Phenomenology of the Neutral-Ionic Valence Instability in Mixed Stack Charge-Transfer Crystals



Abstract
:1. Introduction
2. Temperature Induced NIT
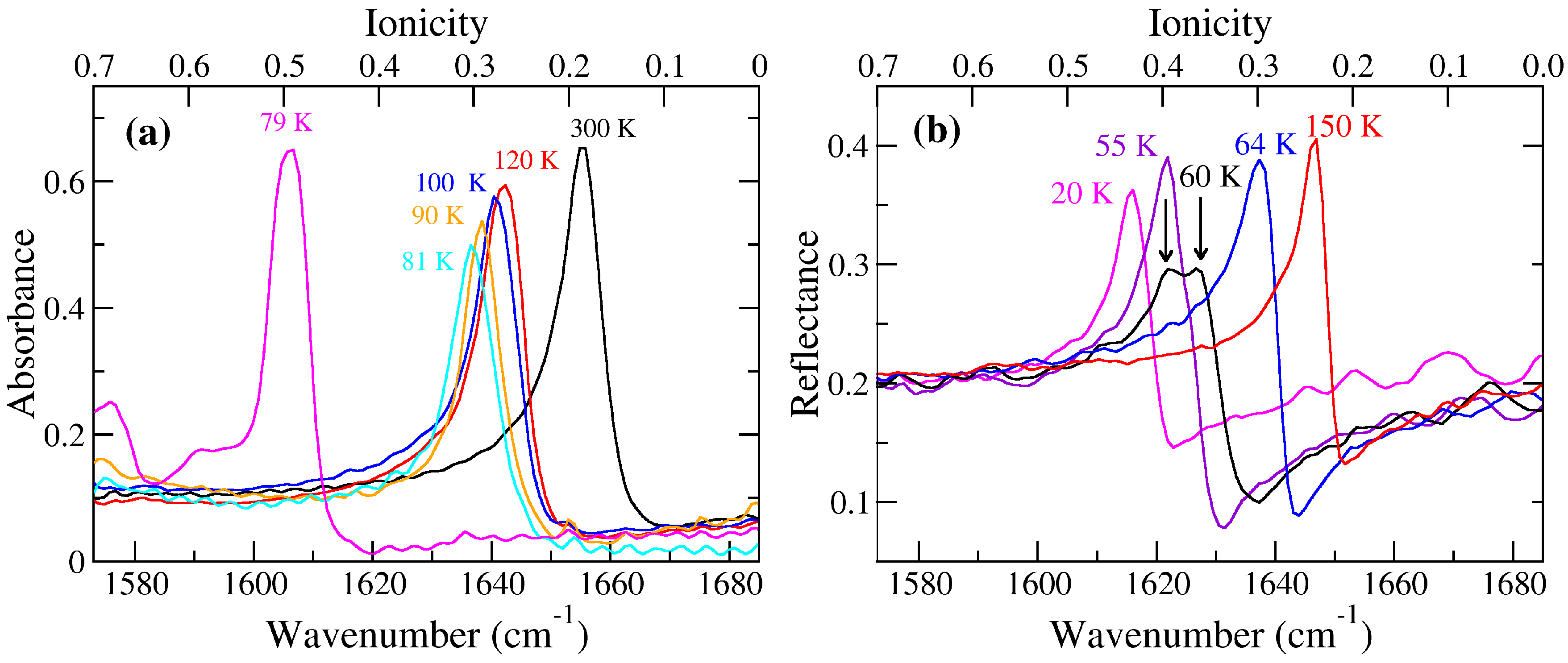
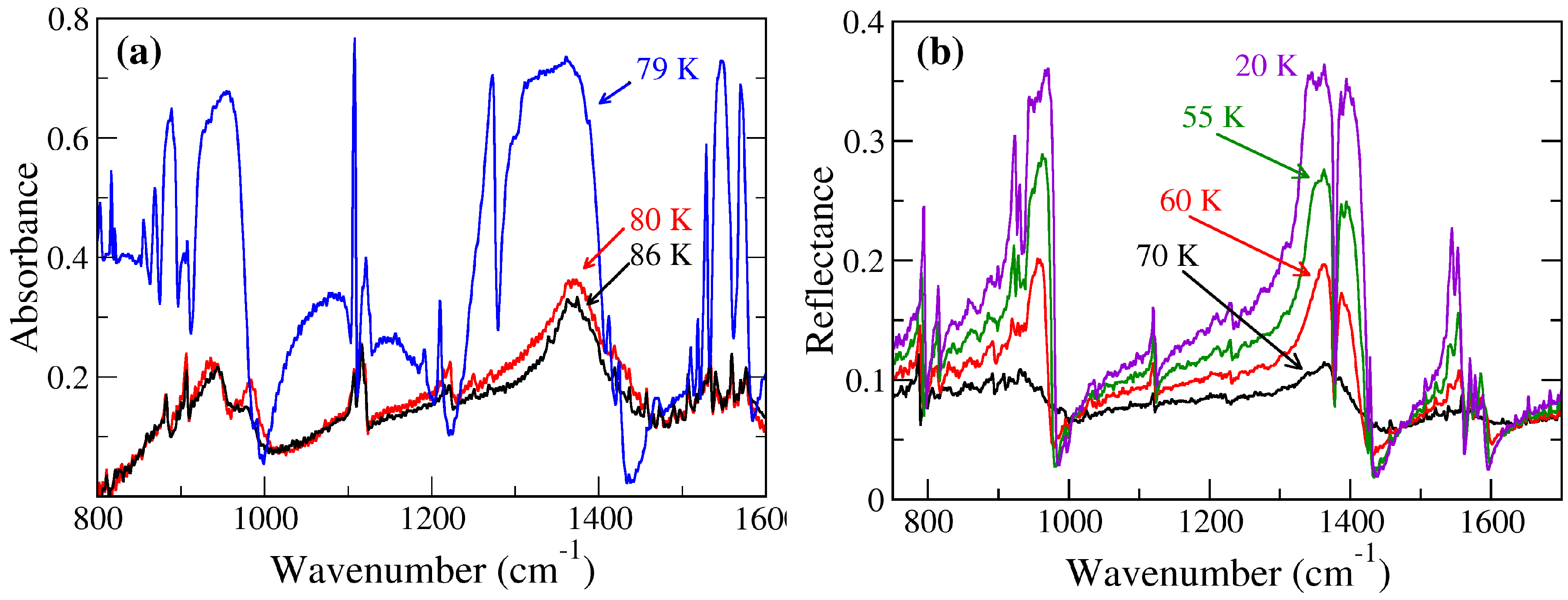
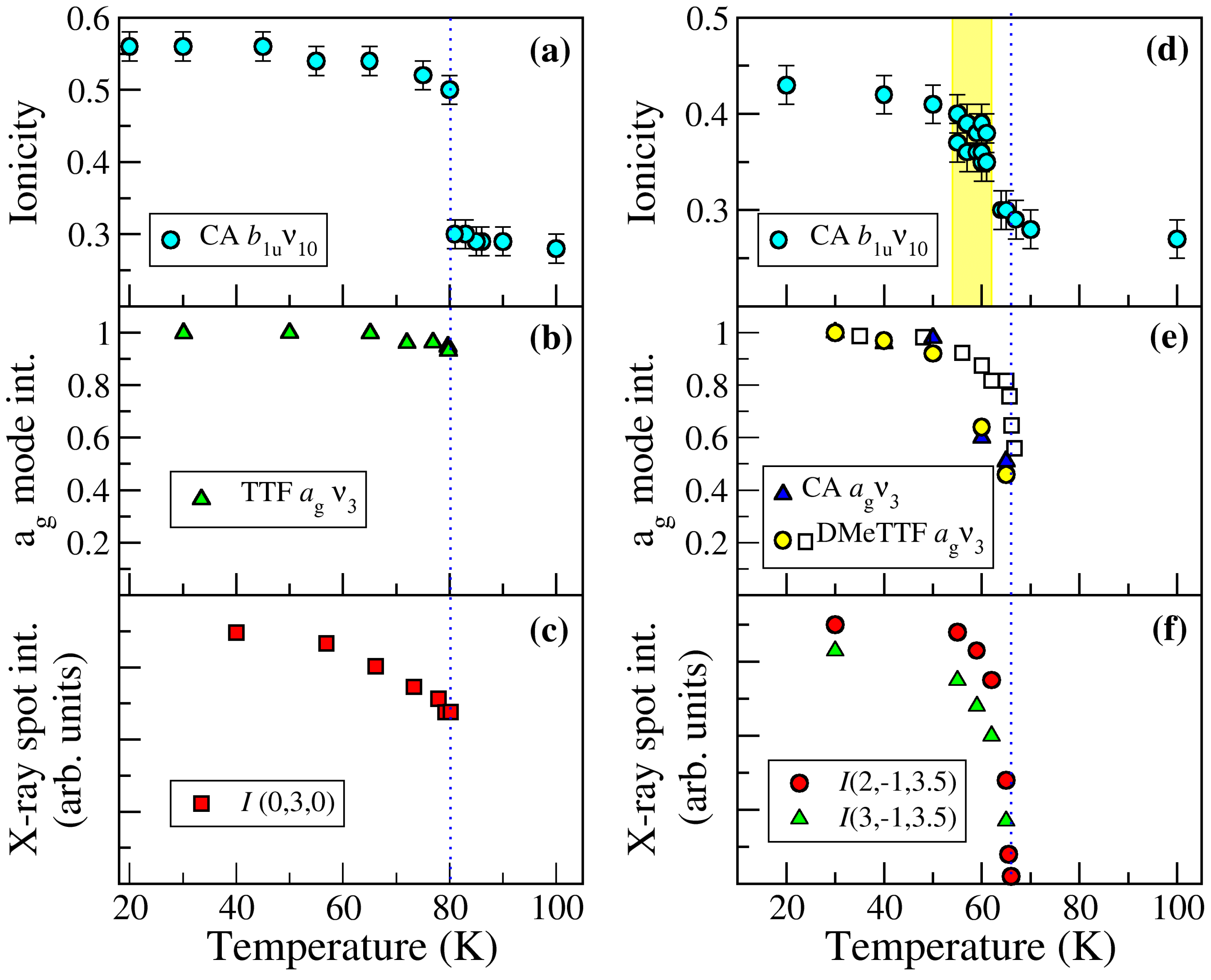
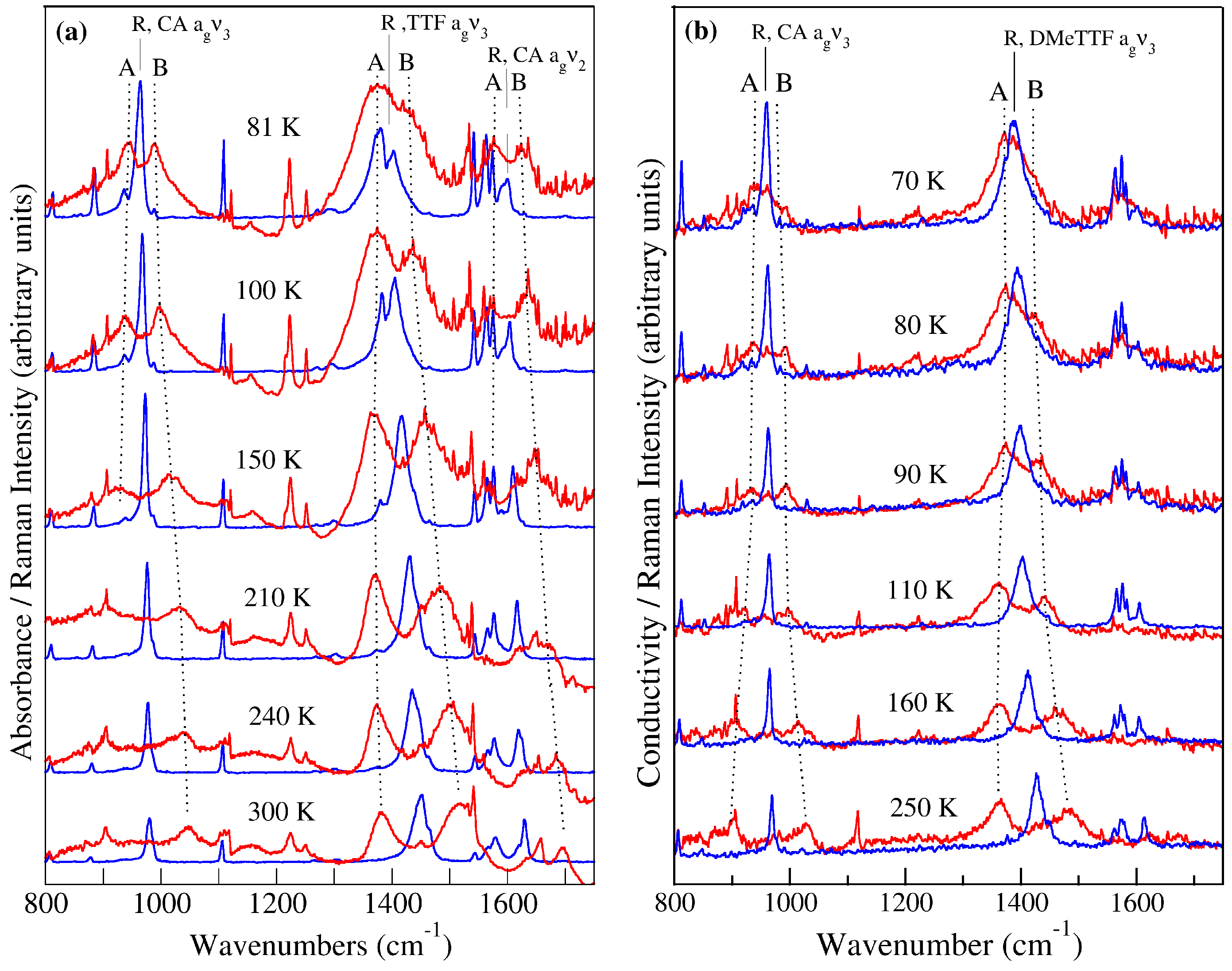
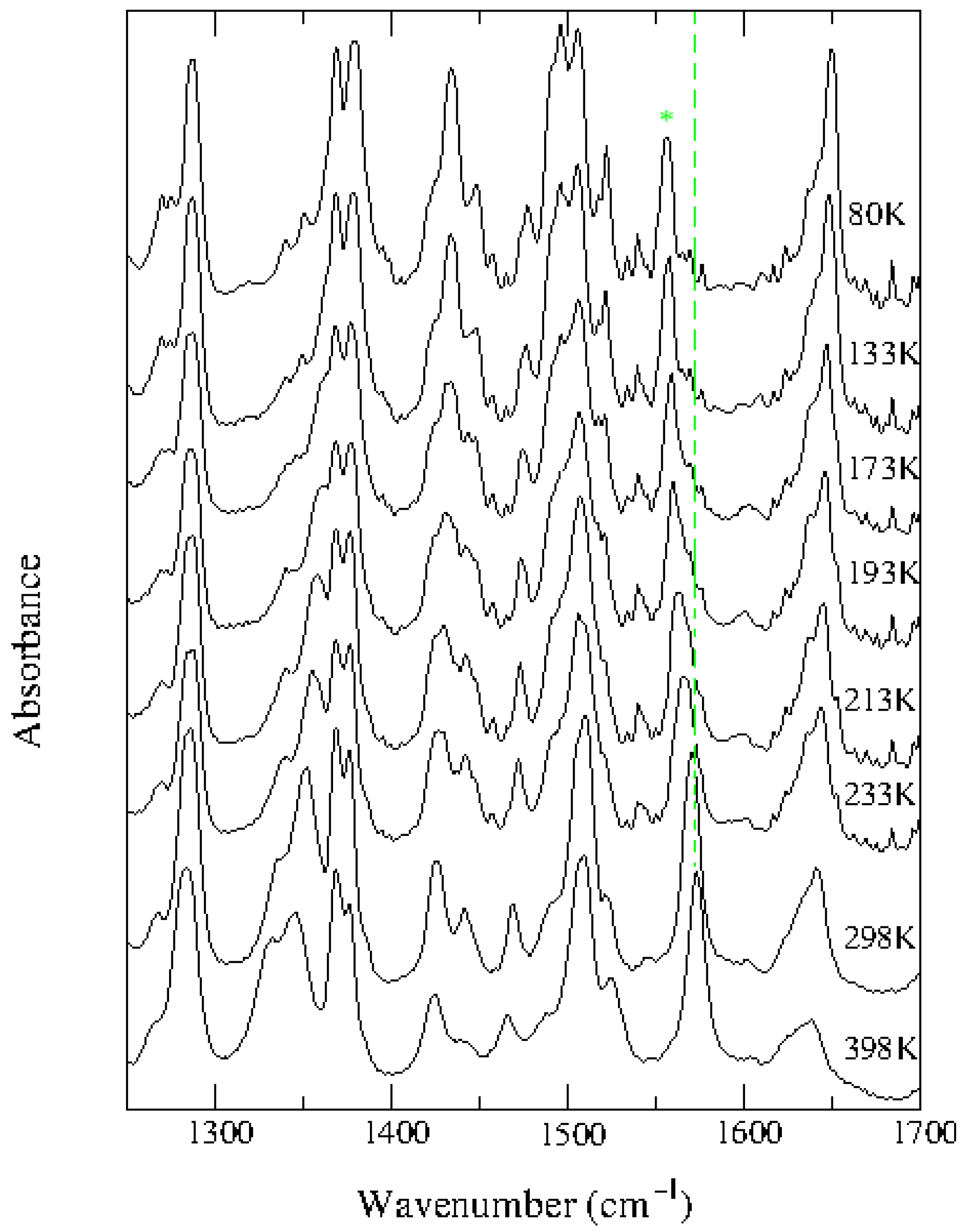
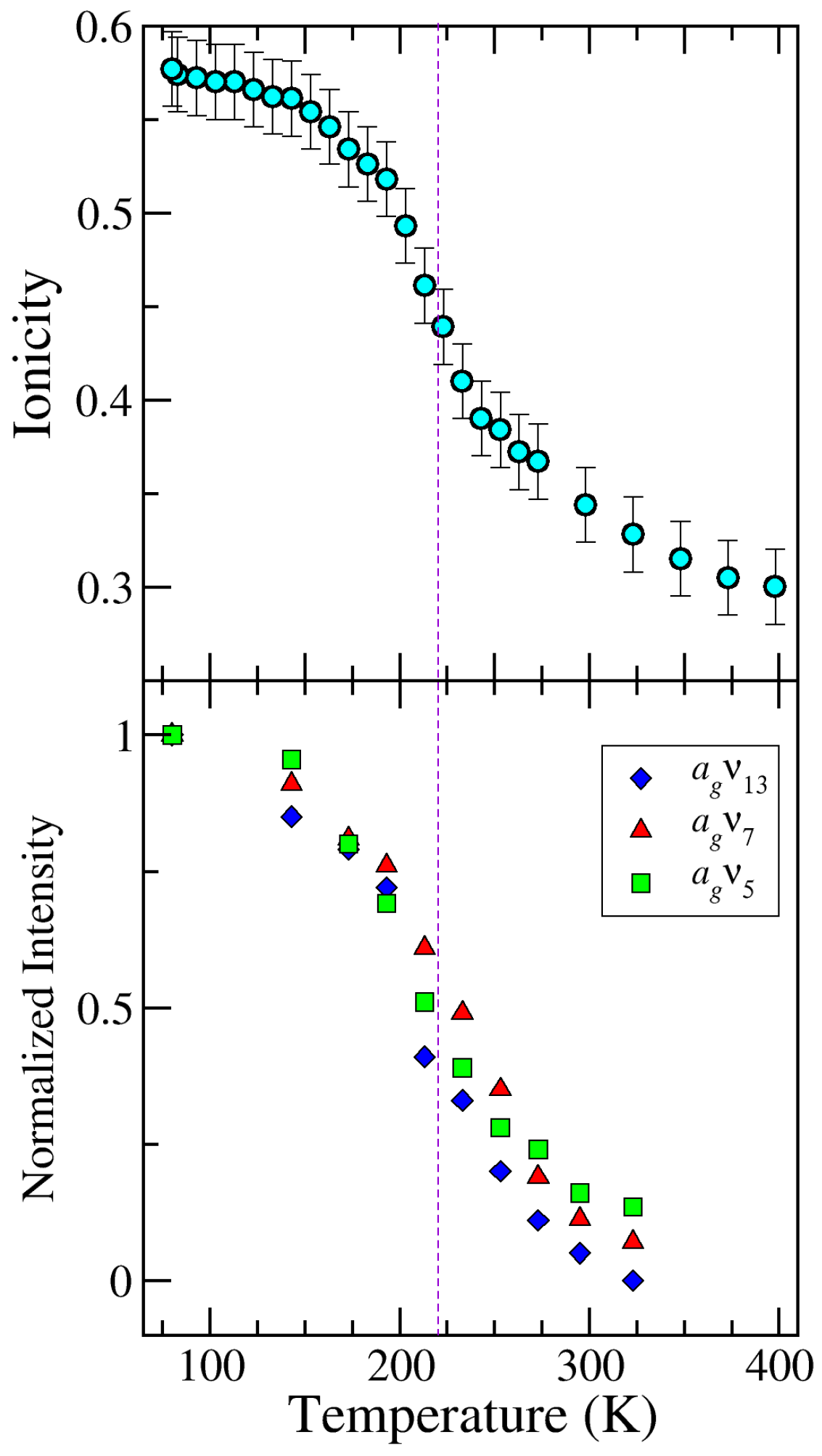
2.1. TTF–CA and DMeTTF–CA
2.1.1. Valence Instability and Stack Dimerizaion
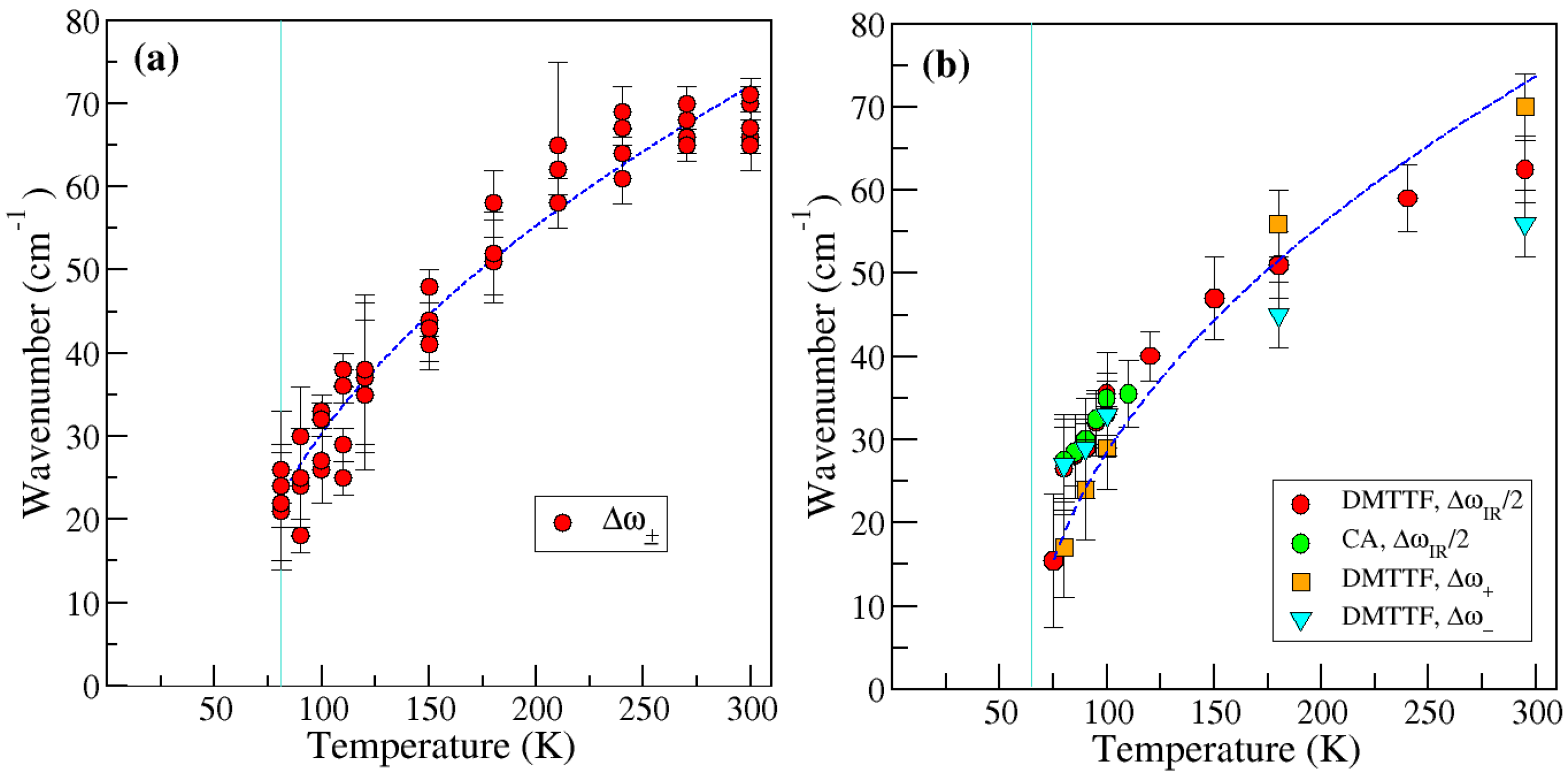
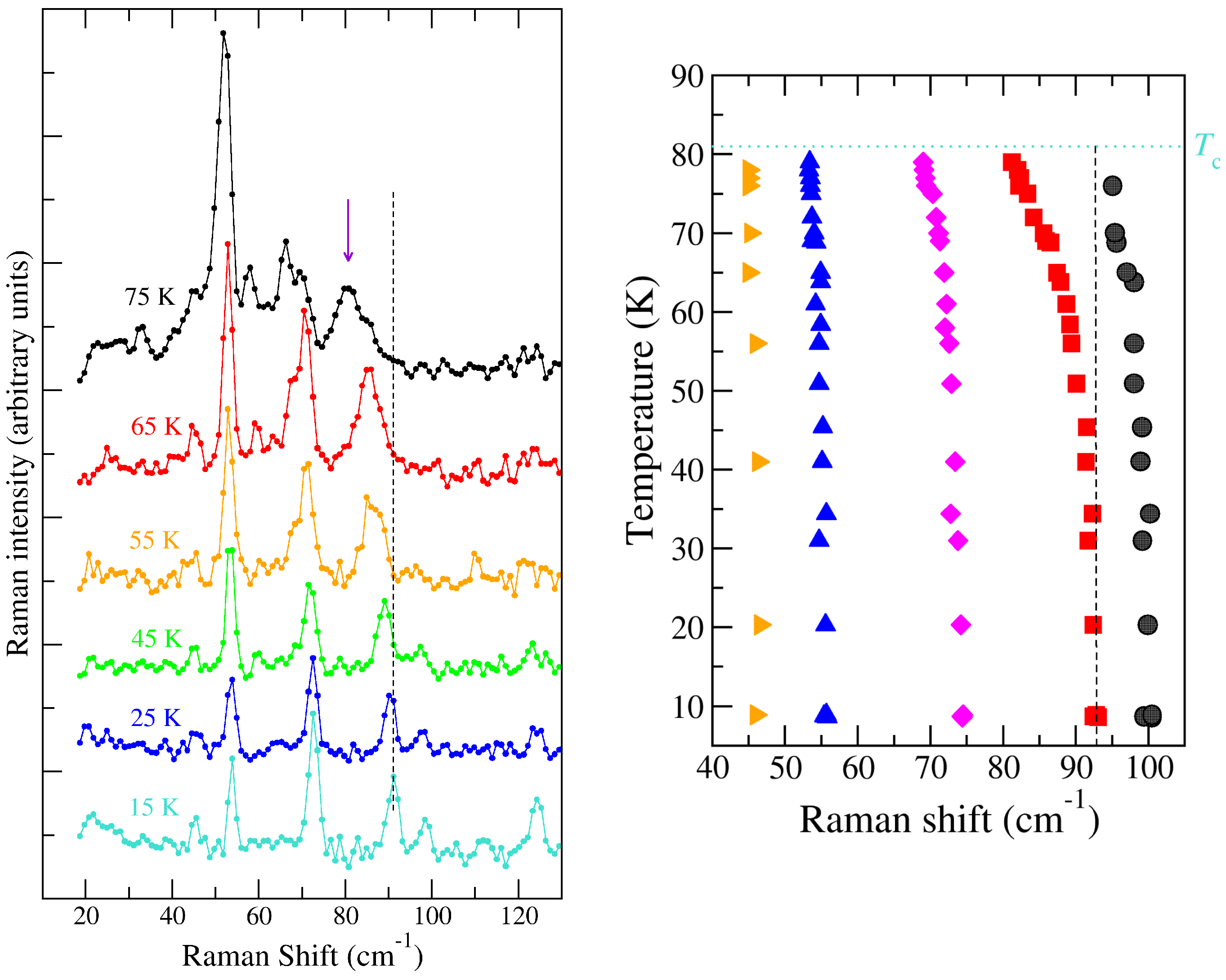
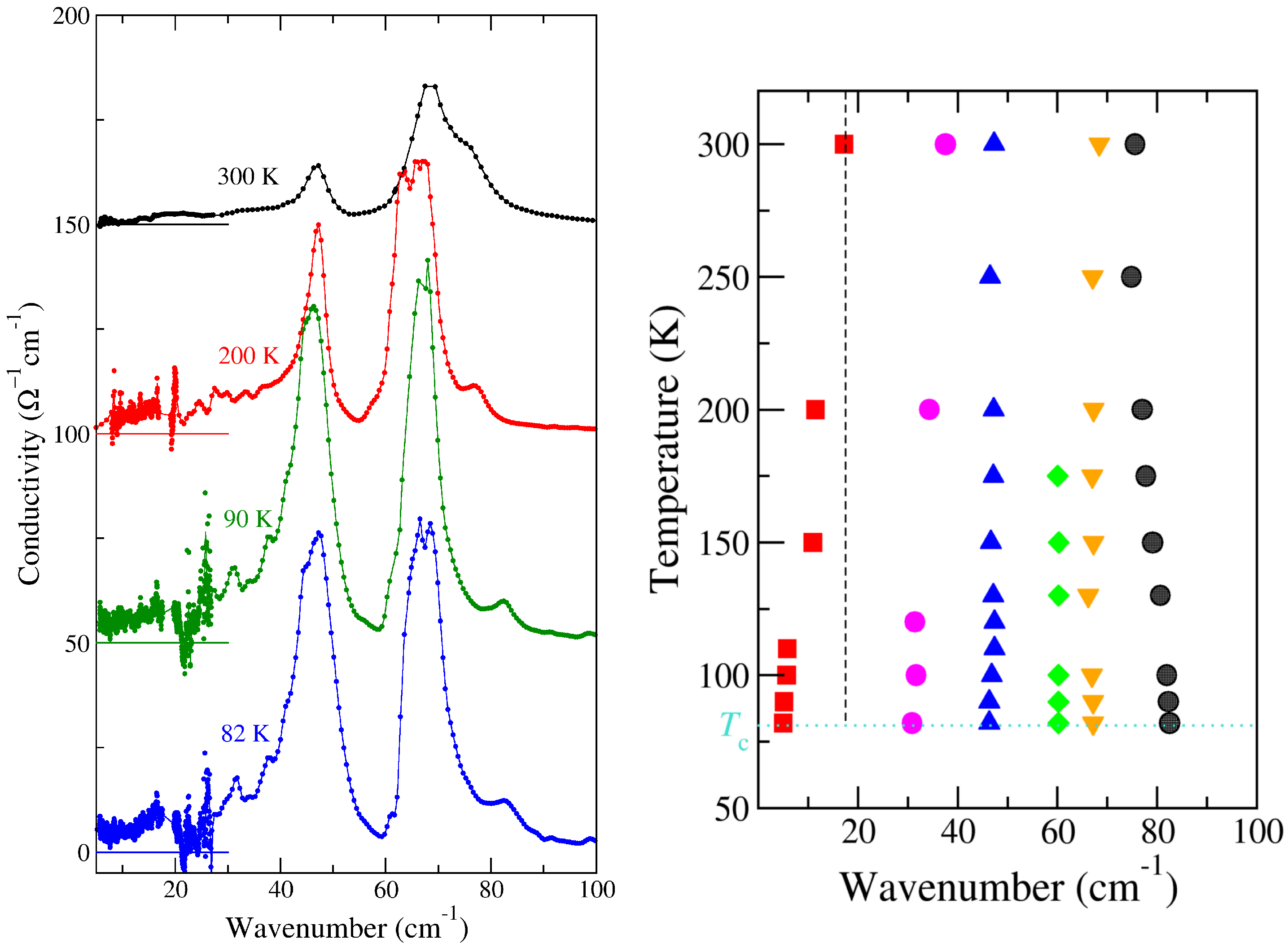
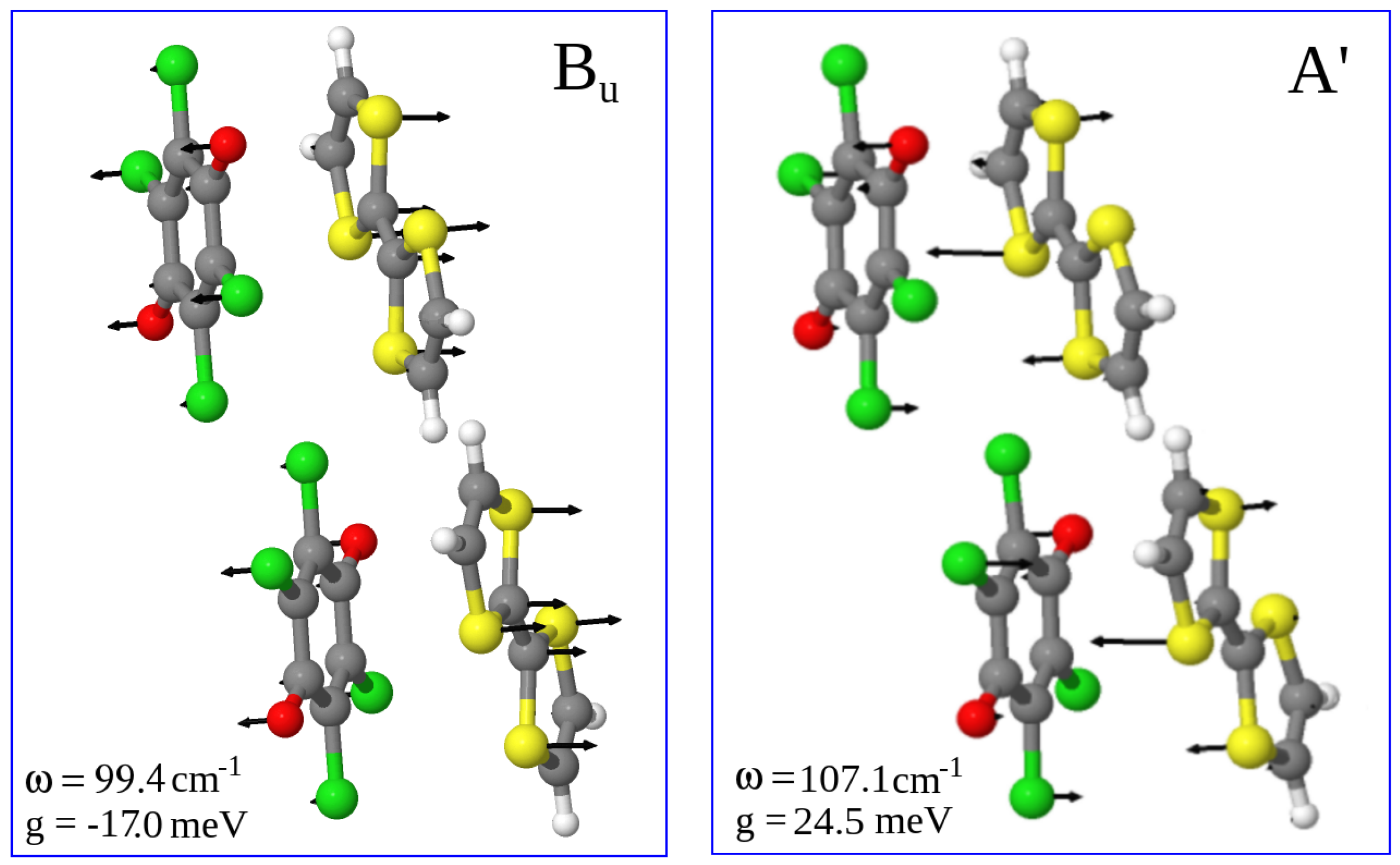
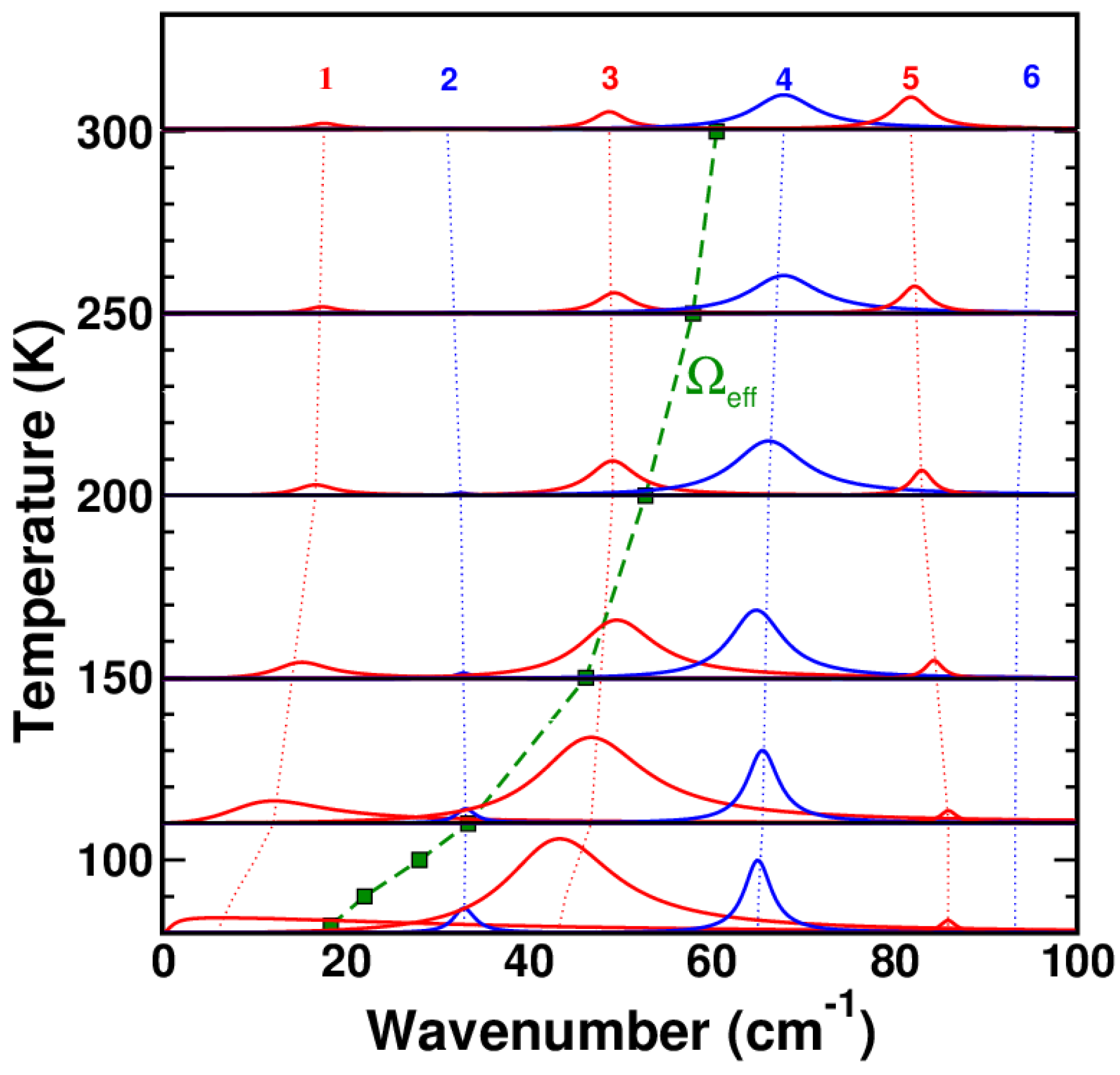
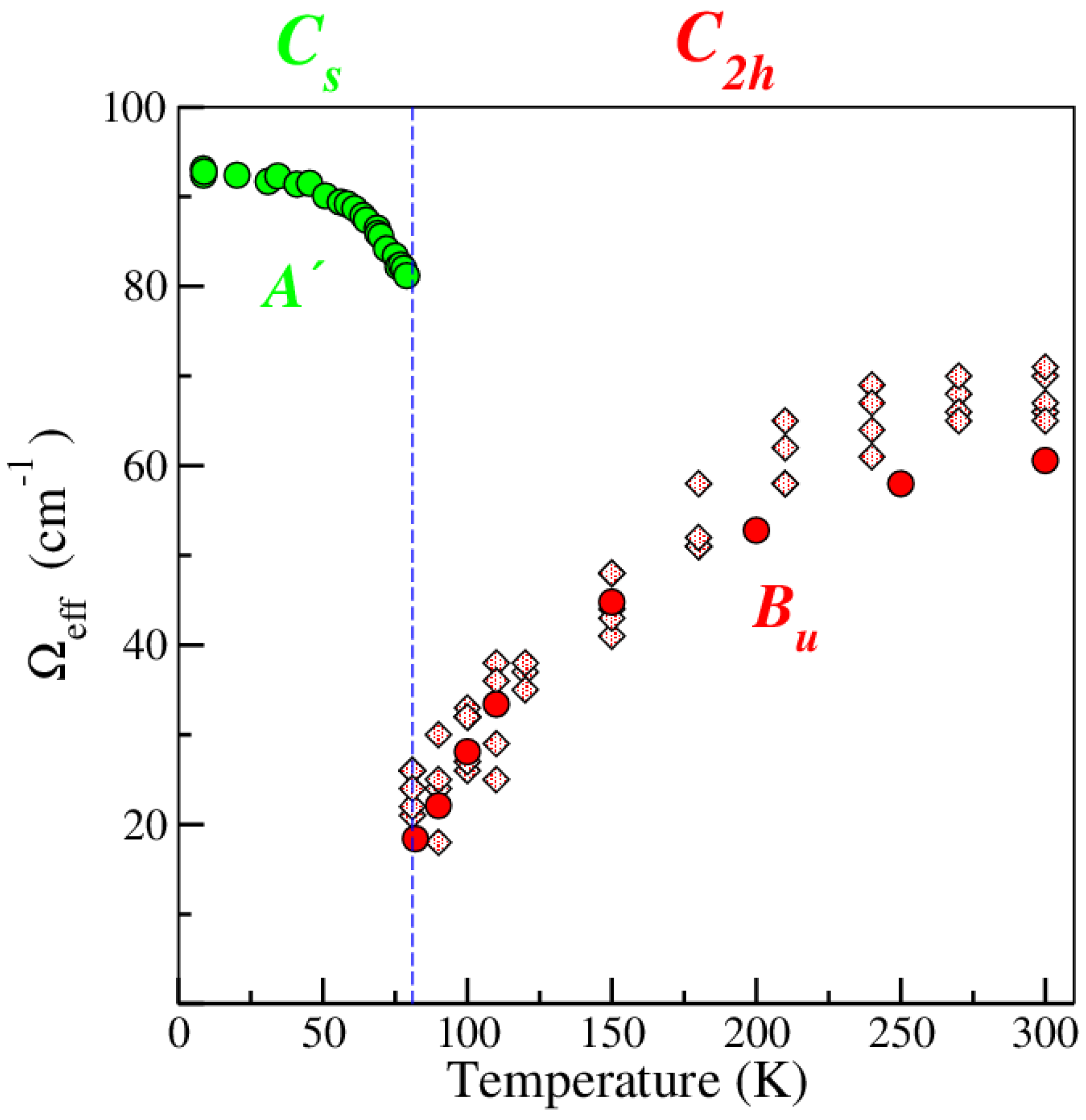
2.1.2. Soft Phonon(s)
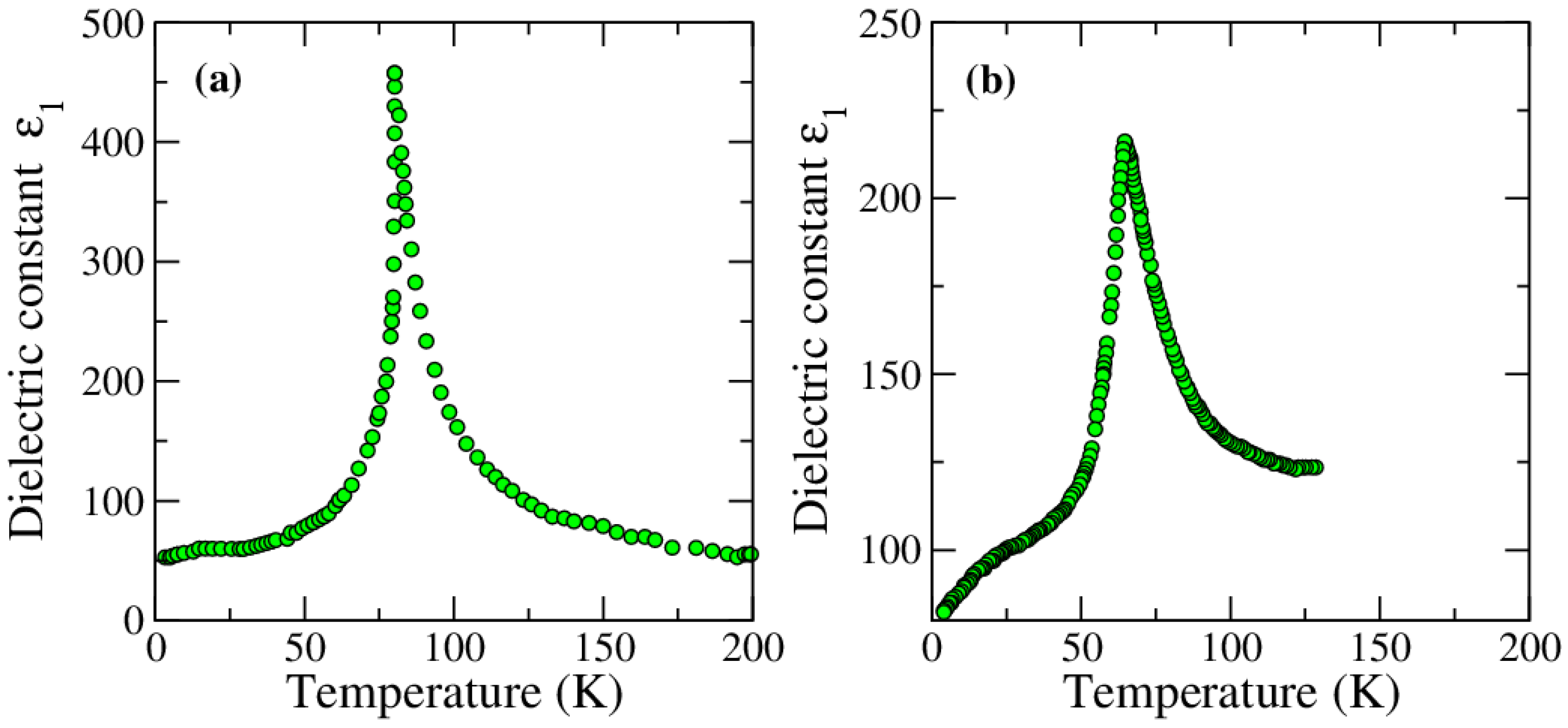
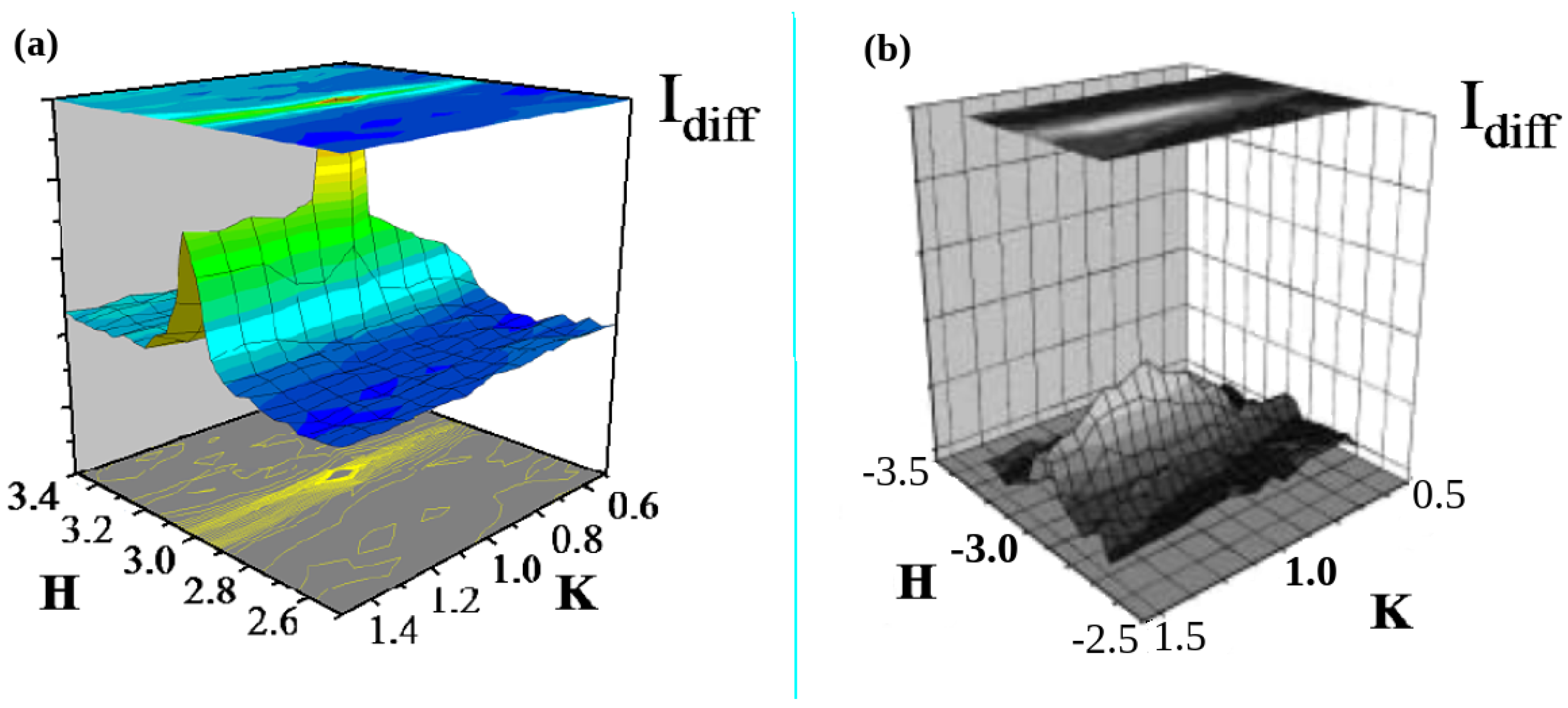
2.1.3. Anomalies in the Proximity of NIT
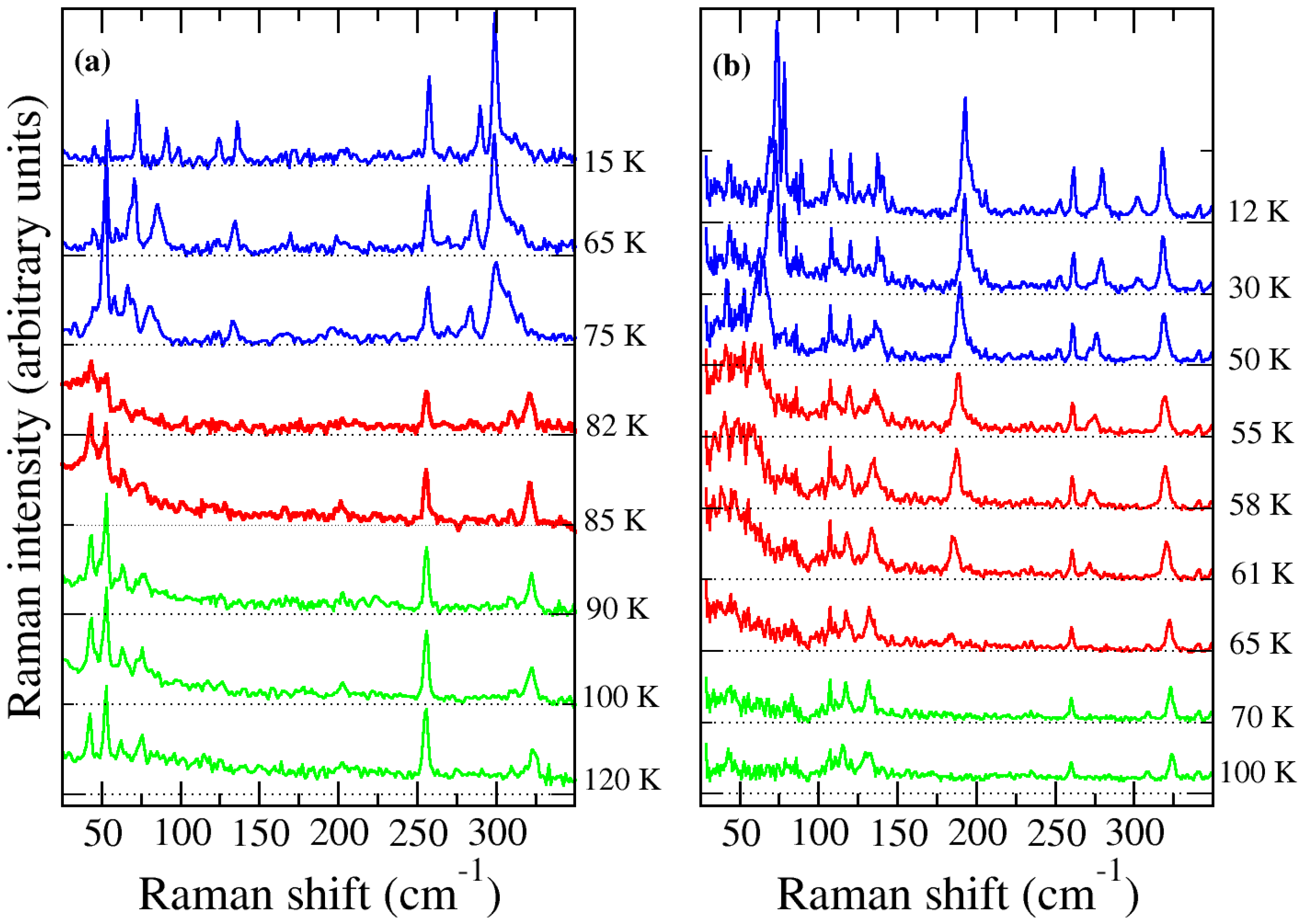
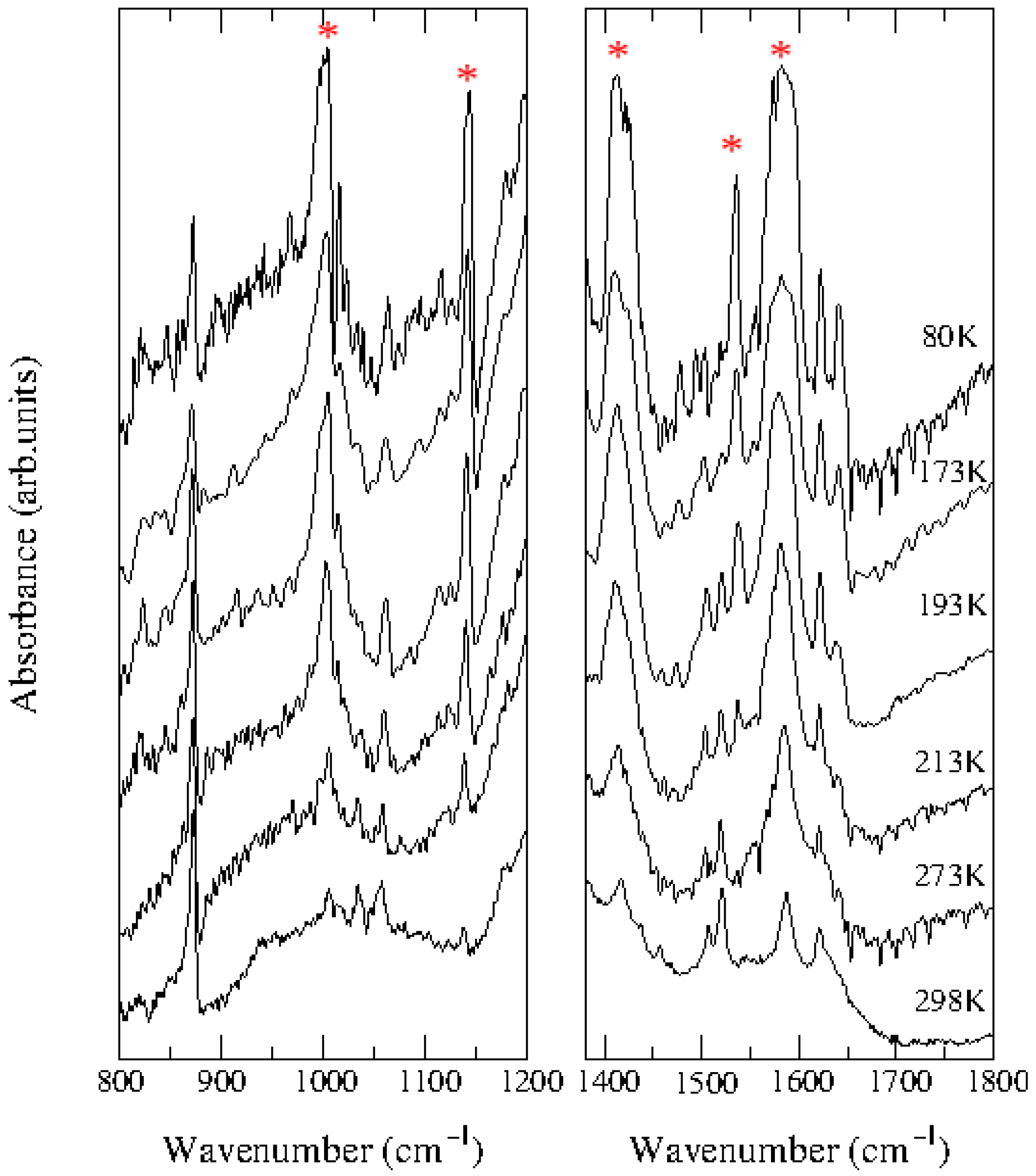
2.2. TMB–TCNQ
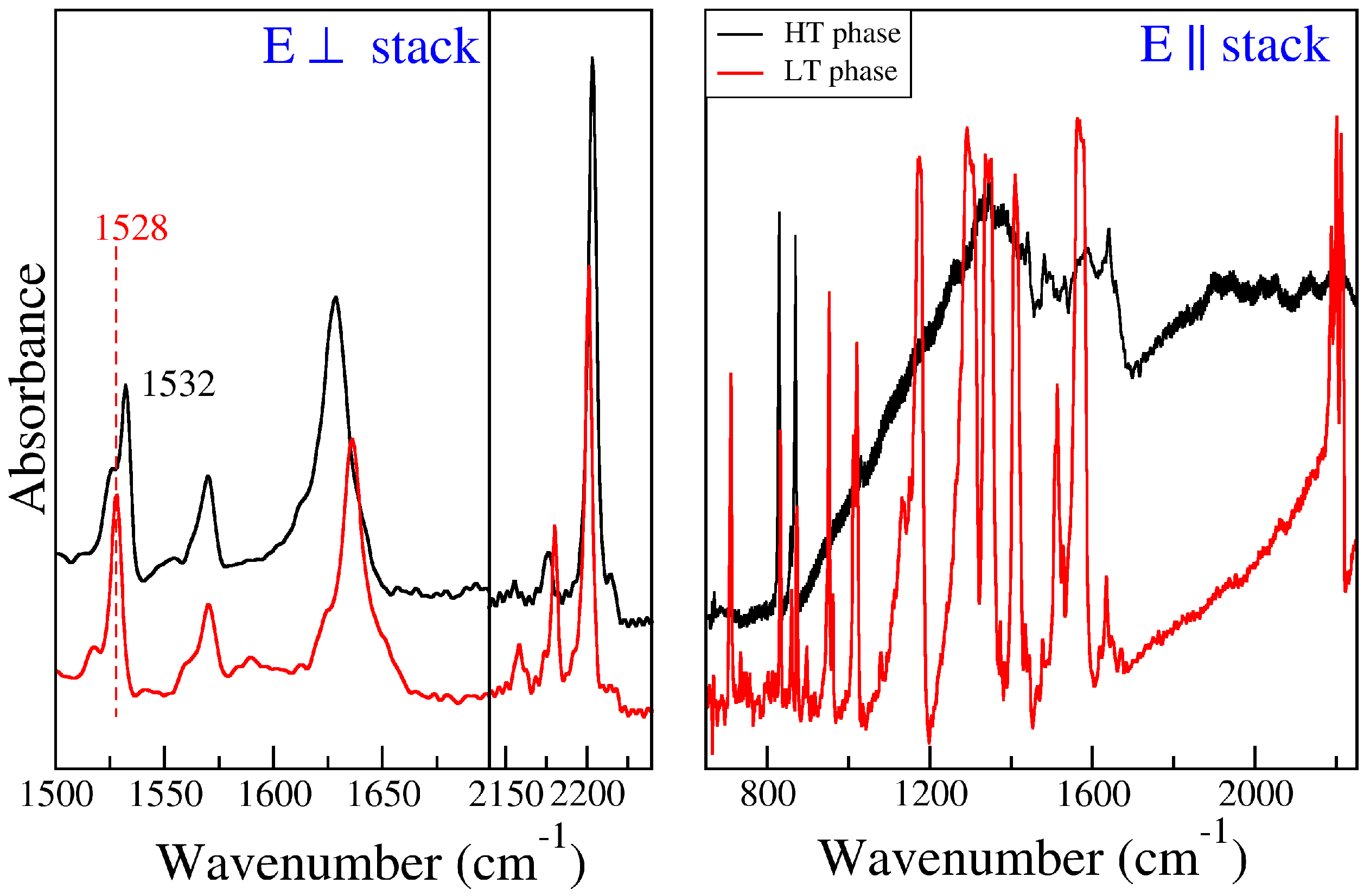
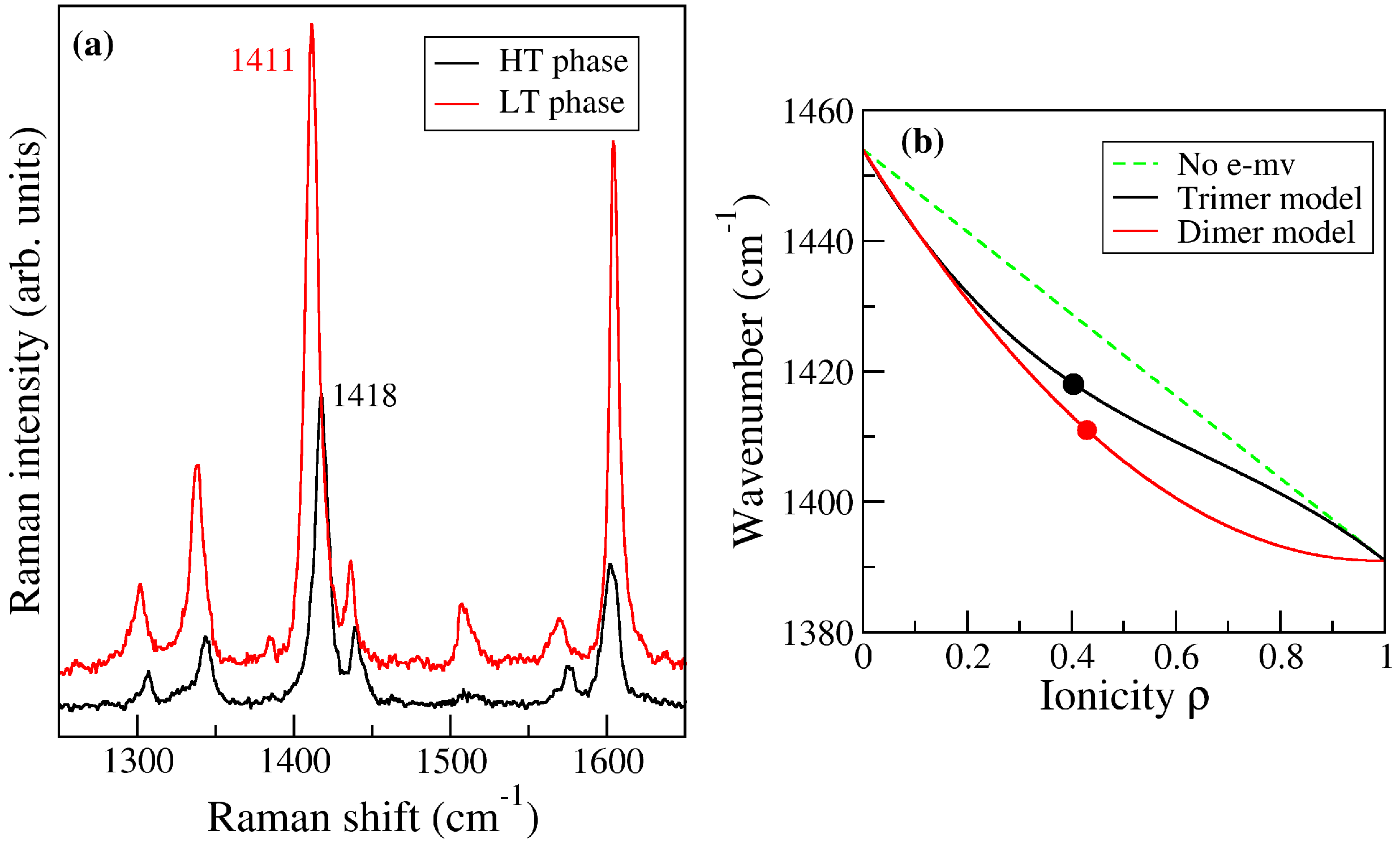
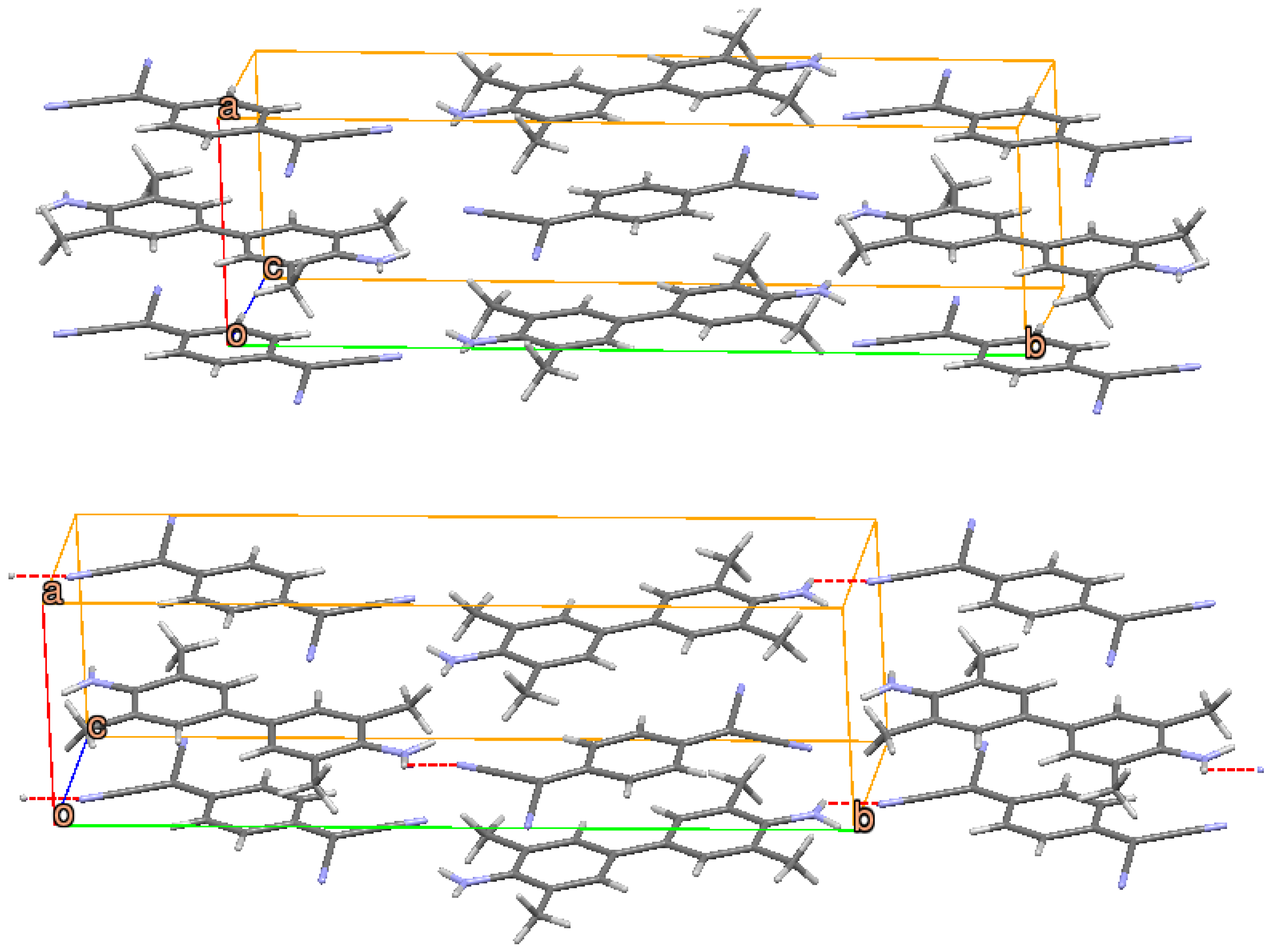
2.3. ClMePd–DMeDCNQI
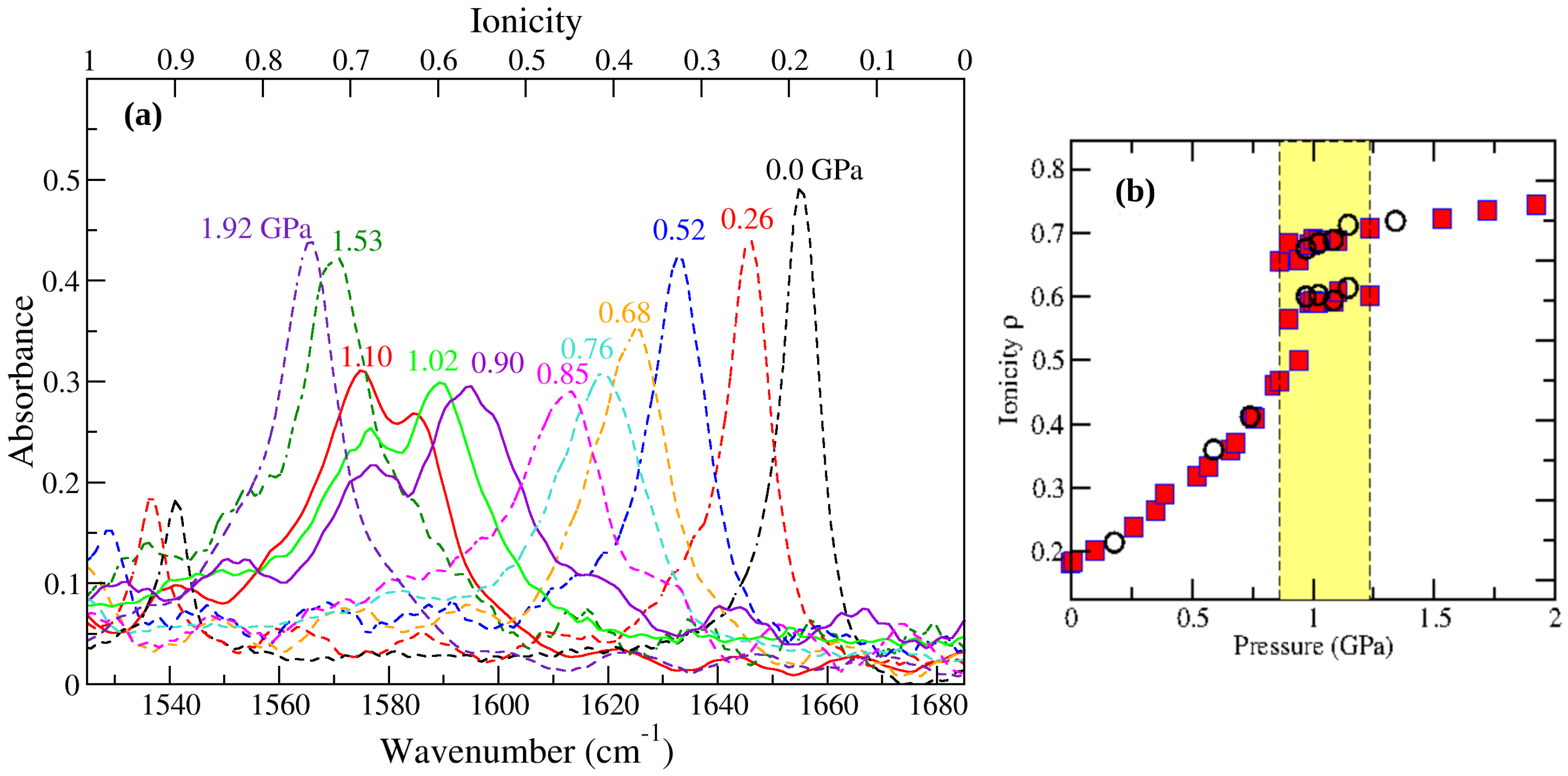
3. Pressure induced NIT
4. Beyond the NIT: Exploring the Phase Space of ms CT Crystals
5. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CT | charge-transfer |
| D | -electron Donor |
| A | -electron Acceptor |
| ms | mixed stack |
| NIT | Neutral-Ionic transition |
| N | Neutral or quasi-Neutral ground state |
| I | Ionic or quasi-Ionic ground state |
| 3D | three-dimensional |
| 1D | one-dimensional |
| CO | Charge Order |
| IR | infrared |
| e-mv | electron-molecular vibration |
| e-ph | electron-(lattice) phonon |
| QHLD | Quasi-Harmonic Lattice Dynamics |
| LR-CT | Lattice-Relaxed exciton strings |
| NIDW | Neutral-Ionic Domain Walls |
| DAC | diamond anvil cell |
References
- Soos, Z.G. Theory of π-molecular charge-transfer crystals. Ann. Rev. Phys. Chem. 1974, 25, 121–153. [Google Scholar] [CrossRef]
- McConnell, H.M.; Hoffman, B.M.; Metzger, R.M. Charge transfer in molecular crystals. Proc. Natl. Acad. Sci. USA 1965, 53, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Torrance, J.B.; Vazquez, J.E.; Mayerle, J.J.; Lee, V.Y. Discovery of a neutral-to-ionic phase transition in organic materials. Phys. Rev. Lett. 1981, 46, 253–257. [Google Scholar] [CrossRef]
- Torrance, J.B.; Girlando, A.; Mayerle, J.J.; Crowley, J.I.; Lee, V.Y.; Batail, P.; LaPlaca, S.J. Anomalous nature of neutral-to-ionic phase transition in tetrathiafulvalene-chloranil. Phys. Rev. Lett. 1981, 47, 1747–1750. [Google Scholar] [CrossRef]
- Koshihara, S.; Tokura, Y.; Mitani, T.; Saito, G.; Koda, T. Photoinduced valence instability in the organic molecular compound tetrathiafulvalene- p-choranil (TTF-CA). Phys. Rev. B 1990, 42, 6853–6856. [Google Scholar] [CrossRef]
- Nad, F.; Monceau, P.; Fabre, J.M. Low frequency dielectric permittivity of quasi-one-dimensional conductor (TMTTF)2Br. Eur. Phys. J. B 1998, 3, 301–306. [Google Scholar] [CrossRef]
- Girlando, A.; Painelli, A.; Bewick, S.A.; Soos, Z.G. Charge fluctuations and electron-phonon coupling in organic charge-transfer salts with neutral-ionic and Peierls transitions. Synth. Met. 2004, 141, 129–138. [Google Scholar] [CrossRef]
- Horiuchi, S.; Kumai, R.; Okimoto, Y.; Tokura, Y. Chemical approach to neutral-ionic valence instability, quantum phase transitions and relaxor ferrolectricity in organic charge-transfer complexes. Chem. Phys. 2006, 325, 78–91. [Google Scholar] [CrossRef]
- Horiuchi, S.; Hasegawa, T.; Tokura, Y. Molecular Donor-Acceptor compounds as prospective organic electronics materials. J. Phys. Soc. Japan 2006, 75, 051016. [Google Scholar] [CrossRef]
- Girlando, A.; Marzola, F.; Pecile, C.; Torrance, J.B. Vibrational spectroscopy of mixed stack organic semiconductors: Neutral and ionic phases of tetrathiafulvalene-chloranil (TTF-CA). J. Chem. Phys. 1983, 79, 1075–1085. [Google Scholar] [CrossRef]
- Girlando, A.; Pecile, C.; Torrance, J.B. A key to understanding ionic mixed stack organic solids: Tetrathiafulvalene-bromanil. Solid State Commun. 1985, 54, 753–759. [Google Scholar] [CrossRef]
- Girlando, A.; Painelli, A. Regular-dimerized stack and and neutral-ionic interfaces in mixed-stack organic charge-transfer crystals. Phys. Rev. B 1986, 34, 2131–2139. [Google Scholar] [CrossRef]
- Painelli, A.; Girlando, A. Zero-temperature phase diagram of mixed-stack charge-transfer crystals. Phys. Rev. B 1988, 37, 5748–5760, and Phys. Rev. B 1989, 39, 9663 (Erratum). [Google Scholar] [CrossRef]
- Horiuchi, S.; Okimoto, Y.; Kumai, R.; Tokura, Y. Quantum phase transition in organic charge-transfer complexes. Science 2003, 299, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Tokura, Y.; Kaneko, Y.; Okamoto, H.; Tanuma, S.; Koda, T.; Mitani, T.; Saito, G. Spectroscopic study of the neutral-to-ionic phase transition in TTF-Chloranil. Mol. Cryst. Liq. Cryst. 1985, 125, 71–80. [Google Scholar] [CrossRef]
- Horiuchi, S.; Okimoto, Y.; Kumai, R.; Tokura, Y. Anomalous charge fluctuations near a ferroelectric transition in an organic charge-transfer complex. J. Phys. Soc. Japan 2000, 69, 1302–1305. [Google Scholar] [CrossRef]
- Ranzieri, P.; Masino, M.; Girlando, A.; Lemée-Cailleau, M.H. Temperature-induced valence and structural instability in DMTTF-CA: Single crystal Raman and infrared measurements. Phys. Rev. B 2007, 76, 134115. [Google Scholar] [CrossRef]
- Horiuchi, S.; Okimoto, Y.; Kumai, R.; Tokura, Y. Ferroelectric valence transition and phase diagram of a series of charge-transfer complexes of 4,4′-dimethyltetrathiafulvalene and tetrahalo-p-benzoquinones. J. Am. Chem. Soc. 2001, 123, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Castagnetti, N.; Kociok-Köhn, G.; Da Como, E.; Girlando, A. Temperature-induced valence instability in the charge-transfer crystal TMB-TCNQ. Phys. Rev. B 2017, 95, 024101. [Google Scholar] [CrossRef]
- Masino, M.; Girlando, A.; Farina, L.; Brillante, A. A new type of neutral-ionic interface in mixed stack organic charge transfer crystals: Temperature induced ionicity change in ClMePD-DMeDCNQI. Phys. Chem. Chem. Phys. 2001, 3, 1904–1910. [Google Scholar] [CrossRef]
- Painelli, A.; Girlando, A. Electron-molecular vibration (e-mv) coupling in charge transfer compounds and its consequences on the optical spectra: A theoretical framework. J. Chem. Phys. 1986, 84, 5655–5671. [Google Scholar] [CrossRef]
- Pecile, C.; Painelli, A.; Girlando, A. Studies of organic semiconductors for 40 years—V. Mol. Cryst. Liq. Cryst. 1989, 171, 69–87. [Google Scholar] [CrossRef]
- Girlando, A.; Bozio, R.; Pecile, C.; Torrance, J.B. Discovery of vibronic effects in the Raman spectra of mixed-stack charge-transfer crystals. Phys. Rev. B 1982, 26, 2306–2309. [Google Scholar] [CrossRef]
- Ranzieri, P.; Masino, M.; Girlando, A. Charge-sensitive vibrations in p-chloranil: The strange case of the C=C antisymmetric stretching. J. Phys. Chem. B 2007, 111, 12844–12848. [Google Scholar] [CrossRef] [PubMed]
- Le Cointe, M.; Lemée-Cailleau, M.H.; Cailleau, H.; Toudic, B.; Toupet, L.; Heger, G.; Moussa, F.; Schweiss, P.; Kraft, K.H.; Karl, N. Symmetry breaking and structural changes at the neutral-to-ionic transition in tetrathiafulvalene-p-chloranil. Phys. Rev. B 1995, 51, 3374–3386. [Google Scholar] [CrossRef]
- Collet, E.; Buron-Le Cointe, M.; Lemée-Cailleau, M.H.; Cailleau, H.; Toupet, L.; Meven, M.; Mattauch, S.; Heger, G.; Karl, N. Structural evidence of ferrielectric neutral-ionic layered ordering in 2,6-dimethyltetrathiafulvalene-p-chloranil. Phys. Rev. B 2001, 63, 054105. [Google Scholar] [CrossRef]
- Oison, V.; Katan, C.; Koenig, C. Intramolecular electronic redistribution coupled to hydrogen bonding: An important mechanism for the “Neutral-to-Ionic” transition. J. Phys. Chem. A 2001, 105, 4300–4307. [Google Scholar] [CrossRef]
- Lemée-Cailleau, M.H.; Le Cointe, M.; Cailleau, H.; Luty, T.; Moussa, F.; Roos, J.; Brinkmann, D.; Toudic, B.; Ayache, C.; Karl, N. Thermodynamics of the Neutral-to-Ionic transition as condensation and crystallization of charge-transfer excitations. Phys. Rev. Lett. 1997, 79, 1690–1693. [Google Scholar] [CrossRef]
- Moreac, A.; Girard, A.; Delugeard, Y.; Marqueton, Y. The neutral-to-ionic phase transition of TTF-CA: A Raman and infrared study versus temperature at atmospheric pressure. J. Phys. Condens. Matter 1996, 8, 3553–3567. [Google Scholar] [CrossRef]
- Masino, M.; Girlando, A.; Soos, Z.G. Evidence for a soft mode in the temperature induced neutral-ionic transition of TTF-CA. Chem. Phys. Lett. 2003, 369, 428–433. [Google Scholar] [CrossRef]
- Masino, M.; Girlando, A.; Brillante, A.; Della Valle, R.G.; Venuti, E.; Drichko, N.; Dressel, M. Lattice dynamics of TTF-CA across the neutral-ionic transition. Chem. Phys. 2006, 325, 71–77. [Google Scholar]
- Girlando, A.; Masino, M.; Painelli, A.; Drichko, N.; Dressel, M.; Brillante, A.; Della Valle, R.G.; Venuti, E. Direct evidence of overdamped Peierls-coupled modes in temperature-induced phase transition in tetrathiafulvalene-chloranil. Phys. Rev. B 2008, 78, 045103. [Google Scholar] [CrossRef]
- Soos, Z.G.; Bewick, S.A.; Peri, A.; Painelli, A. Dielectric response of modified Hubbard models with neutral-ionic and Peierls transitions. J. Chem. Phys. 2004, 120, 6712–6720. [Google Scholar] [CrossRef] [PubMed]
- Masino, M.; Girlando, A. Peierls modes in the ionic phase of TTF-CA. Internal Report 2010 MG10-1 available upon request.
- Tokura, Y.; Okamoto, H.; Koda, T.; Mitani, T.; Saito, G. Nonlinear electric transport and switching phenomenon in the mixed-stack charge-transfer crystal tetrathiafulvalene-p-chloranil. Phys. Rev. B 1988, 38, 2215–2218. [Google Scholar] [CrossRef]
- Heeger, A.J.; Kivelson, S.; Schrieffer, J.R.; Su, W.P. Solitons in conducting polymers. Rev. Mod. Phys. 1988, 60, 781–850. [Google Scholar] [CrossRef]
- Mitani, T.; Saito, G.; Tokura, Y.; Koda, T. Soliton Formation at the Neutral-to-Ionic Phase Transition in the Mixed-Stack Charge-Transfer Crystal Tetrathiafulvalene-p-Chloranil. Phys. Rev. Lett. 1984, 53, 842–845. [Google Scholar] [CrossRef]
- Nagaosa, N. Domain wall picture of the Neutral-Ionic transition in TTF-Chloranil. Solid State Commun. 1986, 57, 179–183. [Google Scholar] [CrossRef]
- D’Avino, G.; Soos, Z.G.; Painelli, A. Modeling the Neutral-Ionic transition with correlated electrons coupled to soft lattices and molecules. Crystals 2017. Submitted. [Google Scholar]
- Buron-Le Cointe, M.; Lemée-Cailleau, M.H.; Cailleau, H.; Ravy, S.; Bérar, J.F.; Rouzière, S.; Elkaïm, E.; Collet, E. One-dimensional fluctuating nanodomains in the charge-transfer molecular system TTF-CA and their first-order crystallization. Phys. Rev. Lett. 2006, 96, 205503. [Google Scholar] [CrossRef] [PubMed]
- Collet, E.; Lemée-Cailleau, M.H.; Buron-Le Cointe, M.; Cailleau, H.; Ravy, S.; Luty, T.; Bérar, J.F.; Czarnecki, P.; Karl, N. Direct evidence of lattice-relaxed charge transfer exciton strings. Europhys. Lett. 2002, 57, 67–73. [Google Scholar] [CrossRef]
- D’Avino, G.; Girlando, A.; Painelli, A.; Lemée-Cailleau, M.H.; Soos, Z.G. Anomalous dispersion of the optical phonons at the Neutral-Ionic transition: Evidence from diffuse X-ray scattering. Phys. Rev. Lett. 2007, 99, 156407. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W. Image of the Fermi surface in the vibration spectrum of a metal. Phys. Rev. Lett. 1959, 2, 393–394. [Google Scholar] [CrossRef]
- Kendziora, C.A.; Sergienko, I.A.; Jin, R.; He, J.; Keppens, V.; Sales, B.C.; Mandrus, D. Goldstone-Mode phonon dynamics in the pyrochlore Cd2Re2O7. Phys. Rev. Lett. 2005, 95, 125503. [Google Scholar] [CrossRef] [PubMed]
- D’Avino, G.; Masino, M.; Girlando, A.; Painelli, A. Correlated electrons in soft lattices: Raman scattering evidence of the nonequilibrium dielectric divergence at the neutral-ionic phase transition. Phys. Rev. B 2011, 83, 161105(R). [Google Scholar] [CrossRef]
- Iwasa, Y.; Koda, T.; Tokura, Y.; Kobayashi, A.; Iwasawa, N.; Saito, G. Temperature-induced neutral-ionic transition in tetramethylbenzidine-tetracyanoquinodimethane (TMB-TCNQ). Phys. Rev. B 1990, 42, 2374–2377. [Google Scholar] [CrossRef]
- Buron-LeCointe, M.; Lemée-Cailleau, M.H.; Cailleau, H.; Toudic, B.; Moréac, A.; Moussa, F.; Ayache, C.; Karl, N. Thermal hysteresis and mesoscopic phase coexistence around the neutral-ionic phase transition in TTF-CA and TMB-TCNQ. Phys. Rev. B 2003, 68, 064103. [Google Scholar]
- Meneghetti, M.; Pecile, C. Charge–transfer organic crystals: Molecular vibrations and spectroscopic effects of electron–molecular vibration coupling of the strong electron acceptor TCNQF4. J. Chem. Phys. 1986, 84, 4149–4162. [Google Scholar] [CrossRef]
- Hanfland, M.; Brillante, A.; Girlando, A.; Syassen, K. Raman study of the pressure induced neutral-to-ionic transition in tetrathiafulvalene-chloranil (TTF-CA). Phys. Rev. B 1988, 38, 1456–1461. [Google Scholar] [CrossRef]
- Iwasa, Y.; Soga, I.; Koda, T.; Tokura, Y.; Saito, G. Neutral-Ionic transition in TTF-Chloranil and TMB-TCNQ. Synth. Met. 1991, 42, 1827–1830. [Google Scholar] [CrossRef]
- Soos, Z.G.; Painelli, A. Metastable domains and potential energy surfaces in organic charge-transfer salts with neutral-ionic phase transitions. Phys. Rev. B 2007, 75, 155119. [Google Scholar] [CrossRef]
- Aoki, S.; Nakayama, T. Temperature-induced neutral-ionic transition in 2-chloro-5-methyl-p- phenylenediamine–2,5-dimethyl-dicyanoquinonediimine. Phys. Rev. B 1997, 56, R2893–R2896. [Google Scholar] [CrossRef]
- Lunardi, G.; Pecile, C. N,N′-dicyanoquinonediimines as a molecular constituent of organic conductors: Vibrational behavior and electron–molecular vibration coupling. J. Chem. Phys. 1991, 95, 6911–6923. [Google Scholar] [CrossRef]
- Bewick, S.A.; Pascal, R.A.; Ho, D.M.; Soos, Z.G.; Masino, M.; Girlando, A. Disorder in organic charge-transfer single crystals: Dipolar disorder in ClMePD-DMeDCNQI. J. Chem. Phys. 2005, 122, 024710. [Google Scholar] [CrossRef] [PubMed]
- Jurgensen, C.W.; Drickamer, H.G. High-pressure studies of the neutral-to-ionic transition in some organic charge-transfer solids. Chem. Phys. Lett. 1986, 125, 554–556. [Google Scholar] [CrossRef]
- Girlando, A.; Pecile, C.; Brillante, A.; Syassen, K. Highe-pressure optical studies of neutral-ionic phase transitions in organic charge-transfer crystals. Synth. Met. 1987, 19, 503–508. [Google Scholar] [CrossRef]
- Brillante, A.; Girlando, A. Pressure-driven phase transition in tetrathiafulvalene-2,5-dichloro-p-benzoquinone: Example of a continuous ionicity change. Phys. Rev. B 1992, 45, 7026–7030. [Google Scholar] [CrossRef]
- Masino, M.; Girlando, A.; Brillante, A. Intermediate regime in the pressure-induced neutral-ionic transition in tetrathiafulvalene-chloranil. Phys. Rev. B 2007, 76, 061114/1–061114/6. [Google Scholar] [CrossRef]
- Okamoto, H.; Koda, T.; Tokura, Y.; Mitani, T.; Saito, G. Pressure-induced neutral-to-ionic phase transition in organic charge-transfer crystals of tetrathiafulvalene-p-benzoquinone derivatives. Phys. Rev. B 1989, 39, 10693–10701. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Hiejima, T.; Sano, M. Pressure-induced neutral-ionic phase transition of a tetrathiafulvalene–iodanil crystal. Bull. Chem. Soc. Jpn. 1991, 65, 2052–2057. [Google Scholar] [CrossRef]
- Basaki, S.; Matsuzaki, S. A new charge-transfer system for neutral-ionic transition: Tetrathiafulvalene–dimethyldicyanoquinonediimine. Solid State Commun. 1994, 91, 865–868. [Google Scholar] [CrossRef]
- Aoki, S.; Nakayama, T.; Miura, A. Neutral-ionic transition in dimethyl-TTF-chloranil. Synth. Met. 1995, 70, 1243–1244. [Google Scholar] [CrossRef]
- Farina, L.; Brillante, A.; Masino, M.; Girlando, A. Pressure-driven neutral-ionic transition in ClMePD–DMeDCNQI. Phys. Rev. B 2001, 64, 144102. [Google Scholar] [CrossRef]
- Hasegawa, T.; Akutagawa, T.; Nakamura, T.; Mochida, T.; Kondo, R.; Kagoshima, S.; Iwasa, Y. Neutral-ionic phase transition of BEDT-TTF–ClMeTCNQ under pressure. Phys. Rev. B 2001, 64, 085106. [Google Scholar] [CrossRef]
- Iwasa, Y.; Watanabe, N.; Koda, T.; Saito, G. Two-step neutral-ionic phase transition in organic charge-transfer compounds: Possible staging effects. Phys. Rev. B 1993, 47, 2920–2923. [Google Scholar] [CrossRef]
- Mitani, T.; Kaneko, Y.; Tanuma, S.; Tokura, Y.; Koda, T.; Saito, G. Electric conductivity and phase diagram of a mixed-stack charge-transfer crystal: Tetrathiafulvalene-p-chloranil. Phys. Rev. B 1987, 35, 427–429. [Google Scholar] [CrossRef]
- Okamoto, H.; Mitani, T.; Tokura, Y.; Koshihara, S.; Komatsu, T.; Iwasa, Y.; Koda, T.; Saito, G. Anomalous dielectric response in tetrathiafulvalene-p-chloranil as observed in temperature- and pressure-induced neutral-to-ionic phase transition. Phys. Rev. B 1991, 43, 8224–8232. [Google Scholar] [CrossRef]
- Lemée-Cailleau, M.H.; Collet, E.; Buron-Le Cointe, M.; Grach, N.; Ouladdiaf, B.; Moussa, F.; Hasegawa, T.; Takahashi, Y.; Roisnel, T.; Cailleau, H. Down to the quantum limit at the Neutral-to-Ionic phase transition of (BEDT-TTF)-(ClMe-TCNQ): Symmetry analysis and phase diagram. J. Phys. Chem. B 2007, 111, 6167–6172. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, F.; Horiuchi, S.; Tokunaga, M.; Fujioka, J.; Tokura, Y. Ferroelectricity in a one-dimensional organic quantum magnet. Nat. Phys. 2010, 6, 169–172. [Google Scholar] [CrossRef]
- Kobayashi, K.; Horiuchi, S.; Kumai, R.; Kagawa, F.; Murakami, Y.; Tokura, Y. Electronic ferroelectricity in a molecular crystal with large polarization directing antiparallel to ionic displacement. Phys. Rev. Lett. 2012, 108, 237601. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.Y.; Yi, Y.P.; Li, Y.; Kim, E.G.; Coropceanu, V.; Bredas, J.-L. Prediction of remarkable ambipolar charge-transport characteristics in organic mixed-stack charge-transfer crystals. J. Am. Chem. Soc. 2012, 134, 2340–2347. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-D.; Wang, F.-X.; Xiao, Y.; Pan, G.-B. Preparation and ambipolar transistor characteristics of co-crystal microrods of dibenzotetrathiafulvalene and tetracyanoquinodimethane. J. Mater. Chem. C 2013, 1, 2286–2289. [Google Scholar] [CrossRef]
- Vermeulen, D.; Zhu, L.Y.; Goetz, K.P.; Hu, P.; Day, C.S.; Jurchescu, O.D.; Coropceanu, V.; Kloc, C.; McNeil, L.E. Charge transport properties of Perylene–TCNQ crystals: The Effect of stoichiometry. J. Phys. Chem. C 2014, 118, 24688–24696. [Google Scholar] [CrossRef]
- Tsutsumi, J.; Matsuoka, S.; Inoue, S.; Minemawari, H.; Yamada, Y.; Hasegawa, T. N-type field-effect transistors based on layered crystalline donor–acceptor semiconductors with dialkylated benzothienobenzothiophenes as electron donors. J. Mater. Chem. C 2015, 3, 1976–1981. [Google Scholar] [CrossRef]
- Hu, P.; Du, K.; Wei, F.; Jiang, H.; Kloc, C. Crystal growth, HOMO–LUMO engineering, and charge transfer degree in Perylene-FxTCNQ (x = 1, 2, 4) organic charge transfer binary compounds. Cryst. Growth Des. 2016, 16, 3019–3027. [Google Scholar] [CrossRef]
- Salzillo, D.; Masino, M.; Kociok-Köhn, G.; di Nuzzo, D.; Venuti, E.; Della Valle, R.G.; Vanossi, D.; Fontanesi, C.; Girlando, A.; Brillante, A.; et al. Structure, stoichiometry, and charge transfer in cocrystals of Perylene with TCNQ-Fx. Cryst. Growth Des. 2016, 16, 3028–3036. [Google Scholar] [CrossRef]
- Morherr, A.; Witt, S.; Chernenkaya, A.; Backer, J.-P.; Schonhense, G.; Bolte, M.; Krellner, C. Crystal growth of new charge-transfer salts based on π-conjugated donor molecules. Phys. B 2016, 496, 98–105. [Google Scholar] [CrossRef]
- Girlando, A.; Painelli, A.; Pecile, C. Molecular vibration analysis of ionicity and phase transition in TMPD-TCNQ (1:1) charge transfer complex. Mol. Cryst. Liq. Cryst. 1984, 112, 325–343. [Google Scholar] [CrossRef]
- Bechgaard, K.; Kistenmacher, T.J.; Bloch, A.N.; Cowan, D.O. The crystal and molecular structure of an organic conductor from 4,4′,5,5′-TetramethylΔ2,2′-bis-1,3-diselenole–7,7,8,8-Tetracyano-p-quinodimethane [TMTSF–TCNQ]. Acta Crystallogr. B 1977, 33, 417–422. [Google Scholar] [CrossRef]
- Kistenmacher, T.J.; Emge, T.J.; Bloch, A.N.; Cowan, D.O. Structure of the red, semiconducting form of 4,4′,5,5′-TetramethylΔ2,2′-bis-1,3-diselenole–7,7,8,8-Tetracyano-p-quinodimethane, TMTSF-TCNQ. Acta Crystallogr. B 1982, 38, 1193–1199. [Google Scholar] [CrossRef]
- Goetz, K.P.; Tsutsumi, J.; Pookpanratana, S.; Chen, J.; Corbin, N.S.; Behera, R.K.; Coropceanu, V.; Richter, C.A.; Hacker, C.A.; Hasegawa, T.; et al. Polymorphism in the 1:1 charge-transfer complex DBTTF– TCNQ and its effects on optical and electronic properties. Adv. Electron. Mater. 2016, 2, 1600203. [Google Scholar] [CrossRef]
- Girlando, A. Comment on Polymorphism in the 1:1 charge-transfer complex DBTTF– TCNQ and its effects on optical and electronic properties. Adv. Electron. Mater. 2017, 3, 1600437. [Google Scholar] [CrossRef]
- Lapidus, S.H.; Naik, A.; Wixtrom, A.; Massa, N.E.; Ta Phuoc, V.; del Campo, L.; Lebègue, S.; Ángyán, J.G.; Abdel-Fattah, T.; Pagola, S. The black polymorph of TTF-CA: TTF polymorphism and solvent effects in mechanochemical and vapor digestion syntheses, FT-IR, crystal packing, and electronic structure. Cryst. Growth Des. 2013, 14, 91–100. [Google Scholar] [CrossRef]
- Kistenmacher, T.J.; Phillips, T.E.; Cowan, D.O. The crystal structure of the 1:1 radical cation–radical anion salt of 2,2′- bis-1,3-dithiole (TTF) and 7,7,8,8-tetracyanoquinodimethane (TCNQ). Acta Crystallogr. B 1974, 30, 763–768. [Google Scholar] [CrossRef]
- Emge, T.J.; Wiygul, F.M.; Ferraris, J.P.; Kistenmacher, T.J. Crystal structure and Madelung energy for the charge-transfer complex tetrathiafulvalene-2,5-difluoro-7,7,8,8-tetracyanoquinodimethane. Observations on a dimerized segregated stack motif. Mol. Cryst. Liq. Cryst. 1981, 78, 295–310. [Google Scholar] [CrossRef]
- Wiygul, F.M.; Emge, T.J.; Kistenmacher, T.J. Elements of the crystal structure of a solvated 1:1 charge-transfer salt derived from tetrathiafulvalene (TTF) and tetrafluorotetracyano-p- quinodimethane (TCNQF4), TTF-TCNQF4. Mol. Cryst. Liq. Cryst. 1982, 90, 163–171. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | T (K) | Jump | Range | Ref. |
|---|---|---|---|---|
| TTF–CA | 81-83 | 0.21 | 0.2–0.6 | [10,15] |
| TTF–BrClBQ | 68 | 0.16 | 0.2–0.6 | [16] |
| DMeTTF–CA | 65 | 0.02 | 0.2–0.4 | [17] |
| DMeTTF–BrClBQ | 54 | 0.1 | 0.2–0.6 | [18] |
| DMeTTF–2,5BrClBQ | 49 | 0.06 | 0.3–0.4 | [18] |
| TMB–TCNQ | 205–235 | 0.1 | 0.3–0.4 | [19] |
| ClMePD–DCNQI | 200 | absent | 0.3–0.6 | [20] |
| System | p (GPa) | Jump | Range | Ref. |
|---|---|---|---|---|
| TTF–CA | 0.86–1.24 | 0.2 | 0.2–0.8 | [58] |
| TTF–2,5ClBQ | 3.8 | 0 | 0.2–0.8 | [57] |
| TTF–2,3,5ClBQ | 0.6–2.5 | 0.2 | 0.3–0.8 | [59] |
| TTF–IA | 1.9–2.2 | 0.1 | 0.0–0.6 | [60] |
| TTF-DMeDCNQI | 1.0 | 0.8 | 0.8 | [61] |
| DMeTTF–CA | 0.7–1.2 | 0.2 | ? | [62] |
| DMeTTF–BA | 1.2 | ? | ? | [14] |
| ClMePD–DMeDCNQI | 0.75 | 0 | 0.4–0.7 | [63] |
| DBTTF–TCNQ | 1.1 | 0.1 | 0.3–0.4 | [56] |
| BEDT-TTF–ClMeTCNQ | 0.8–1.2 | 0.2 ? | 0.3–1.0 | [64] |
| TMB–TCNQ | 0.49–2.7 | ? | ? | [65] |
| System | Space Group | (eV) | Ref. | |
|---|---|---|---|---|
| Per–TCNQ | () | 0.01 | 1.34, onset 1.05 | [76] |
| Per–TCNQF | () | 0.08 | – | [75] |
| Per–TCNQF | () | 0.13 | onset 0.88 | [76] |
| Per–TCNQF | () | 0.29 | 0.98, onset 0.71 | [76] |
| System | Space Group | (eV) | Ref. | |
|---|---|---|---|---|
| TMB–CA | () | 0.14 | onset 0.67 | This work |
| TMB–BA | () | 0.16 | onset 0.61 | This work |
| TMB–TCNQ | () | 0.29 | 0.94, onset 0.46 | [19] |
| TMB–TCNQF | () | 0.65 | onset 0.57 | This work |
| TMB–TCNQF | () | 0.89 | onset 0.47 | This work |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masino, M.; Castagnetti, N.; Girlando, A. Phenomenology of the Neutral-Ionic Valence Instability in Mixed Stack Charge-Transfer Crystals. Crystals 2017, 7, 108. https://doi.org/10.3390/cryst7040108
Masino M, Castagnetti N, Girlando A. Phenomenology of the Neutral-Ionic Valence Instability in Mixed Stack Charge-Transfer Crystals. Crystals. 2017; 7(4):108. https://doi.org/10.3390/cryst7040108
Chicago/Turabian StyleMasino, Matteo, Nicola Castagnetti, and Alberto Girlando. 2017. "Phenomenology of the Neutral-Ionic Valence Instability in Mixed Stack Charge-Transfer Crystals" Crystals 7, no. 4: 108. https://doi.org/10.3390/cryst7040108
APA StyleMasino, M., Castagnetti, N., & Girlando, A. (2017). Phenomenology of the Neutral-Ionic Valence Instability in Mixed Stack Charge-Transfer Crystals. Crystals, 7(4), 108. https://doi.org/10.3390/cryst7040108