034. [Google Scholar] [CrossRef]
- Bidault, X.; Chaudhuri, S. Improved Predictions of Thermomechanical Properties of Molecular Crystals from Energy and Dispersion Corrected DFT. J. Chem. Phys. 2021, 154, 164105. [Google Scholar] [CrossRef]
- Kapil, V.; Engel, E.A. A Complete Description of Thermodynamic Stabilities of Molecular Crystals. Proc. Natl. Acad. Sci. USA 2022, 119, e2111769119. [Google Scholar] [CrossRef]
- Hunnisett, L.M.; Nyman, J.; Francia, N.; Abraham, N.S.; Adjiman, C.S.; Aitipamula, S.; Alkhidir, T.; Almehairbi, M.; Anelli, A.; Anstine, D.M.; et al. The Seventh Blind Test of Crystal Structure Prediction: Structure Generation Methods. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2024, 80, 517–547. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Reilly, A.M. Sixth Blind Test of Organic Crystal-Structure Prediction Methods. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 776–777. [Google Scholar] [CrossRef]
- Bardwell, D.A.; Adjiman, C.S.; Arnautova, Y.A.; Bartashevich, E.; Boerrigter, S.X.M.; Braun, D.E.; Cruz-Cabeza, A.J.; Day, G.M.; Della Valle, R.G.; Desiraju, G.R.; et al. Towards Crystal Structure Prediction of Complex Organic—A Report on the Fifth Blind Test. Acta Crystallogr. Sect. B Struct. Sci. 2011, 67, 535–551. [Google Scholar] [CrossRef]
- Hoja, J.; Reilly, A.M.; Tkatchenko, A. First-Principles Modeling of Molecular Crystals: Structures and Stabilities, Temperature and Pressure. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1294. [Google Scholar] [CrossRef]
- Hasan, S.; Rulis, P.; Ching, W.Y. First-Principles Calculations of the Structural, Electronic, Optical, and Mechanical Properties of 21 Pyrophosphate Crystals. Crystals 2022, 12, 1139. [Google Scholar] [CrossRef]
- Dubok, A.S.; Rychkov, D. Deformcell: A Python Script to Simplify and Fasten Mechanical Properties Calculations of Molecular Crystals in VASP Package for Research and Teaching Purposes. J. Struct. Chem. 2024, 65, 132571. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty Years of Density Functional Theory in Computational Chemistry: An Overview and Extensive Assessment of 200 Density Functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density Functional Theory Is Straying from the Path toward the Exact Functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Kamencek, T.; Wieser, S.; Kojima, H.; Bedoya-Martínez, N.; Dürholt, J.P.; Schmid, R.; Zojer, E. Evaluating Computational Shortcuts in Supercell-Based Phonon Calculations of Molecular Crystals: The Instructive Case of Naphthalene. J. Chem. Theory Comput. 2020, 16, 2716–2735. [Google Scholar] [CrossRef]
- Duong, T.C.; Paulson, N.H.; Stan, M.; Chaudhuri, S. An Efficient Approximation of the Supercell Approach to the Calculation of the Full Phonon Spectrum. Calphad 2021, 72, 102215. [Google Scholar] [CrossRef]
- Shang, H.; Carbogno, C.; Rinke, P.; Scheffler, M. Lattice Dynamics Calculations Based on Density-Functional Perturbation Theory in Real Space. Comput. Phys. Commun. 2017, 215, 26–46. [Google Scholar] [CrossRef]
- Running Phonon Calculations. Available online: https://www.tcm.phy.cam.ac.uk/castep/Phonons_Guide/2-sec:examples.html#sec:dfpt-gamma (accessed on 10 March 2025).
- Sholl, D.S.; Steckel, J.A. Density Functional Theory; Wiley: Hoboken, NJ, USA, 2009; ISBN 9780470373170. [Google Scholar]
- Lee, J.G. Computational Materials Science, 2nd ed.; CRC Press, Taylor & Francis: Boca Raton, FL, USA, 2016; ISBN 9781315368429. [Google Scholar]
- Martin, R.M. Electronic Structure; Cambridge University Press: Cambridge, UK, 2020; ISBN 9781108555586. [Google Scholar]
- Takaki, Y.; Sasada, Y.; Watanabé, T. The Crystal Structure of α-Pyrazinamide. Acta Crystallogr. 1960, 13, 693–702. [Google Scholar] [CrossRef]
- Rø, G.; Sørum, H. The Crystal and Molecular Structure of β-Pyrazinecarboxamide. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1972, 28, 991–998. [Google Scholar] [CrossRef]
- Tamura, C.; Kuwano, H. Crystallographic Data of Carboxylic Acids and Carboxyamides of Picoline and Pyrazine Derivatives. Acta Crystallogr. 1961, 14, 693–694. [Google Scholar] [CrossRef]
- Rø, G.; Sørum, H. The Crystal and Molecular Structure of δ-Pyrazinecarboxamide. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1972, 28, 1677–1684. [Google Scholar] [CrossRef]
- Cherukuvada, S.; Thakuria, R.; Nangia, A. Pyrazinamide Polymorphs: Relative Stability and Vibrational Spectroscopy. Cryst. Growth Des. 2010, 10, 3931–3941. [Google Scholar] [CrossRef]
- Castro, R.A.E.; Maria, T.M.R.; Évora, A.O.L.; Feiteira, J.C.; Silva, M.R.; Beja, A.M.; Canotilho, J.; Eusébio, M.E.S. A New Insight into Pyrazinamide Polymorphic Forms and Their Thermodynamic Relationships. Cryst. Growth Des. 2010, 10, 274–282. [Google Scholar] [CrossRef]
- Borba, A.; Albrecht, M.; Gómez-Zavaglia, A.; Suhm, M.A.; Fausto, R. Low Temperature Infrared Spectroscopy Study of Pyrazinamide: From the Isolated Monomer to the Stable Low Temperature Crystalline Phase. J. Phys. Chem. A 2010, 114, 151–161. [Google Scholar] [CrossRef]
- Rajalakshmi, G.; Hathwar, V.R.; Kumaradhas, P. Intermolecular Interactions, Charge-Density Distribution and the Electrostatic Properties of Pyrazinamide Anti-TB Drug Molecule: An Experimental and Theoretical Charge-Density Study. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, N.; Ciochoń, P.; Petriĉek, V.; Madsen, A.Ø. Polymorph Stability Prediction: On the Importance of Accurate Structures: A Case Study of Pyrazinamide. Cryst. Growth Des. 2014, 14, 381–388. [Google Scholar] [CrossRef]
- Hoser, A.A.; Rekis, T.; Madsen, A.Ø. Dynamics and Disorder: On the Stability of Pyrazinamide Polymorphs. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2022, 78, 416–424. [Google Scholar] [CrossRef]
- David, S.; Nidhin, P.V.; Srinivasan, P. Ab Initio Prediction of the Polymorphic Structures of Pyrazinamide: A Validation Study. J. Serbian Chem. Soc. 2016, 81, 763–776. [Google Scholar] [CrossRef]
- Li, K.; Gbabode, G.; Vergé-Depré, M.; Robert, B.; Barrio, M.; Itié, J.-P.; Tamarit, J.-L.; Rietveld, I.B. The Pressure–temperature Phase Diagram of Tetramorphic Pyrazinamide. CrystEngComm 2022, 24, 5041–5051. [Google Scholar] [CrossRef]
- Dubok, A.S.; Rychkov, D.A. Relative Stability of Pyrazinamide Polymorphs Revisited: A Computational Study of Bending and Brittle Forms Phase Transitions in a Broad Temperature Range. Crystals 2023, 13, 617. [Google Scholar] [CrossRef]
- Nangia, A.; Srinivasulu, A. CSD Communication (Private Communication); CCDC: Boston, MA, USA, 2005. [Google Scholar] [CrossRef]
- Björkman, T. CIF2Cell: Generating Geometries for Electronic Structure Programs. Comput. Phys. Commun. 2011, 182, 1183–1186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Hamada, I. Van Der Waals Density Functional Made Accurate. Phys. Rev. B 2014, 89, 121103. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Armiento, R.; Mattsson, A.E. Functional Designed to Include Surface Effects in Self-Consistent Density Functional Theory. Phys. Rev. B 2005, 72, 085108. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Armiento, R.; Paier, J.; Kresse, G.; Wills, J.M.; Mattsson, T.R. The AM05 Density Functional Applied to Solids. J. Chem. Phys. 2008, 128, 084714. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Armiento, R. Implementing and Testing the AM05 Spin Density Functional. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 79, 155101. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, W. Comment on “Generalized Gradient Approximation Made Simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Steinmann, S.N.; Corminboeuf, C. A Generalized-Gradient Approximation Exchange Hole Model for Dispersion Coefficients. J. Chem. Phys. 2011, 134, 044117. [Google Scholar] [CrossRef]
- Steinmann, S.N.; Corminboeuf, C. Comprehensive Benchmarking of a Density-Dependent Dispersion Correction. J. Chem. Theory Comput. 2011, 7, 3567–3577. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type Density Functional Constructed with a Long-range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Tkatchenko, A.; DiStasio, R.A.; Car, R.; Scheffler, M. Accurate and Efficient Method for Many-Body van Der Waals Interactions. Phys. Rev. Lett. 2012, 108, 236402. [Google Scholar] [CrossRef]
- Ambrosetti, A.; Reilly, A.M.; DiStasio, R.A.; Tkatchenko, A. Long-Range Correlation Energy Calculated from Coupled Atomic Response Functions. J. Chem. Phys. 2014, 140, 18A508. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]
- Bučko, T.; Lebègue, S.; Hafner, J.; Ángyán, J.G. Improved Density Dependent Correction for the Description of London Dispersion Forces. J. Chem. Theory Comput. 2013, 9, 4293–4299. [Google Scholar] [CrossRef] [PubMed]
- Bučko, T.; Lebègue, S.; Ángyán, J.G.; Hafner, J. Extending the Applicability of the Tkatchenko-Scheffler Dispersion Correction via Iterative Hirshfeld Partitioning. J. Chem. Phys. 2014, 141, 034114. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First Principles Phonon Calculations in Materials Science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Togo, A. First-Principles Phonon Calculations with Phonopy and Phono3py. J. Phys. Soc. Jpn. 2023, 92, 012001. [Google Scholar] [CrossRef]
- Borioni, J.L.; Puiatti, M.; Vera, D.M.A.; Pierini, A.B. In Search of the Best DFT Functional for Dealing with Organic Anionic Species. Phys. Chem. Chem. Phys. 2017, 19, 9189–9198. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, V.; Abburu, S.; Alsberg, B.K. Can Chemometrics Be Used to Guide the Selection of Suitable DFT Functionals? Chemom. Intell. Lab. Syst. 2015, 142, 87–94. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Grimme, S. Organic Crystal Polymorphism: A Benchmark for Dispersion-Corrected Mean-Field Electronic Structure Methods. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 502–513. [Google Scholar] [CrossRef]
- Terentjev, A.V.; Constantin, L.A.; Pitarke, J.M. Dispersion-Corrected PBEsol Exchange-Correlation Functional. Phys. Rev. B 2018, 98, 214108. [Google Scholar] [CrossRef]
- Csonka, G.I.; Perdew, J.P.; Ruzsinszky, A.; Philipsen, P.H.T.; Lebègue, S.; Paier, J.; Vydrov, O.A.; Ángyán, J.G. Assessing the Performance of Recent Density Functionals for Bulk Solids. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 79, 155107. [Google Scholar] [CrossRef]
- Beran, G.J.O. Modeling Polymorphic Molecular Crystals with Electronic Structure Theory. Chem. Rev. 2016, 116, 5567–5613. [Google Scholar] [CrossRef] [PubMed]
- Nyman, J.; Day, G.M. Static and Lattice Vibrational Energy Differences between Polymorphs. CrystEngComm 2015, 17, 5154–5165. [Google Scholar] [CrossRef]
- Fedorov, A.Y.; Rychkov, D.A.; Losev, E.A.; Zakharov, B.A.; Stare, J.; Boldyreva, E.V. Effect of Pressure on Two Polymorphs of Tolazamide: Why No Interconversion? CrystEngComm 2017, 19, 2243–2252. [Google Scholar] [CrossRef]
- Dolgonos, G.A.; Hoja, J.; Boese, A.D. Revised Values for the X23 Benchmark Set of Molecular Crystals. Phys. Chem. Chem. Phys. 2019, 21, 24333–24344. [Google Scholar] [CrossRef]
- O’Connor, D.; Bier, I.; Hsieh, Y.-T.; Marom, N. Performance of Dispersion-Inclusive Density Functional Theory Methods for Energetic Materials. J. Chem. Theory Comput. 2022, 18, 4456–4471. [Google Scholar] [CrossRef]
- Li, K.; Gbabode, G.; Barrio, M.; Tamarit, J.-L.; Vergé-Depré, M.; Robert, B.; Rietveld, I.B. The Phase Relationship between the Pyrazinamide Polymorphs α and γ. Int. J. Pharm. 2020, 580, 119230. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | VASP (5.4.4/6.4.3) | CASTEP (23.1.1) | Quantum Espresso (7.3) | ABINIT (10.2.3) |
|---|---|---|---|---|
| DFPT implementation | q = 0 | q = 0 and q ≠ 0 | q = 0 and q ≠ 0 | q = 0 and q ≠ 0 |
| Calculation Method | α | β | γ(A) | γ(B) | γ | δ |
|---|---|---|---|---|---|---|
| PBE-D3BJ, kJ/mol * | 2.7 (III) | 0 (I) | 3.1 | 10.0 | 4.1 (IV) | 2.1 (II) |
| rev-vdW-DF2, kJ/mol * | 2.1 (III) | 0 (I) | 2.0 | 7.0 | 2.6 (IV) | 1.1 (II) |
| Calculation Method | A | Β | γ(A) | γ(B) | γ | δ |
|---|---|---|---|---|---|---|
| FD/PBE-D3BJ supercell, kJ/mol | 2.0 (II–III) | 0 (I) | 3.1 | 9.6 | 4.0 (IV) | 2.0 (II–III) |
| FD/PBE-D3BJ primitive cell, kJ/mol | 1.8 (II) | 0 (I) | 2.9 | 9.9 | 3.8 (IV) | 2.2 (III) |
| DFPT/rev-vdW-DF2 supercell, kJ/mol | 1.5 (III) | 0 (I) | 1.9 | 6.8 | 2.6 (IV) | 1.0 (II) |
| DFPT/rev-vdW-DF2 primitive cell, kJ/mol | 1.3 (II–III) | 0 (I) | 1.8 | 7.1 | 2.5 (IV) | 1.3 (II–III) |
| FD/rev-vdW-DF2 primitive cell, kJ/mol | 1.3(II) | 0 (I) | 1.9 | 7.2 | 2.6 (IV) | 1.5 (III) |
| Method | Δ → α | Β → γ | Δ → γ | A → γ |
|---|---|---|---|---|
| 1 Ttrans, K | 299 | 314 | 326 | 399 |
| 1 Exp. ΔH, kJ/mol | 0.4 | 2.2 | 2.1 | 1.6 |
| FD/PBE-D3BJ supercell, kJ/mol | 0.2 | –4.5 | 1.7 | 1.6 |
| FD/PBE-D3BJ primitive cell, kJ/mol | 2.0 | 3.0 | 4.9 | 2.9 |
| DFPT/rev-vdW-DF2 supercell, kJ/mol | 0.7 | 2.1 | 1.1 | 0.3 |
| DFPT/rev-vdW-DF2 primitive cell, kJ/mol | 2.1 | 1.8 | 4.7 | 2.8 |
| FD/rev-vdW-DF2 primitive cell, kJ/mol | 1.8 | 1.9 | 4.6 | 3.1 |
| FD/PBE-D3BJ Supercell | FD/PBE-D3BJ Primitive Cell | DFPT/Rev-Vdw-DF2 Supercell | DFPT/Rev-vdW-DF2 Primitive Cell | FD/Rev-Vdw-DF2 Primitive Cell | |
| FD/PBE-D3BJ supercell | 1 | 0.19 | 0.85 | −0.26 | −0.16 |
| FD/PBE-D3BJ primitive cell | 0.19 | 1 | 0.18 | 0.90 | 0.91 |
| DFPT/rev-vdW-DF2 supercell | 0.85 | 0.18 | 1 | −0.24 | −0.24 |
| DFPT/rev-vdW-DF2 primitive cell | −0.26 | 0.90 | −0.24 | 1 | 0.98 |
| FD/rev-vdW-DF2 primitive cell | −0.16 | 0.91 | −0.24 | 0.98 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubok, A.S.; Rychkov, D.A. What Is More Important When Calculating the Thermodynamic Properties of Organic Crystals, Density Functional, Supercell, or Energy Second-Order Derivative Method Choice? Crystals 2025, 15, 274. https://doi.org/10.3390/cryst15030274
Dubok AS, Rychkov DA. What Is More Important When Calculating the Thermodynamic Properties of Organic Crystals, Density Functional, Supercell, or Energy Second-Order Derivative Method Choice? Crystals. 2025; 15(3):274. https://doi.org/10.3390/cryst15030274
Chicago/Turabian StyleDubok, Aleksandr S., and Denis A. Rychkov. 2025. "What Is More Important When Calculating the Thermodynamic Properties of Organic Crystals, Density Functional, Supercell, or Energy Second-Order Derivative Method Choice?" Crystals 15, no. 3: 274. https://doi.org/10.3390/cryst15030274
APA StyleDubok, A. S., & Rychkov, D. A. (2025). What Is More Important When Calculating the Thermodynamic Properties of Organic Crystals, Density Functional, Supercell, or Energy Second-Order Derivative Method Choice? Crystals, 15(3), 274. https://doi.org/10.3390/cryst15030274