
The Role of Halogen Bonding in Controlling Assembly and Organization of Cu(II)-Acac Based Coordination Complexes

 ,
, 
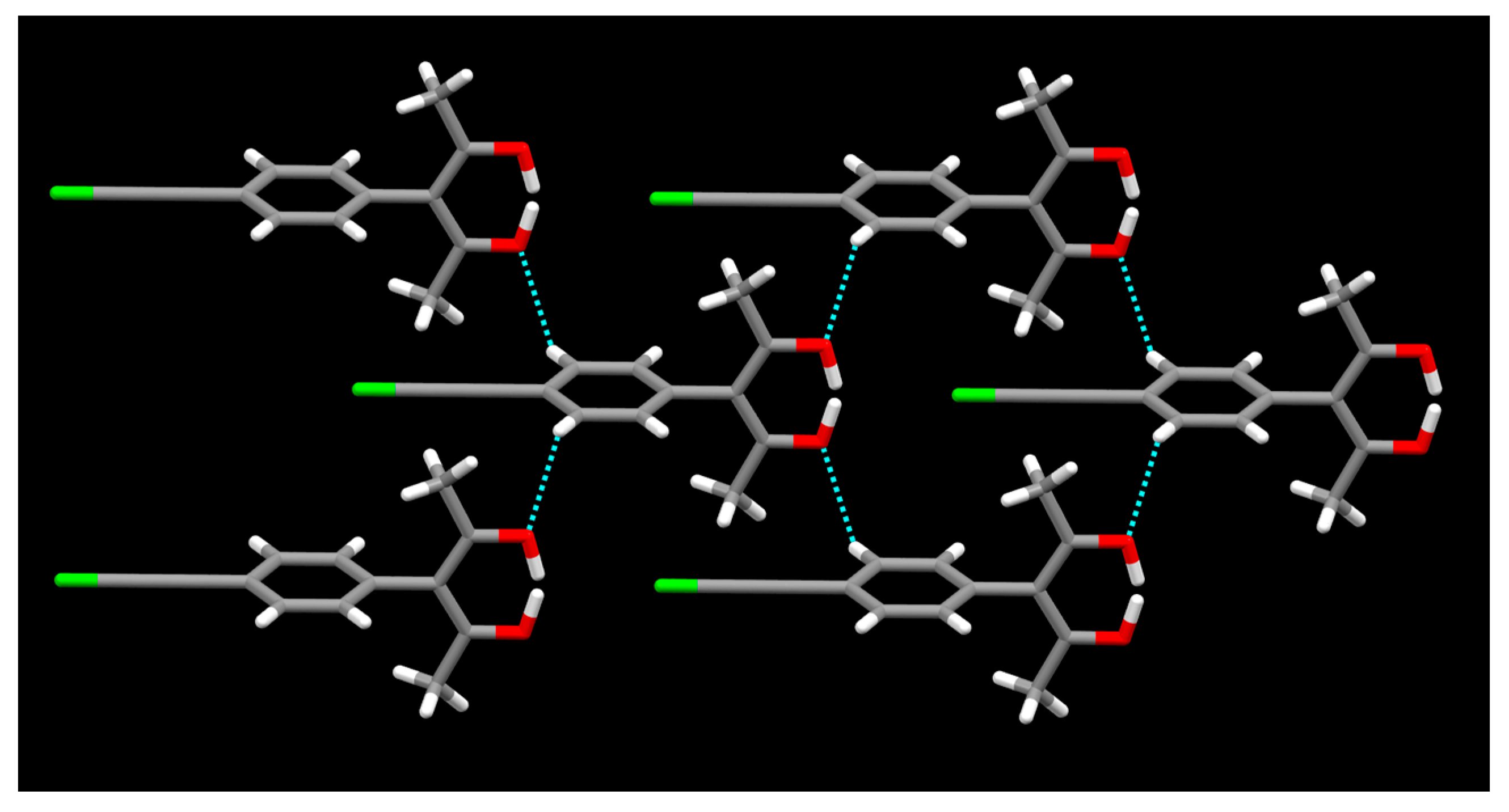{kind=link}
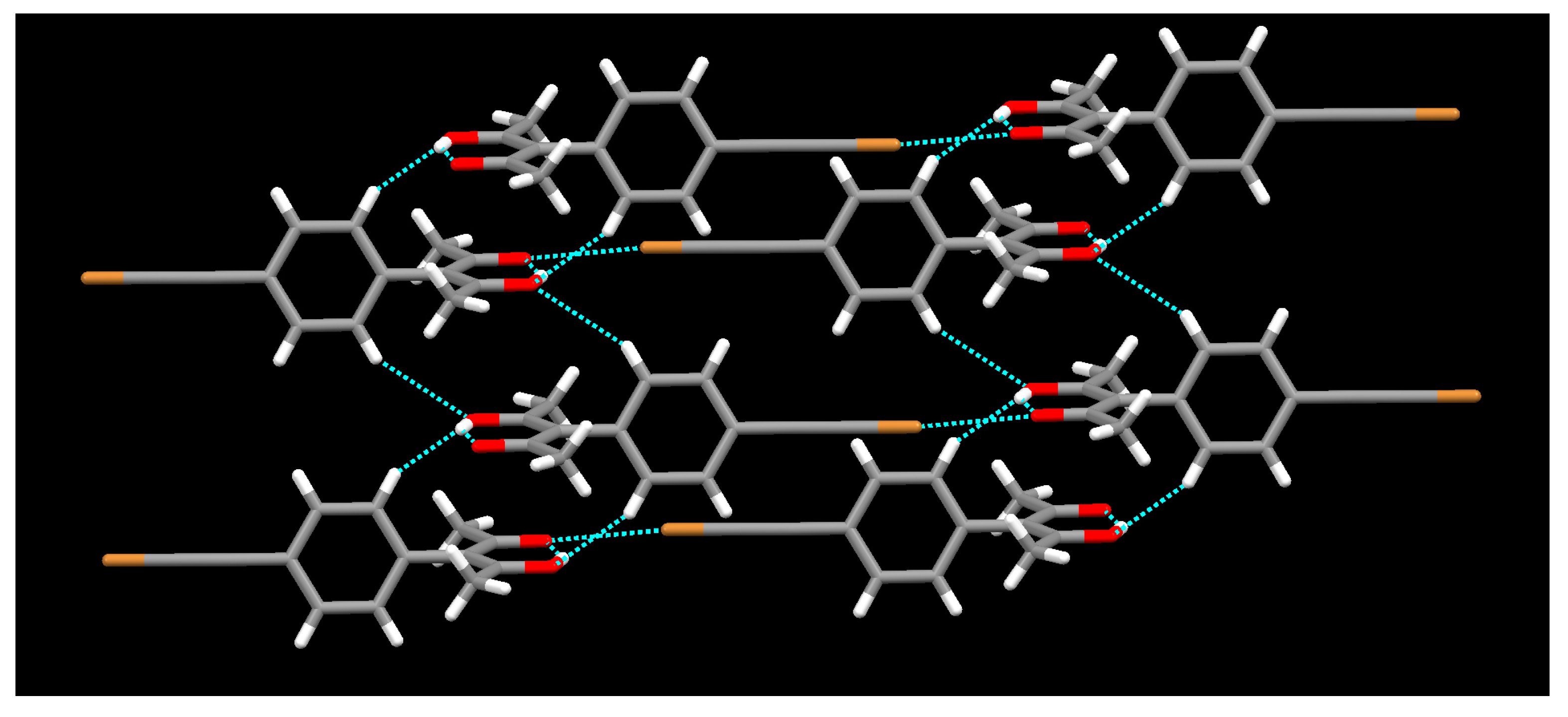{kind=link}
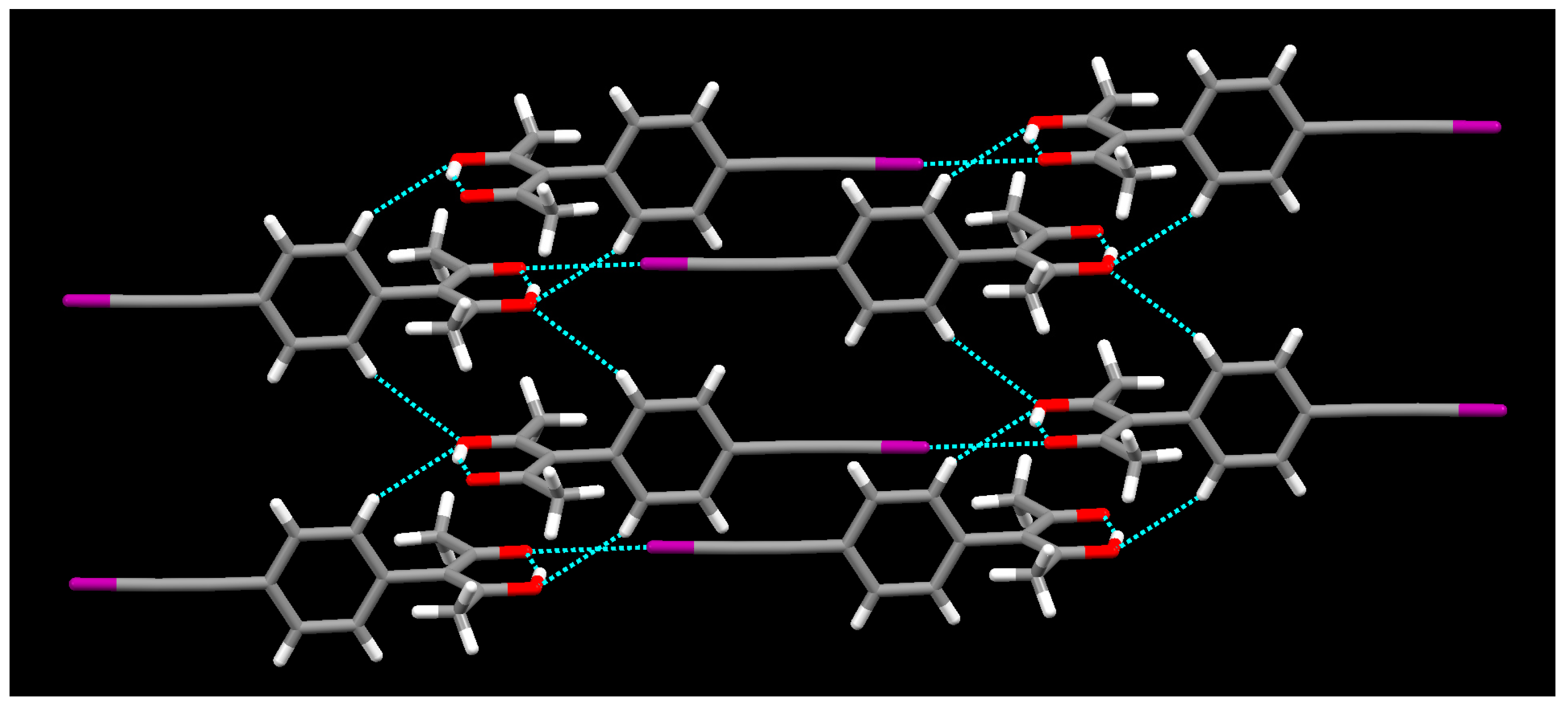{kind=link}
{kind=link}
{kind=link}
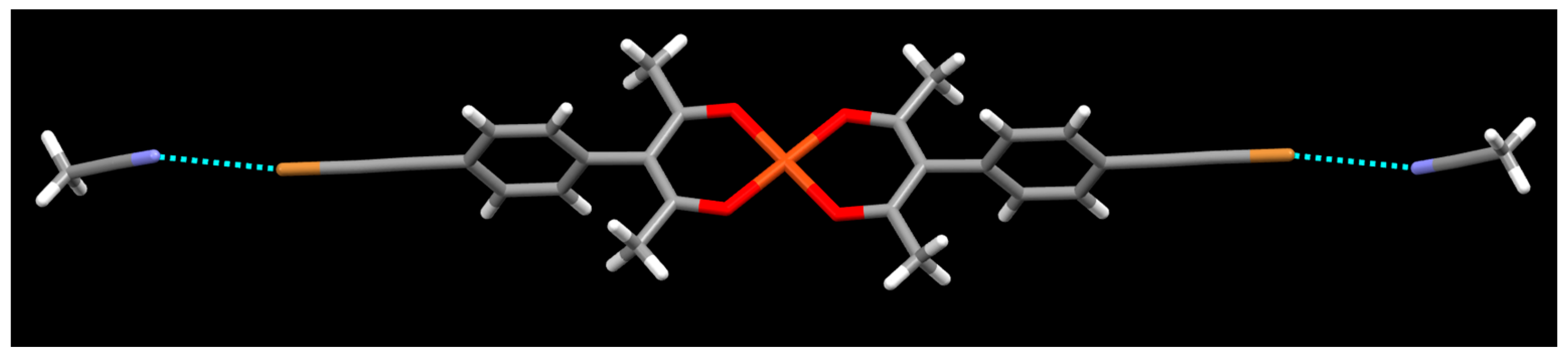{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
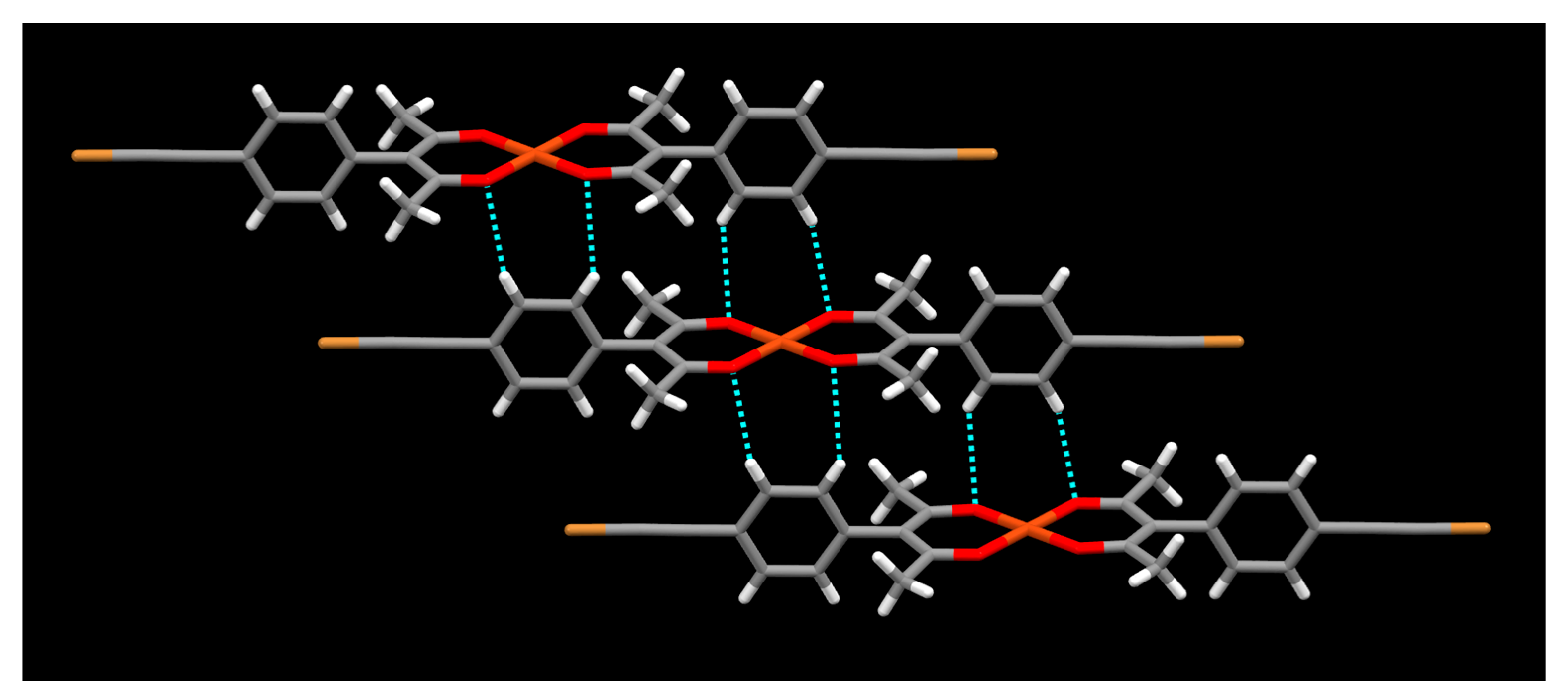
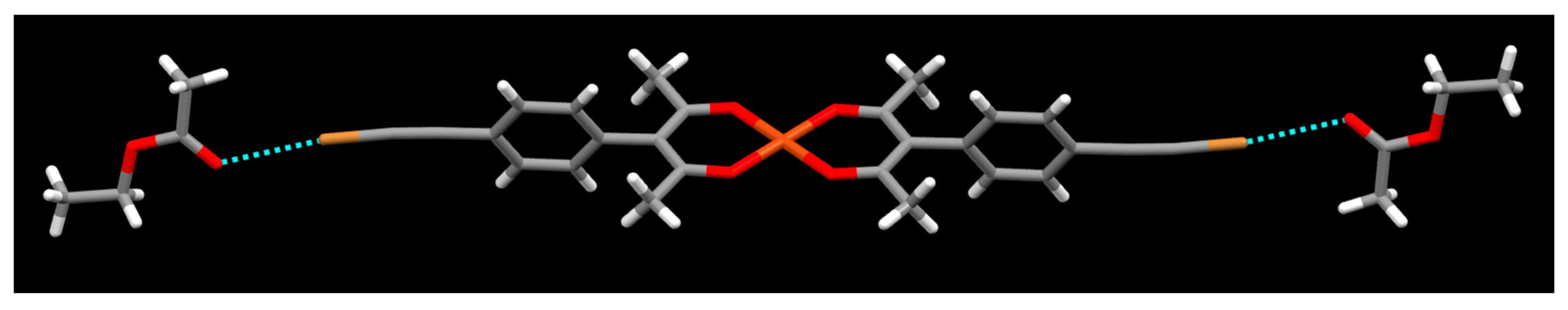
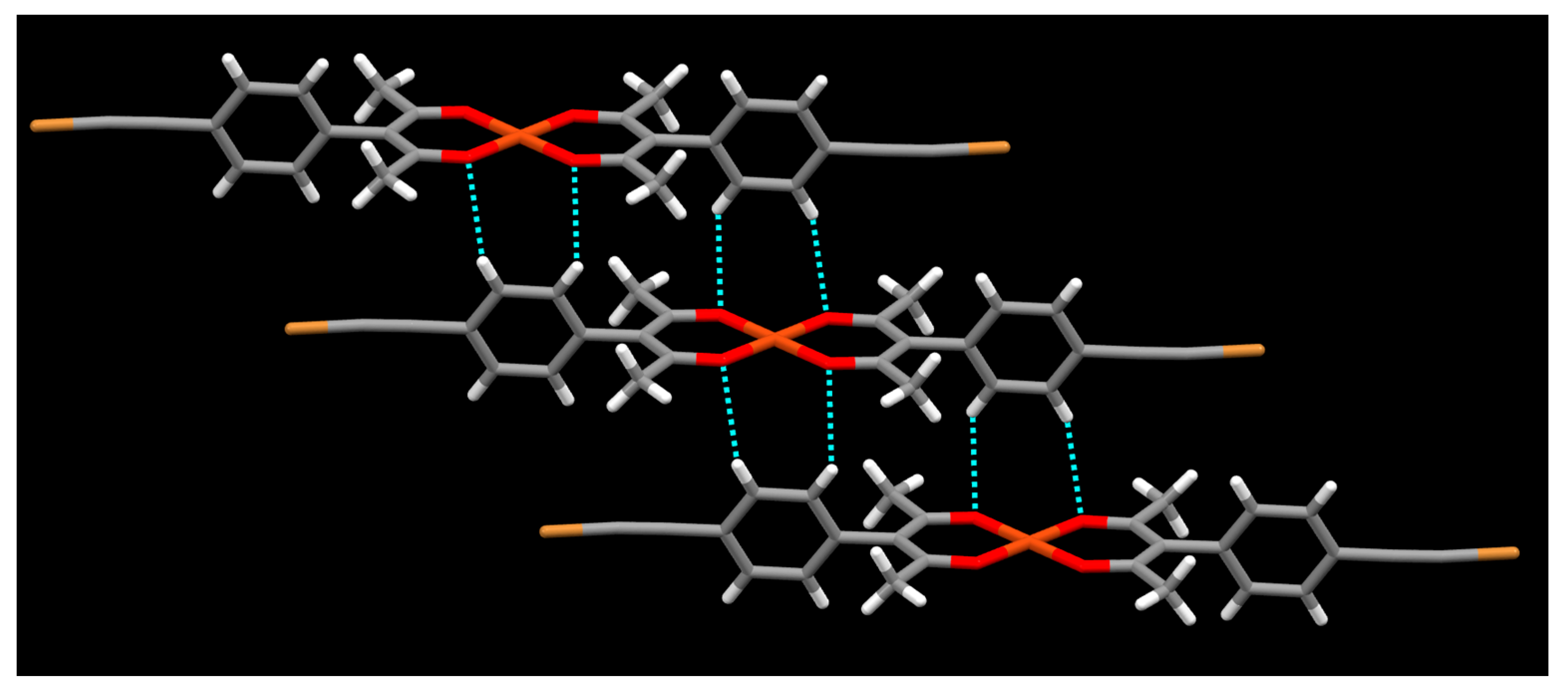
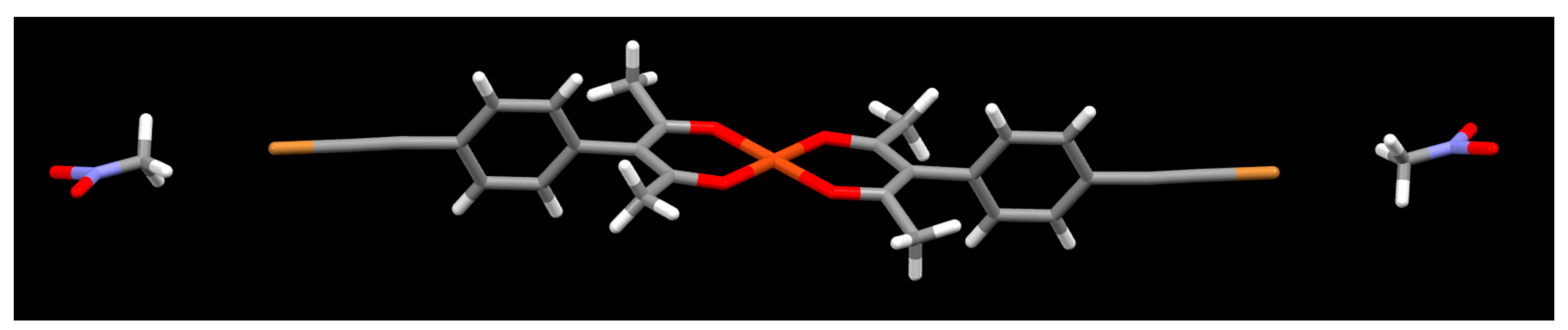
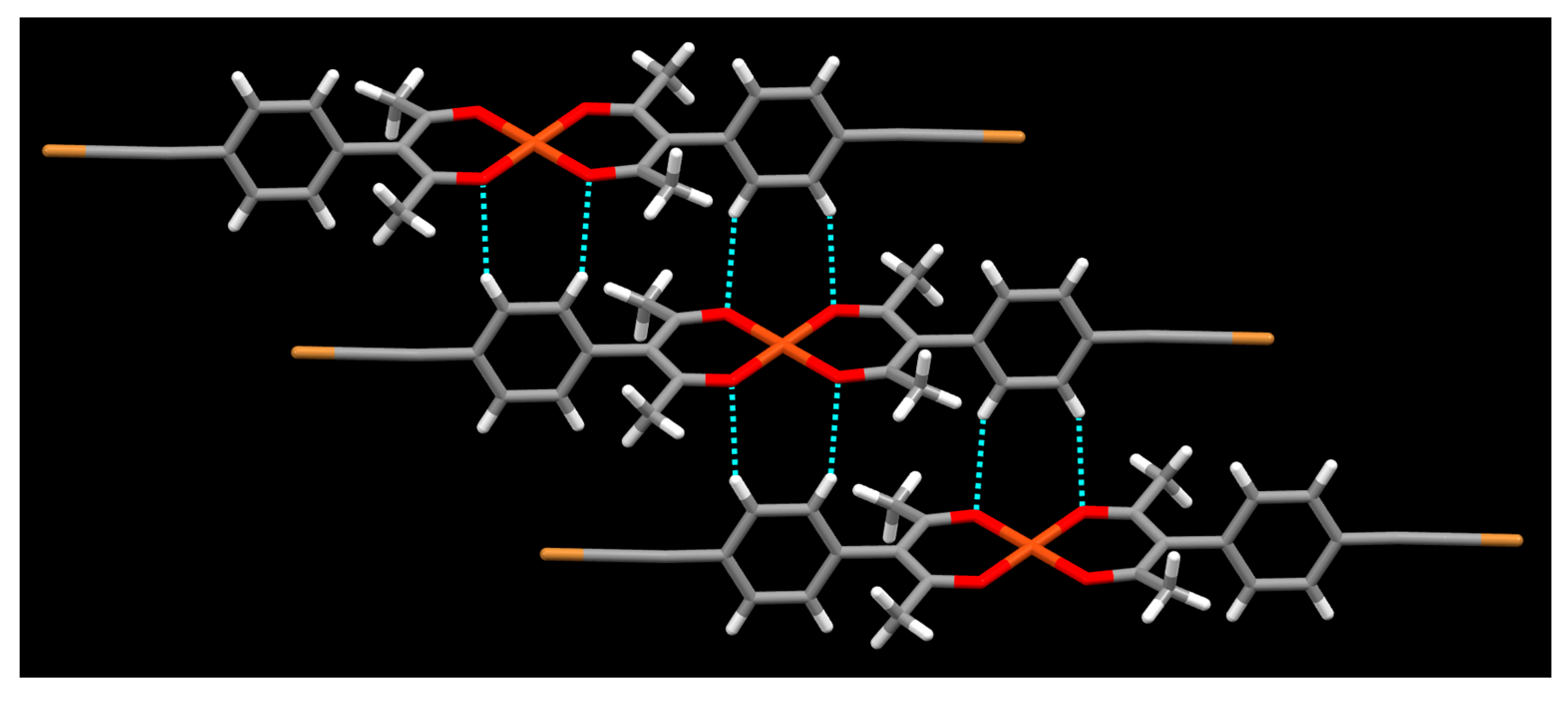
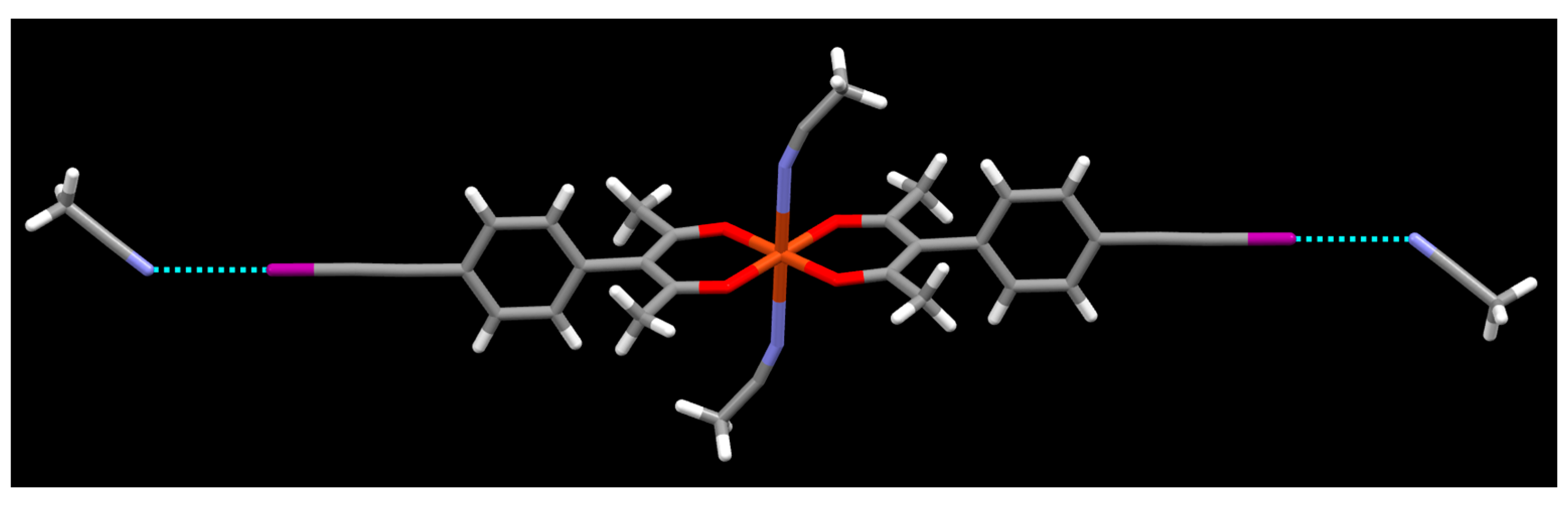
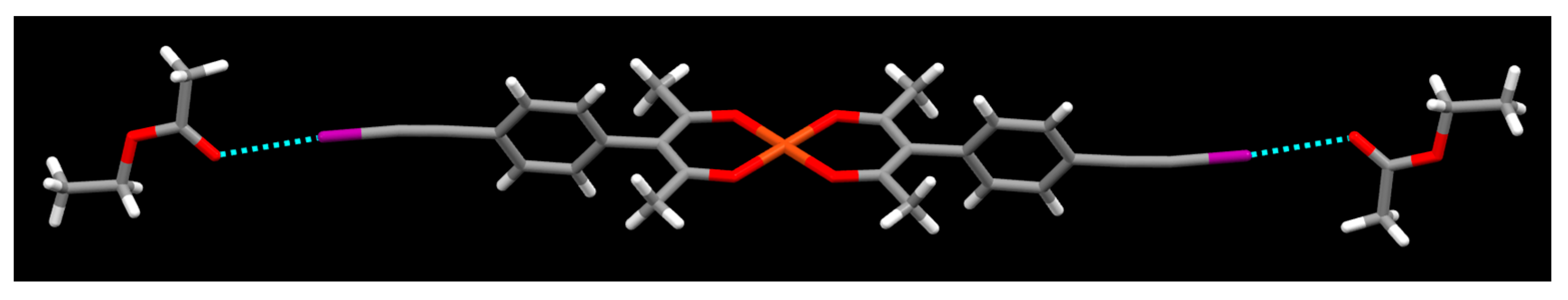
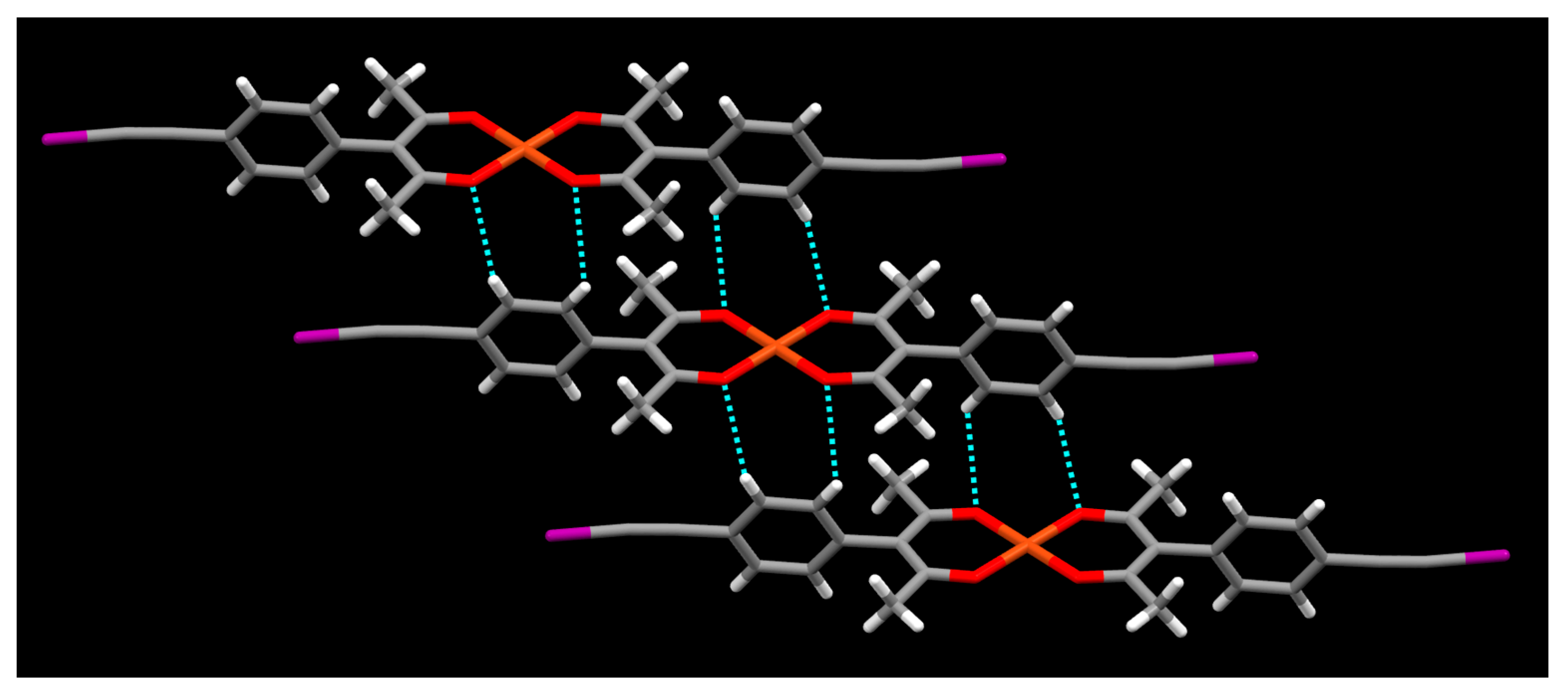
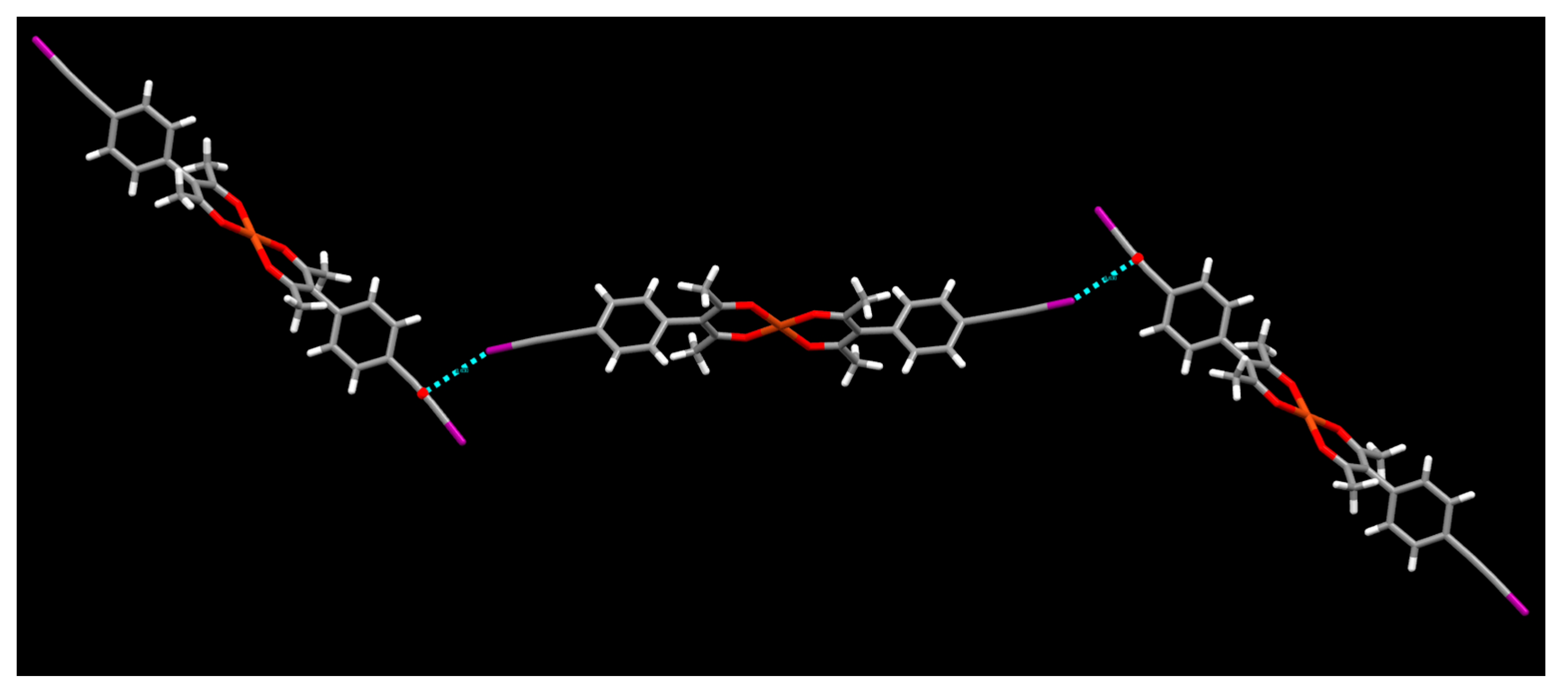
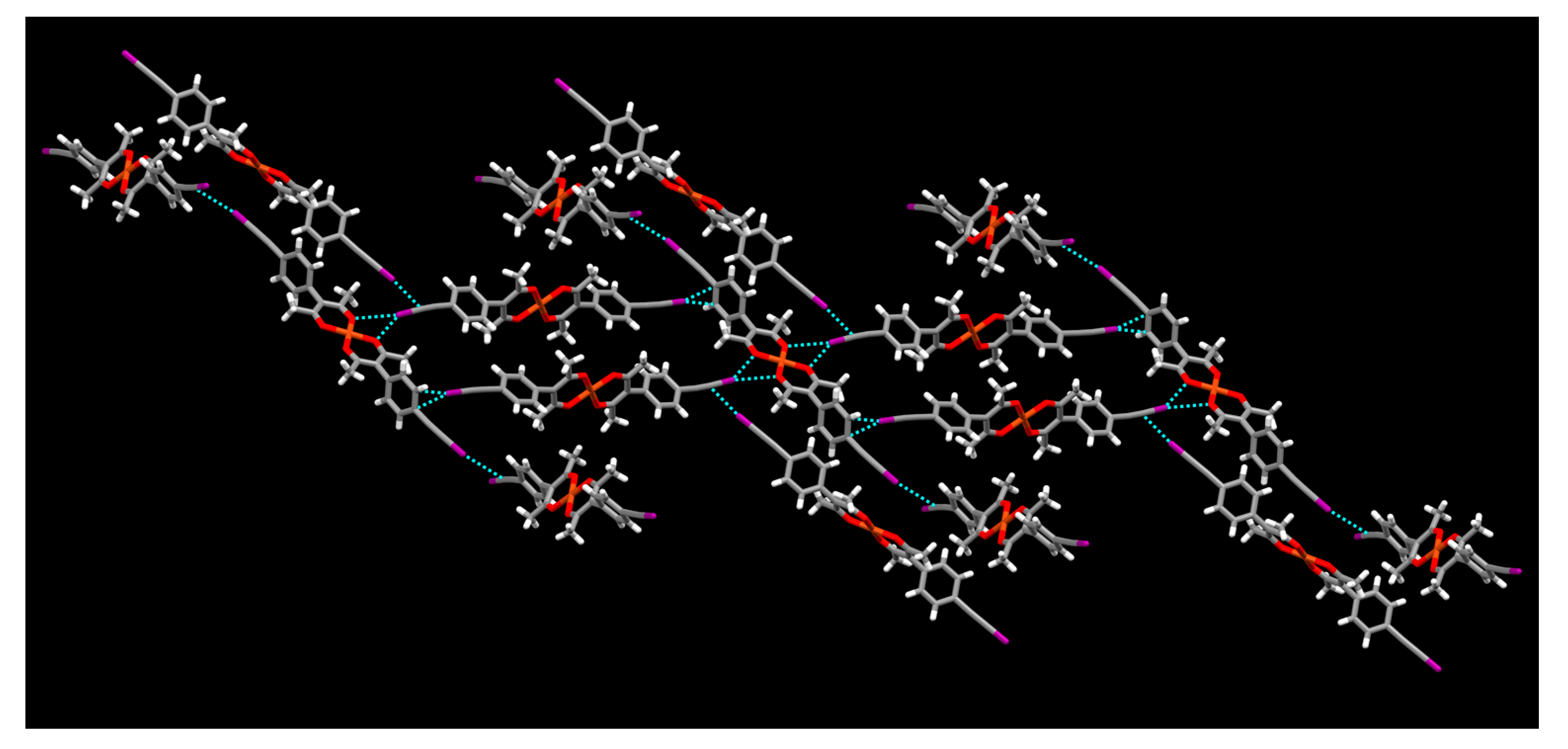
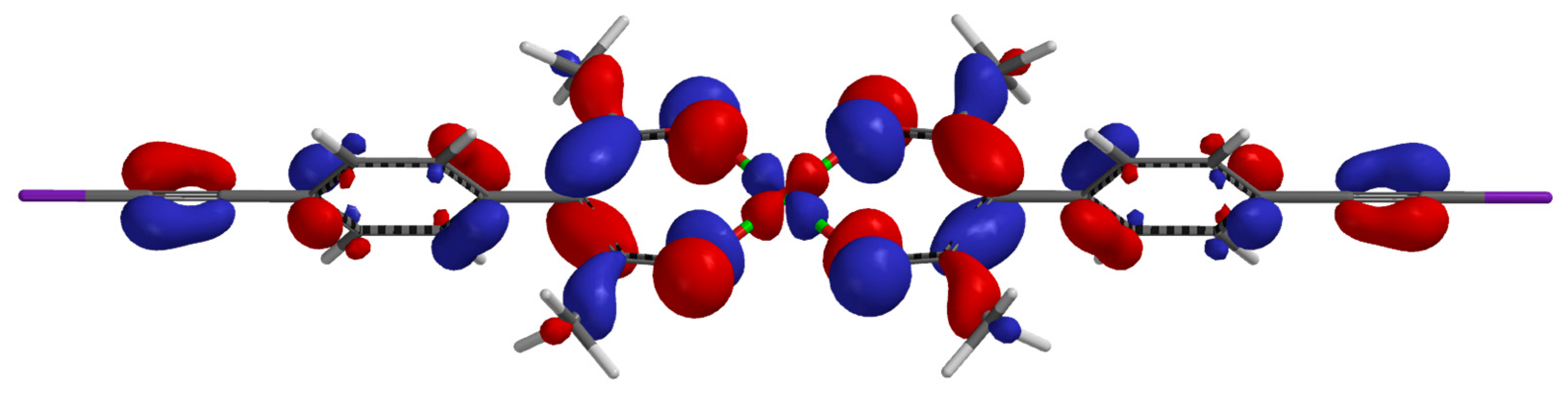
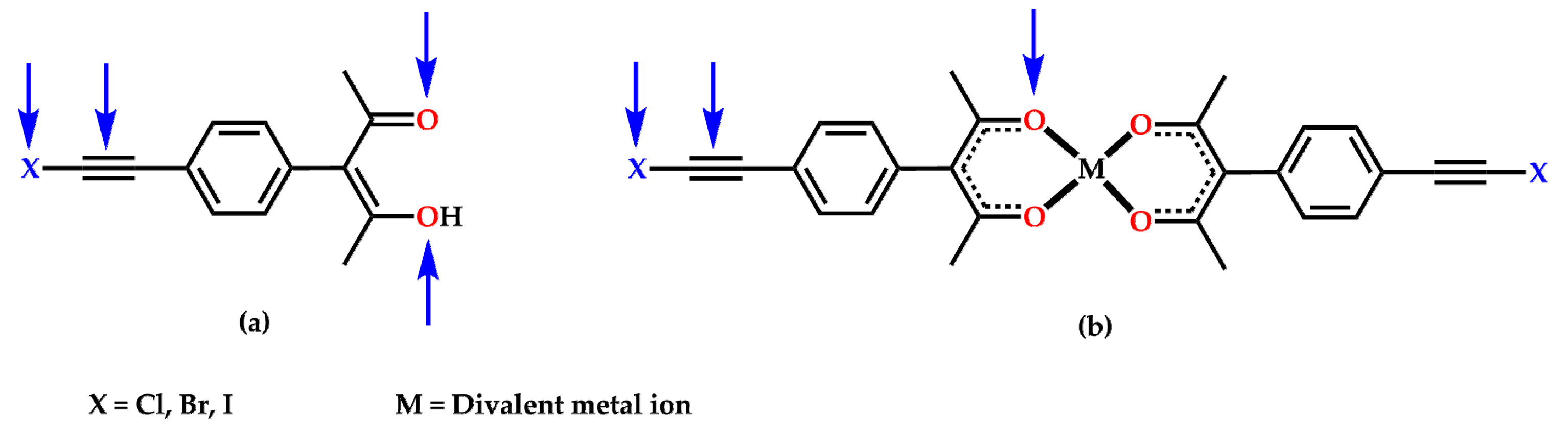
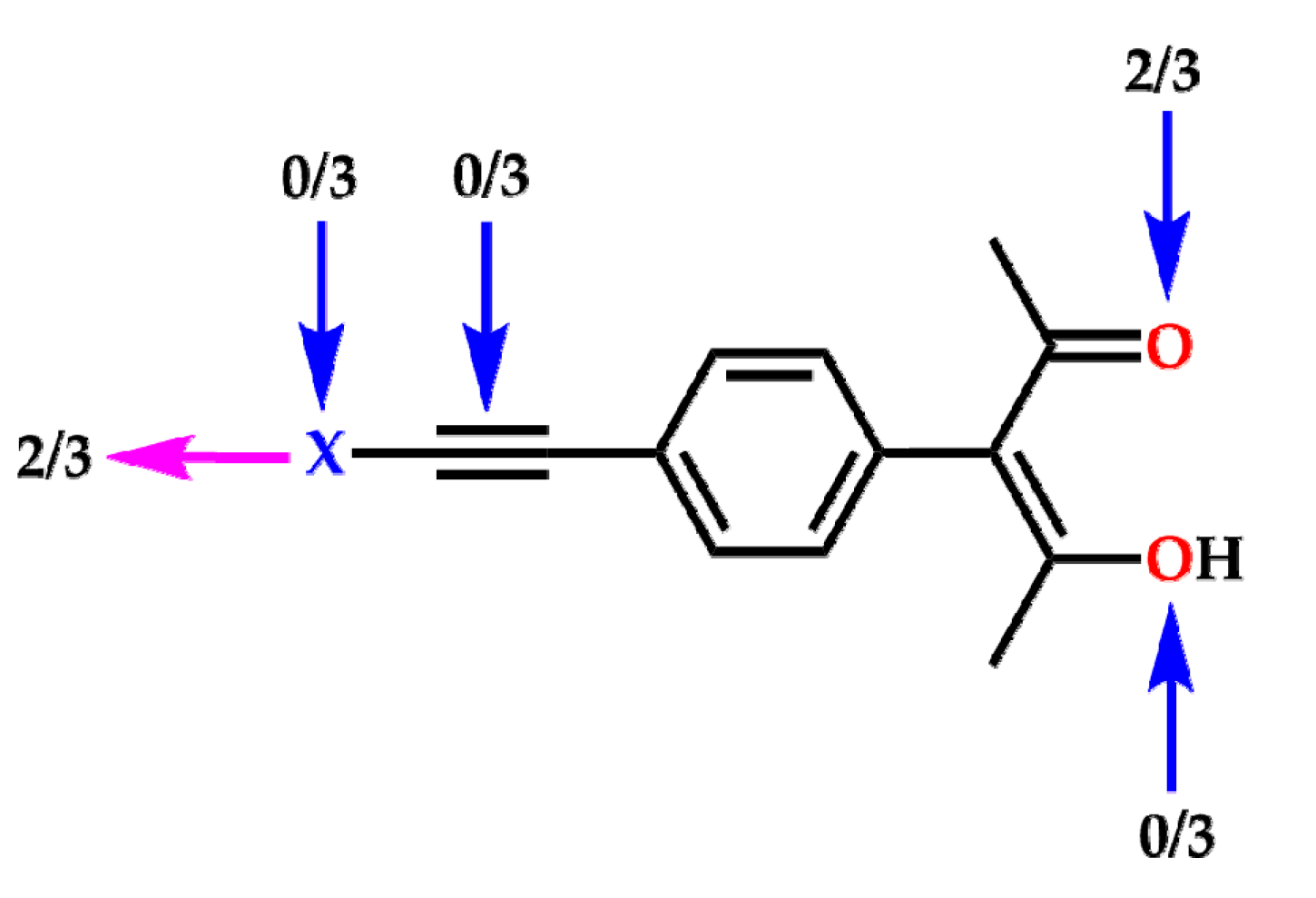
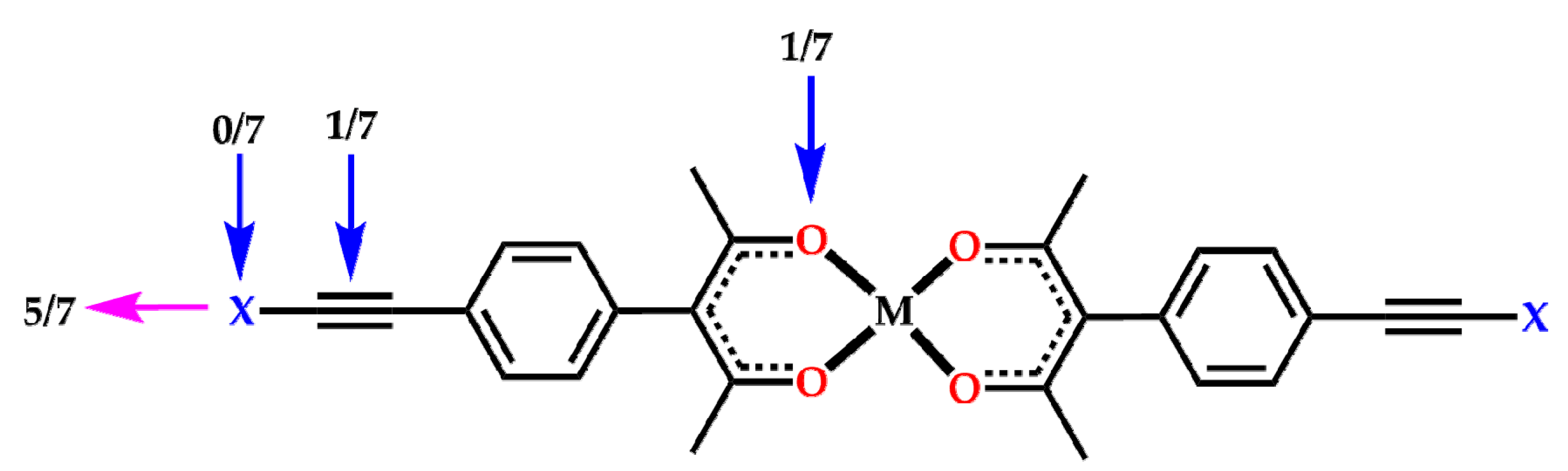
2. Results
3. Discussion
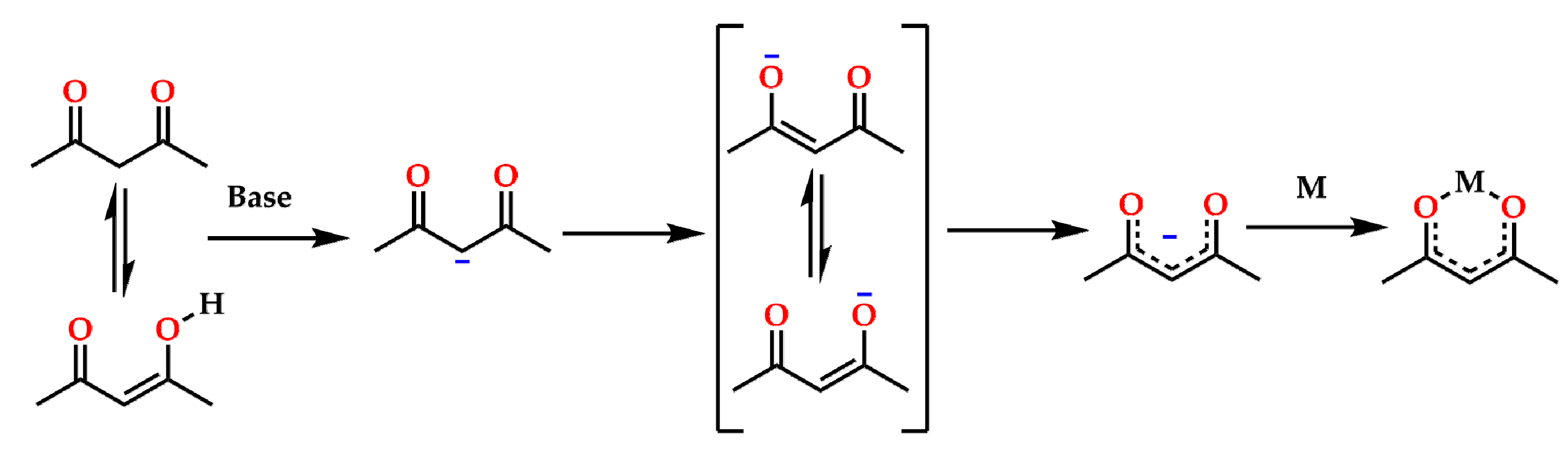
4. Materials and Methods
4.1. Synthesis
4.1.1. Synthesis of 4-(trimethylsilylethynyl)benzaldehyde
4.1.2. Synthesis of 4-(chloroethynyl)benzaldehyde
4.1.3. Synthesis of 4-(bromoethynyl)benzaldehyde
4.1.4. Synthesis of 4-(iodoethynyl)benzaldehyde
4.1.5. Synthesis of 3-(4-(chloroethynyl)phenyl)-4-Hydroxypent-3-en-2-one, L1
4.1.6. Synthesis of 3-(4-(bromoethynyl)phenyl)-4-Hydroxypent-3-en-2-one, L2
4.1.7. Synthesis of 3-(4-(iodoethynyl)phenyl)-4-Hydroxypent-3-en-2-one, L3
4.1.8. Synthesis of [Cu(L1)2]
4.1.9. Synthesis of [Cu(L2)2]·2CH3CN
4.1.10. Synthesis of [Cu(L2)2]·2C4H8O2
4.1.11. Synthesis of [Cu(L2)2]·2CH3NO2
4.1.12. Synthesis of [Cu(L3)2]·4CH3CN
4.1.13. Synthesis of [Cu(L3)2]·2C4H8O2
4.1.14. Synthesis of [Cu(L3)2]
4.2. X-ray Crystallography
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aakeröy, C.B.; Desper, J.; Smith, M.M. Constructing, deconstructing, and reconstructing ternary supermolecules. Chem. Commun. 2007, 3936–3938. [Google Scholar] [CrossRef] [PubMed]
- Amombo Noa, F.M.; Bourne, S.A.; Su, H.; Weber, E.; Nassimbeni, L.R. Hydrogen bonding versus halogen bonding in host–guest compounds. Cryst. Growth Des. 2016, 16, 4765–4771. [Google Scholar] [CrossRef]
- Gawade, R.L.; Chakravarty, D.K.; Kotmale, A.; Sangtani, E.; Joshi, P.V.; Ahmed, A.; Mane, M.V.; Das, S.; Vanka, K.; Rajamohanan, P.R.; et al. Additive mediated syn-anti conformational tuning at nucleation to capture elusive polymorphs: Remarkable role of extended π-stacking interactions in driving the self-assembly. Cryst. Growth Des. 2016, 16, 2416–2428. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Wijethunga, T.K.; Desper, J. Constructing molecular polygons using halogen bonding and bifurcated n-oxides. CrystEngComm 2014, 16, 28–31. [Google Scholar] [CrossRef]
- Bauza, A.; Quinonero, D.; Deya, P.M.; Frontera, A. Halogen bonding versus chalcogen and pnicogen bonding: A combined cambridge structural database and theoretical study. CrystEngComm 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Takemura, A.; McAllister, L.J.; Karadakov, P.B.; Pridmore, N.E.; Whitwood, A.C.; Bruce, D.W. Competition and cooperation: Hydrogen and halogen bonding in co-crystals involving 4-iodotetrafluorobenzoic acid, 4-iodotetrafluorophenol and 4-bromotetrafluorophenol. CrystEngComm 2014, 16, 4254–4264. [Google Scholar] [CrossRef]
- Maugeri, L.; Lébl, T.; Cordes, D.B.; Slawin, A.M.Z.; Philp, D. Cooperative binding in a phosphine oxide-based halogen bonded dimer drives supramolecular oligomerization. J. Org. Chem. 2017, 82, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Kukovec, B.M.; Malik, M.; Kodrin, I.; Aakeröy, C.B.; Đaković, M. Directed assembly of acac-based complexes by deliberately fine-tuning electrostatic molecular-recognition events. Cryst. Growth Des. 2016, 16, 7308–7317. [Google Scholar] [CrossRef]
- Bauza, A.; Frontera, A. Supramolecular nanotubes based on halogen bonding interactions: Cooperativity and interaction with small guests. Phys. Chem. Chem. Phys. 2017, 19, 12936–12941. [Google Scholar] [CrossRef] [PubMed]
- Pizzi, A.; Lascialfari, L.; Demitri, N.; Bertolani, A.; Maiolo, D.; Carretti, E.; Metrangolo, P. Halogen bonding modulates hydrogel formation from fmoc amino acids. CrystEngComm 2017, 19, 1870–1874. [Google Scholar] [CrossRef]
- Béziau, A.; Baudron, S.A.; Rogez, G.; Hosseini, M.W. Assembly, disassembly, and reassembly: Conversion of homometallic coordination networks into mixed metal–organic frameworks. Inorg. Chem. 2015, 54, 2032–2039. [Google Scholar] [CrossRef] [PubMed]
- Durá, G.; Carrión, M.C.; Jalón, F.A.; Manzano, B.R.; Rodríguez, A.M.; Mereiter, K. Robust 2d coordination networks from a two-step assembly process with predesigned silver cyclic dimers and hexamethylenetetramine. Cryst. Growth Des. 2015, 15, 3321–3331. [Google Scholar] [CrossRef]
- Durá, G.; Carrión, M.C.; Jalón, F.A.; Manzano, B.R.; Rodríguez, A.M. Formation of mono-, di- and trinuclear species in the self-assembly of Bis(pyraz-olyl)(pyridin-3-yl)methane ligands and metals with different coordination geometries. Eur. J. Inorg. Chem. 2015, 2015, 5874–5885. [Google Scholar] [CrossRef]
- He, L.; Ma, D.; Duan, L.; Wei, Y.; Qiao, J.; Zhang, D.; Dong, G.; Wang, L.; Qiu, Y. Control of intramolecular π–π stacking interaction in cationic iridium complexes via fluorination of pendant phenyl rings. Inorg. Chem. 2012, 51, 4502–4510. [Google Scholar] [CrossRef] [PubMed]
- Khavasi, H.R.; Kavand, S. [small pi]-stacking synthon repetitivity in coordination compounds. CrystEngComm 2016, 18, 4760–4764. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Dey, B.; Das, S.; Gamez, P.; Robertazzi, A.; Chan, K.-T.; Lee, H.M.; Mukhopadhyay, S. Supramolecular lone pair-π/π-π/π-anion assembly in a Mg(II)-malonate-2-aminopyridine-nitrate ternary system. J. Phys. Chem. A 2009, 113, 1623–1627. [Google Scholar] [CrossRef] [PubMed]
- Kumari, H.; Jin, P.; Teat, S.J.; Barnes, C.L.; Dalgarno, S.J.; Atwood, J.L. Strong cation⋯π interactions promote the capture of metal ions within metal-seamed nanocapsule. J. Am. Chem. Soc. 2014, 136, 17002–17005. [Google Scholar] [CrossRef] [PubMed]
- Safin, D.A.; Robeyns, K.; Babashkina, M.G.; Vande Velde, C.M.L.; Filinchuk, Y. Ligand-driven anion-π interaction-induced silver(i) coordination chemistry. Cryst. Growth Des. 2016, 16, 3763–3770. [Google Scholar] [CrossRef]
- Manna, P.; Seth, S.K.; Das, A.; Hemming, J.; Prendergast, R.; Helliwell, M.; Choudhury, S.R.; Frontera, A.; Mukhopadhyay, S. Anion induced formation of supramolecular associations involving lone pair-π and anion-π interactions in Co(II) malonate complexes: Experimental observations, hirshfeld surface analyses and dft studies. Inorg. Chem. 2012, 51, 3557–3571. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.K.; Saha, I.; Estarellas, C.; Frontera, A.; Kar, T.; Mukhopadhyay, S. Supramolecular self-assembly of m-ida complexes involving lone-pair⋯π interactions: Crystal structures, hirshfeld surface analysis, and DFT calculations [H2IDA = iminodiacetic acid, M = Cu(II), Ni(II)]. Cryst. Growth Des. 2011, 11, 3250–3265. [Google Scholar] [CrossRef]
- Tiekink, E.R.T.; Zukerman-Schpector, J. Gold[three dots, centered][small pi] aryl interactions as supramolecular synthons. CrystEngComm 2009, 11, 1176–1186. [Google Scholar] [CrossRef]
- Cheng, B.; Tehrani, A.A.; Hu, M.-L.; Morsali, A. Supramolecular assemblies of Ru(II) organometallic half-sandwich complexes. CrystEngComm 2014, 16, 9125–9134. [Google Scholar] [CrossRef]
- Dong, H.; Wang, D.; Sun, G.; Hinestroza, J.P. Assembly of metal nanoparticles on electrospun nylon 6 nanofibers by control of interfacial hydrogen-bonding interactions. Chem. Mater. 2008, 20, 6627–6632. [Google Scholar] [CrossRef]
- Piñón, V.; Weck, M. Patterned polymeric multilayered assemblies through hydrogen bonding and metal coordination. Langmuir 2012, 28, 3279–3284. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.J.; de Mendoza, J. Self-assembled squares and triangles by simultaneous hydrogen bonding and metal coordination. Org. Lett. 2013, 15, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Sommer, S.K.; Zakharov, L.N.; Pluth, M.D. Design, synthesis, and characterization of hybrid Metal-Ligand Hydrogen-Bonded (MLHB) supramolecular architectures. Inorg. Chem. 2015, 54, 1912–1918. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhu, L.; Yin, P.; Fu, W.; Chen, J.; Hao, J.; Xiao, F.; Lv, C.; Zhang, J.; Shi, L.; et al. From 0 d dimer to 2 d network—Supramolecular assembly of organic derivatized polyoxometalates with remote hydroxyl via hydrogen bonding. Inorg. Chem. 2009, 48, 9222–9235. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Priimagi, A.; Cavallo, G.; Metrangolo, P.; Resnati, G. The halogen bond in the design of functional supramolecular materials: Recent advances. Acc. Chem. Res. 2013, 46, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Desiraju Gautam, R.; Ho, P.S.; Kloo, L.; Legon Anthony, C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC recommendations 2013). Pure Appl. Chem. 2013, 85, 1711. [Google Scholar] [CrossRef]
- Meyer, F.; Dubois, P. Halogen bonding at work: Recent applications in synthetic chemistry and materials science. CrystEngComm 2013, 15, 3058–3071. [Google Scholar] [CrossRef]
- Berger, G.; Soubhye, J.; Meyer, F. Halogen bonding in polymer science: From crystal engineering to functional supramolecular polymers and materials. Polym. Chem. 2015, 6, 3559–3580. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Kirina, Y.V.; Kukushkin, V.Y. Halogen bonding between metal centers and halocarbons. Chem. Commun. 2016, 52, 5565–5568. [Google Scholar] [CrossRef] [PubMed]
- Khavasi, H.R.; Ghanbarpour, A.; Tehrani, A.A. The role of intermolecular interactions involving halogens in the supramolecular architecture of a series of Mn(II) coordination compounds. RSC Adv. 2016, 6, 2422–2430. [Google Scholar] [CrossRef]
- Bertani, R.; Sgarbossa, P.; Venzo, A.; Lelj, F.; Amati, M.; Resnati, G.; Pilati, T.; Metrangolo, P.; Terraneo, G. Halogen bonding in metal–organic–supramolecular networks. Coord. Chem. Rev. 2010, 254, 677–695. [Google Scholar] [CrossRef]
- Li, B.; Zang, S.-Q.; Wang, L.-Y.; Mak, T.C.W. Halogen bonding: A powerful, emerging tool for constructing high-dimensional metal-containing supramolecular networks. Coord. Chem. Rev. 2016, 308, 1–21. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Sinha, A.S.; Chopade, P.D.; Desper, J. Halogen bonding or close packing? Examining the structural landscape in a series of Cu(II)-acac complexes. Dalton Trans. 2011, 40, 12160–12168. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Murray, J.S.; Fanfrlík, J.; Řezáč, J.; Solá, R.J.; Concha, M.C.; Ramos, F.M.; Politzer, P. Halogen bond tunability i: The effects of aromatic fluorine substitution on the strengths of halogen-bonding interactions involving chlorine, bromine, and iodine. J. Mol. Model. 2011, 17, 3309–3318. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Wijethunga, T.K.; Desper, J. Practical crystal engineering using halogen bonding: A hierarchy based on calculated molecular electrostatic potential surfaces. J. Mol. Struct. 2014, 1072, 20–27. [Google Scholar] [CrossRef]
- Nguyen, S.T.; Rheingold, A.L.; Tschumper, G.S.; Watkins, D.L. Elucidating the effects of fluoro and nitro substituents on halogen bond driven assemblies of pyridyl-capped π-conjugated molecules. Cryst. Growth Des. 2016, 16, 6648–6653. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Baldrighi, M.; Desper, J.; Metrangolo, P.; Resnati, G. Supramolecular hierarchy among halogen-bond donors. Chem. A Eur. J. 2013, 19, 16240–16247. [Google Scholar] [CrossRef] [PubMed]
- Carlucci, L.; Gavezzotti, A. A quantitative measure of halogen bond activation in cocrystallization. Phys. Chem. Chem. Phy. 2017. [Google Scholar] [CrossRef] [PubMed]
- Thalladi, V.R.; Goud, B.S.; Hoy, V.J.; Allen, F.H.; Howard, J.A.K.; Desiraju, G.R. Supramolecular synthons in crystal engineering. Structure simplification, synthon robustness and supramolecular retrosynthesis. Chem. Commun. 1996, 3, 401–402. [Google Scholar] [CrossRef]
- Desiraju, G.R. Designer crystals: Intermolecular interactions, network structures and supramolecular synthons. Chem. Commun. 1997, 16, 1475–1482. [Google Scholar] [CrossRef]
- Dumele, O.; Wu, D.; Trapp, N.; Goroff, N.; Diederich, F. Halogen bonding of (iodoethynyl)benzene derivatives in solution. Org. Lett. 2014, 16, 4722–4725. [Google Scholar] [CrossRef] [PubMed]
- Baldrighi, M.; Bartesaghi, D.; Cavallo, G.; Chierotti, M.R.; Gobetto, R.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Polymorphs and co-crystals of haloprogin: An antifungal agent. CrystEngComm 2014, 16, 5897–5904. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Welideniya, D.; Desper, J. Ethynyl hydrogen bonds and iodoethynyl halogen bonds: A case of synthon mimicry. CrystEngComm 2017, 19, 11–13. [Google Scholar] [CrossRef]
- Ramirez, F.; Patwardhan, A.V.; Ramanathan, N.; Desai, N.B.; Greco, C.V.; Heller, S.R. A new synthesis of α,β-dihydroxy ketones via oxyphosphoranes. Condensation of aliphatic α-diketones with aldehydes by means of trialkyl phosphites. P31 and H1 nuclear magnetic resonance spectra1,2. J. Am. Chem. Soc. 1965, 87, 543–548. [Google Scholar] [CrossRef]
- Osowska, K.; Lis, T.; Szafert, S. Protection/deprotection-free syntheses and structural analysis of (keto-aryl)diynes. Eur. J. Org. Chem. 2008, 2008, 4598–4606. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Sinha, A.S.; Epa, K.N.; Spartz, C.L.; Desper, J. A versatile and green mechanochemical route for aldehyde-oxime conversions. Chem. Commun. 2012, 48, 11289–11291. [Google Scholar] [CrossRef] [PubMed]
- Dumele, O.; Schreib, B.; Warzok, U.; Trapp, N.; Schalley, C.A.; Diederich, F. Halogen-bonded supramolecular capsules in the solid state, in solution, and in the gas phase. Angew. Chem. Int. Ed. 2017, 56, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Bruker. APEX2 v2013.10-0, © 2013, Bruker Analytical X-ray Systems; Bruker: Madison, WI, USA, 2013. [Google Scholar]
- Bruker. COSMO v1.61, © 1999–2009, Bruker Analytical X-ray Systems; Bruker: Madison, WI, USA, 1999. [Google Scholar]
- Bruker. SAINT v8.34a, © 1997–2013, Bruker Analytical X-ray Systems; Bruker: Madison, WI, USA, 1997. [Google Scholar]
- Bruker. SADABS v2012/1, © 2012, Bruker Analytical X-ray Systems; Bruker: Madison, WI, USA, 2012. [Google Scholar]
- Bruker. SHELXTL v2008/4, © 2008, Bruker Analytical X-ray Systems; Bruker: Madison, WI, USA, 2008. [Google Scholar]
- Oxford Diffraction Ltd. Oxford Diffraction, Xcalibur CCD System, CrysAlis Software System, Version 171.31; Oxford Diffraction Ltd.: Oxford, UK, 2004. [Google Scholar]
- Sheldrick, G. A short history of shelx. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]























© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamekkanda, J.C.; Sinha, A.S.; Desper, J.; Ðaković, M.; Aakeröy, C.B. The Role of Halogen Bonding in Controlling Assembly and Organization of Cu(II)-Acac Based Coordination Complexes. Crystals 2017, 7, 226. https://doi.org/10.3390/cryst7070226
Gamekkanda JC, Sinha AS, Desper J, Ðaković M, Aakeröy CB. The Role of Halogen Bonding in Controlling Assembly and Organization of Cu(II)-Acac Based Coordination Complexes. Crystals. 2017; 7(7):226. https://doi.org/10.3390/cryst7070226
Chicago/Turabian StyleGamekkanda, Janaka C., Abhijeet S. Sinha, John Desper, Marijana Ðaković, and Christer B. Aakeröy. 2017. "The Role of Halogen Bonding in Controlling Assembly and Organization of Cu(II)-Acac Based Coordination Complexes" Crystals 7, no. 7: 226. https://doi.org/10.3390/cryst7070226
APA StyleGamekkanda, J. C., Sinha, A. S., Desper, J., Ðaković, M., & Aakeröy, C. B. (2017). The Role of Halogen Bonding in Controlling Assembly and Organization of Cu(II)-Acac Based Coordination Complexes. Crystals, 7(7), 226. https://doi.org/10.3390/cryst7070226