New Polymorphs of the Phase-Change Material Sodium Acetate


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Experimental
2.2. Least-Squares Refinement
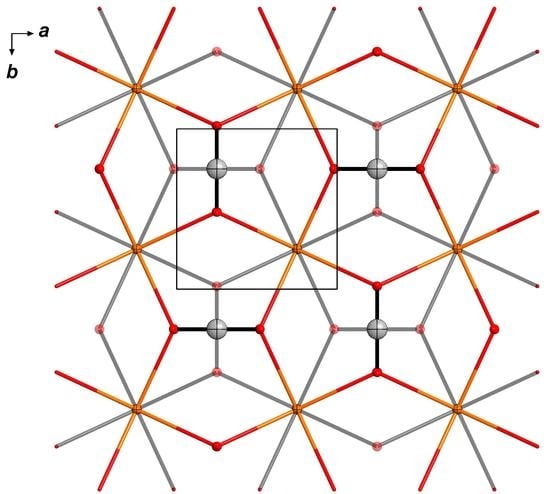
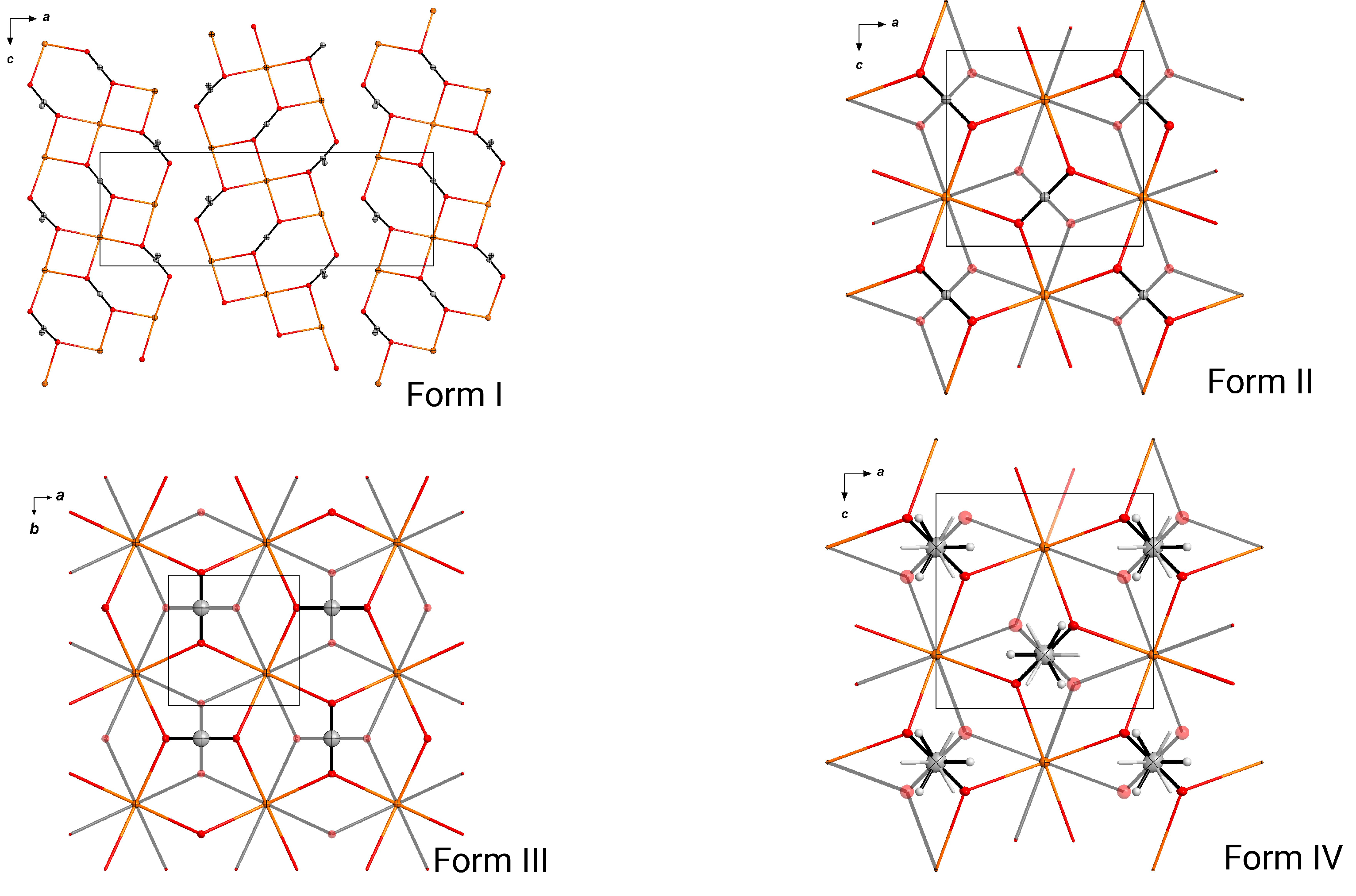
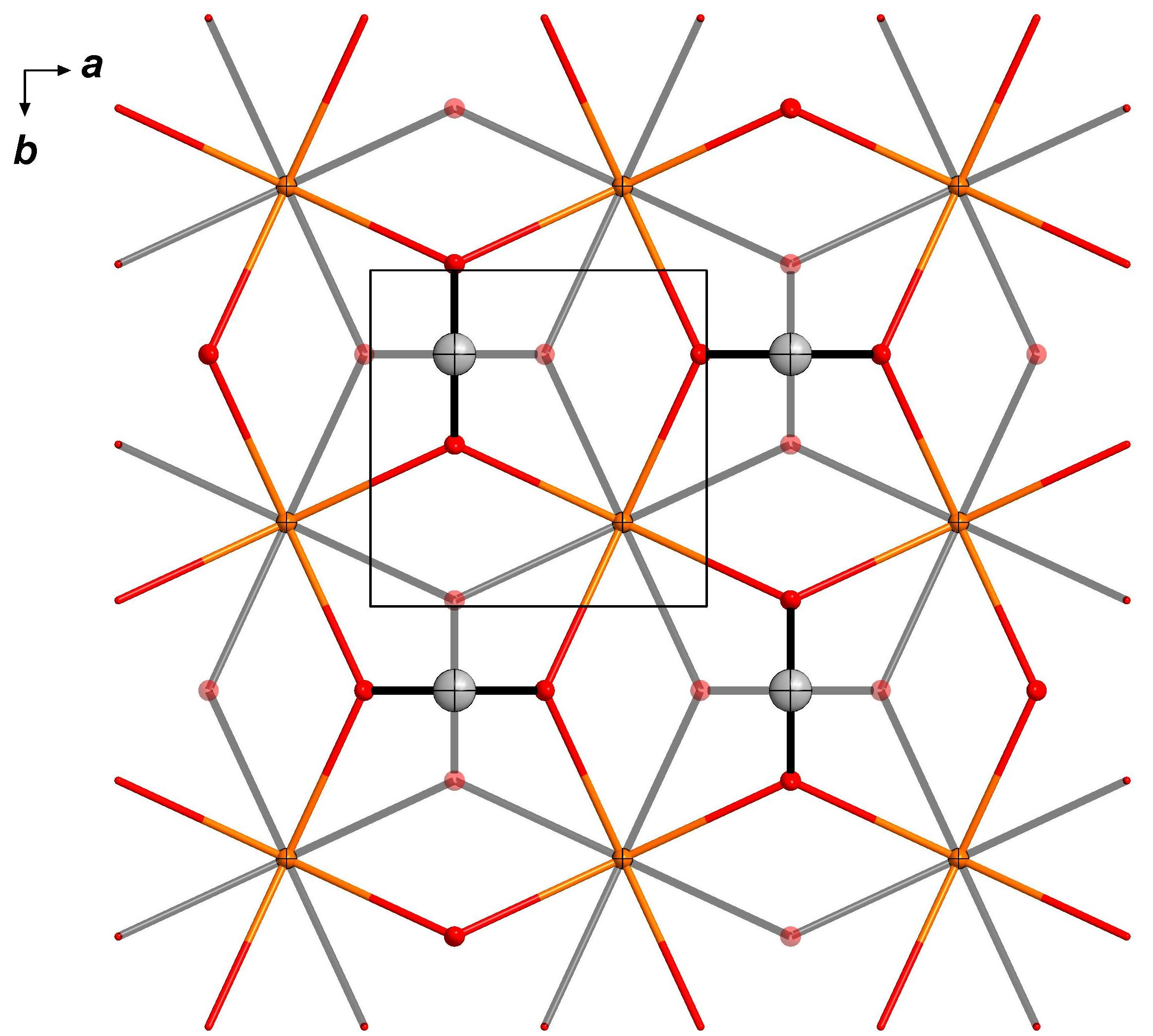
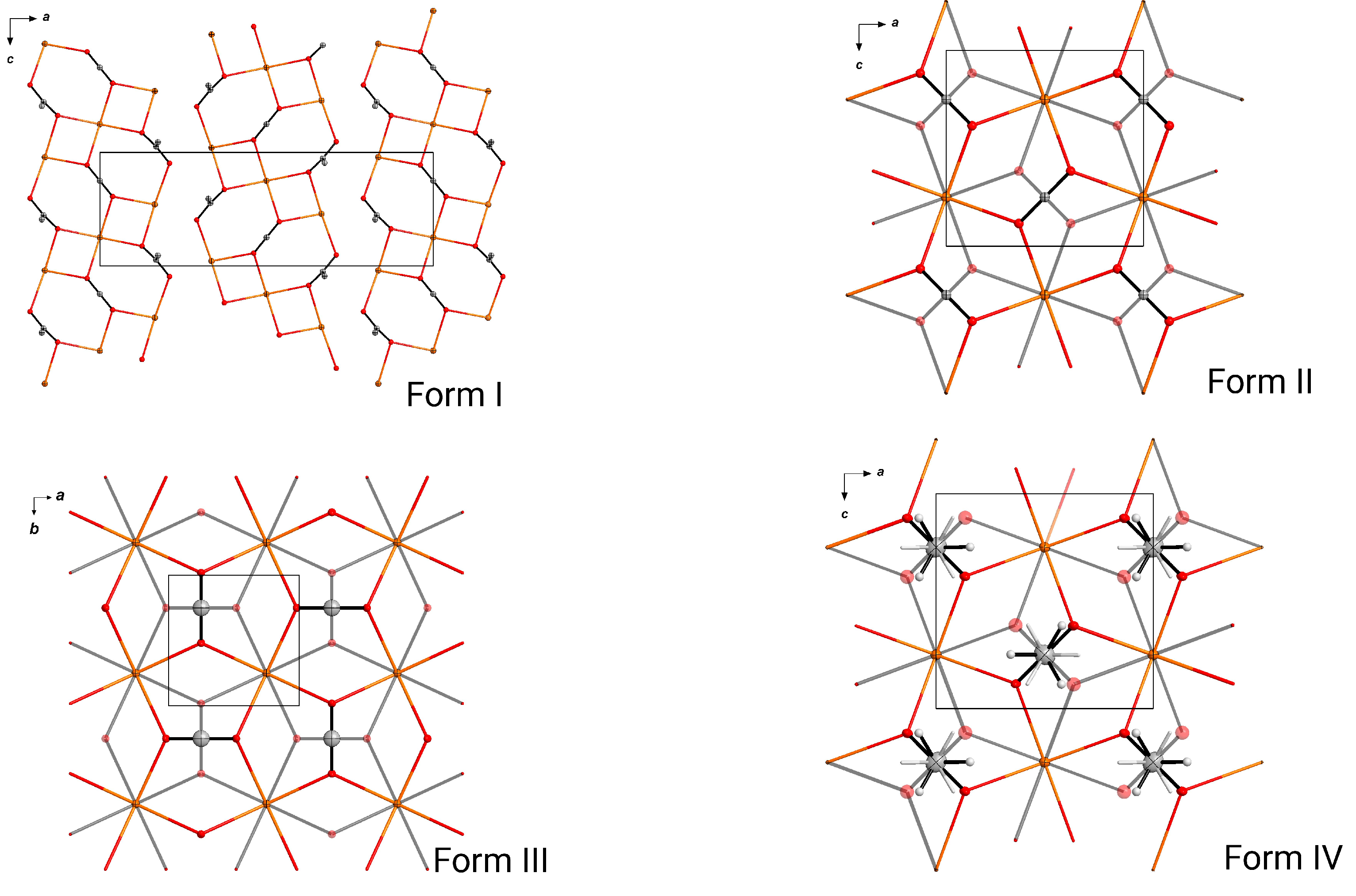
3. Results
3.1. Crystallization and Space Group Changes
3.2. Figures of Merit and Hirshfeld Test
3.3. Structural Details
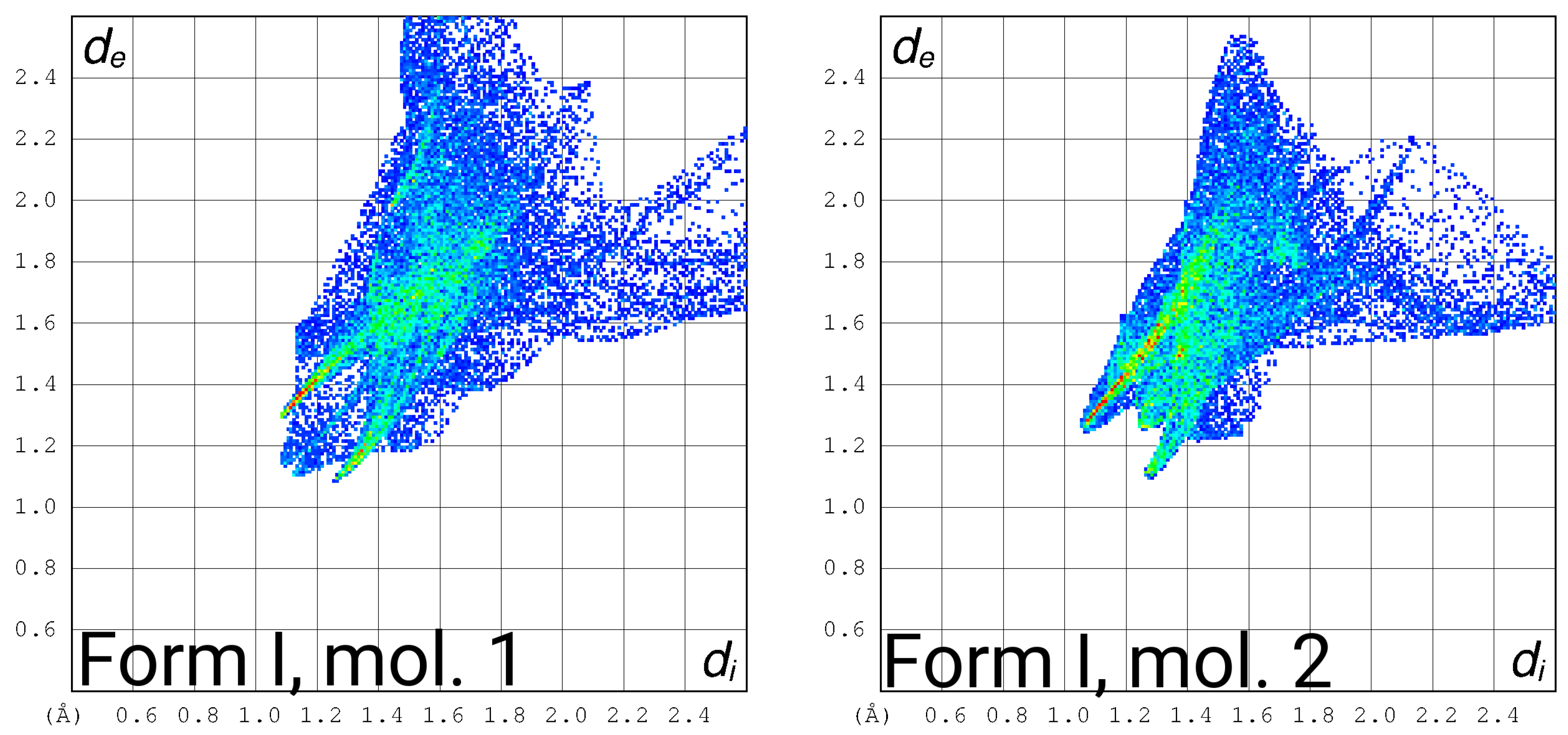
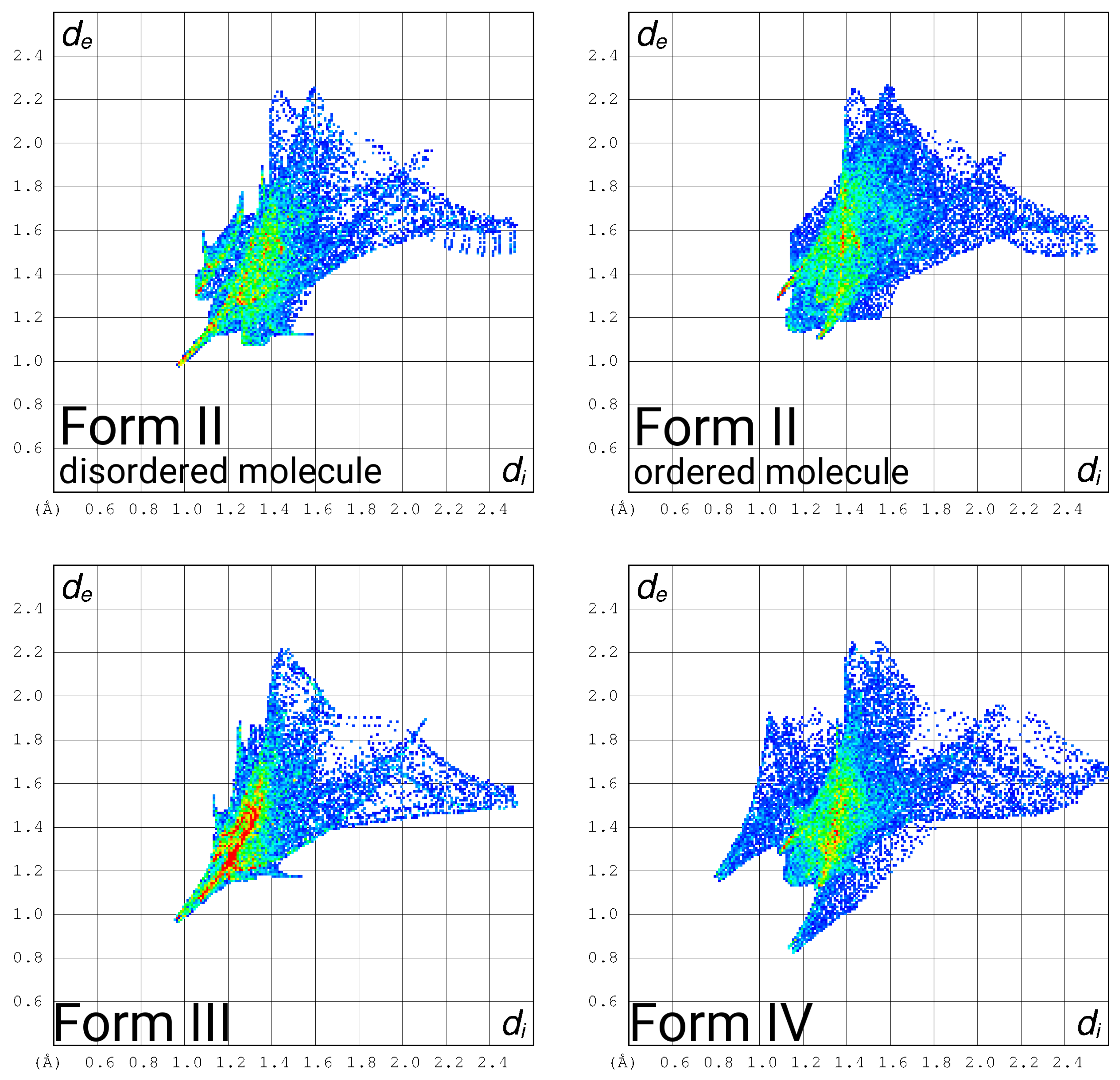
3.4. Hirshfeld Surface Fingerprint Plots
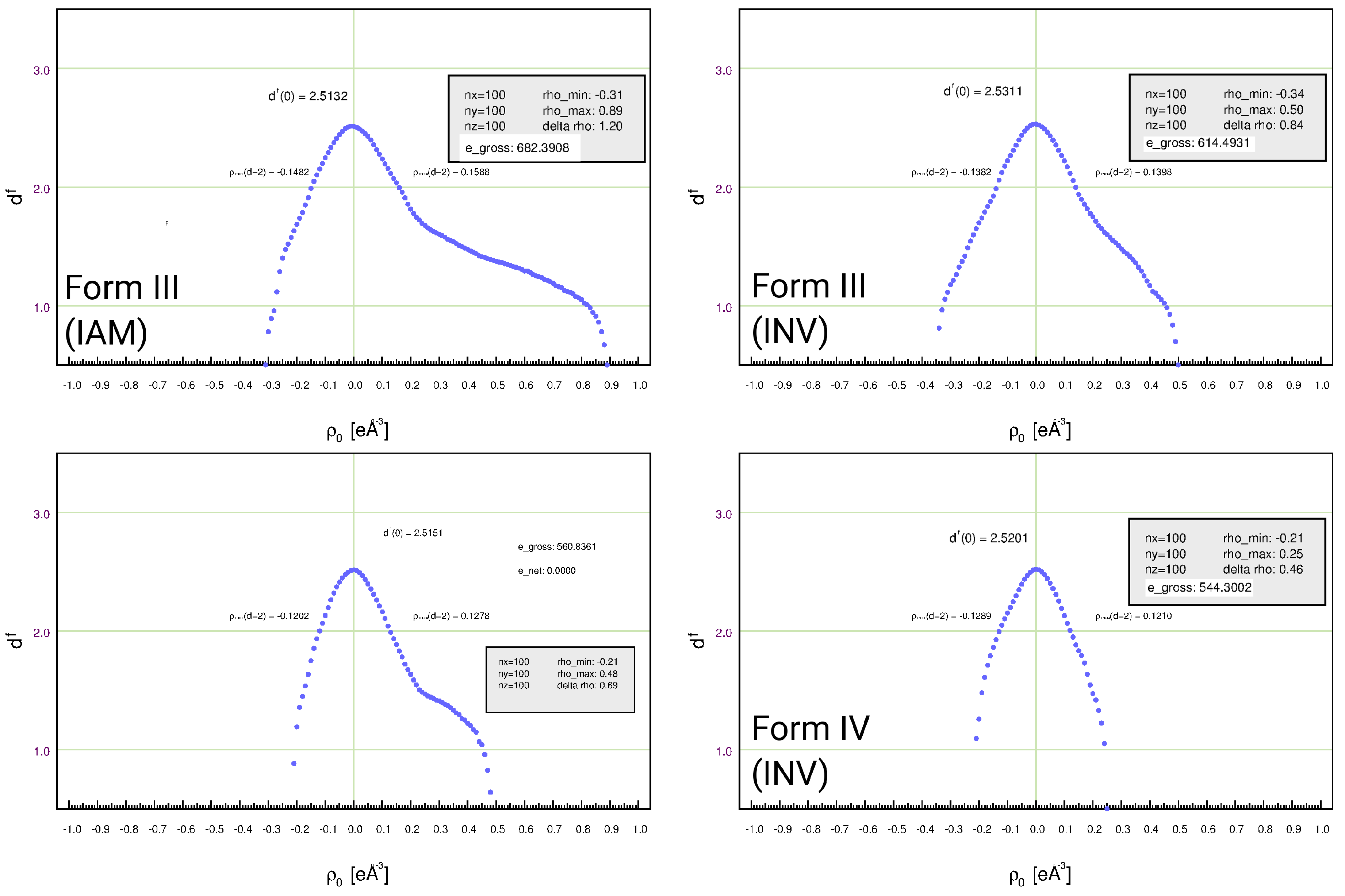
3.5. Residual-Density Analysis
4. Discussion
Speculations on the Causes of Supersaturation
5. Conclusions and Outlook
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zalba, B.; Marín, J.; Cabeza, L.F.; Mehling, H. Review on thermal energy storage with phase change: Materials, heat transfer analysis and applications. Appl. Therm. Eng. 2003, 23, 251–283. [Google Scholar] [CrossRef]
- Agyenim, F.; Hewitt, N.; Eames, P.; Smyth, M. A review of materials, heat transfer and phase change problem formulation for latent heat thermal energy storage systems (LHTESS). Renew. Sustain. Energy Rev. 2010, 14, 615–628. [Google Scholar] [CrossRef]
- Mohamed, S.A.; Al-Sulaiman, F.A.; Ibrahim, N.I.; Zahir, H.; Al-Ahmed, A.; Saidur, R.; Yilbas, B.S.; Sahin, A.Z. A review on current status and challenges of inorganic phase change materials for thermal energy storage systems. Renew. Sustain. Energy Rev. 2017, 70, 1072–1089. [Google Scholar] [CrossRef]
- Kumar, R.; Vyas, S.; Kumar, R.; Dixit, A. Development of sodium acetate trihydrate-ethylene glycol composite phase change materials with enhanced thermophysical properties for thermal comfort and therapeutic applications. Sci. Rep. 2017, 7, 5203–5214. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.-Y.; Nordman, C.E. Structures of two forms of sodium acetate, Na+·C2H3O2−. Acta Cryst. C 1983, 39, 690–694. [Google Scholar] [CrossRef]
- Cameron, T.S.; Mannan, K.M.; Obaidur Rahman, M. The crystal structure of sodium acetate trihydrate. Acta Cryst. B 1976, 32, 87–90. [Google Scholar] [CrossRef]
- Wei, K.-T.; Ward, D.L. Sodium acetate trihydrate: A redetermination. Acta Cryst. B 1977, 33, 522–526. [Google Scholar] [CrossRef]
- Dittrich, B.; Hübschle, C.B.; Pröpper, K.; Dietrich, F.; Stolper, T.; Holstein, J.J. The generalized invariom database (GID). Acta Cryst. B 2013, 69, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Coquerel, G. Crystallization of molecular systems from solution: Phase diagrams, supersaturation and other basic concepts. Chem. Soc. Rev. 2014, 43, 2286–2300. [Google Scholar] [CrossRef] [PubMed]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Blessing, R.H. An empirical correction for absorption anisotrophy. Acta Cryst. A 1995, 51, 33–38. [Google Scholar] [CrossRef]
- Macchi, P.; Bürgi, H.-B.; Chimpri, A.S.; Hauser, J.; Gál, Z. Low-energy contamination of Mo microsource X-ray radiation: Analysis and solution of the problem. J. Appl. Cryst. 2011, 44, 763–771. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Shelxt-integrated space-group and crystal structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with shelxl. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Volkov, A.; Macchi, P.; Farrugia, L.J.; Gatti, C.; Mallinson, P.; Richter, T.; Koritsánszky, T. XD2006—A Computer Program Package for Multipole Refinement, Topological Analysis of Charge Densities and Evaluation of Intermolecular Energies from Experimental or Theoretical Structure Factors; University at Buffalo, State University of New York: New York, NY, USA, 2006. [Google Scholar]
- Dittrich, B.; Munshi, P.; Spackman, M.A. Re-determination and invariom model refinement of l-ornithine hydrochloride. Acta Cryst. B 2007, 63, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Nelyubina, Y.V.; Lyssenko, K.A. Probing ionic crystals by the invariom approach: An electron density study of guanidinium chloride and carbonate. Chem. Eur. J. 2015, 21, 9733–9741. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.F. Electron population analysis with rigid pseudoatoms. Acta Cryst. A 1976, 32, 565–574. [Google Scholar] [CrossRef]
- Hansen, N.K.; Coppens, P. Testing aspherical atom refinements on small-molecule data sets. Acta Cryst. A 1978, 34, 909–921. [Google Scholar] [CrossRef]
- Dittrich, B.; Schürmann, C.; Hübschle, C.B. Invariom modeling of disordered structures: Case studies on a dipeptide, an amino acid, and cefaclor, a cephalosporin antibiotic. Z. Kristallogr. 2016, 231, 725–736. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Luger, P.; Dittrich, B. Automation of invariom and of experimental charge density modelling of organic molecules with the preprocessor program invariomtool. J. Appl. Cryst. 2007, 40, 623–627. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Can X-ray data distinguish bonding effects from vibrational smearing? Acta Cryst. A 1976, 32, 239–244. [Google Scholar] [CrossRef]
- Brandenburg, K.; Berndt, M. DIAMOND—Visual Crystal Structure Information System. J. Appl. Cryst. 1999, 32, 1028–1029. [Google Scholar] [CrossRef]
- Hahn, T. (Ed.) International Tables for X-ray Crystallography Volume A: Space Group Symmetry, 5th ed.; Springer: Dordrecht, The Netherlands, 2005. [Google Scholar]
- McKinnon, J.J.; Mitchell, A.S.; Spackman, M.A. Hirshfeld surfaces: A new tool for visualising and exploring molecular crystals. Chem. Eur. J. 1998, 4, 2136–2141. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Meindl, K.; Henn, J. Foundations of residual-density analysis. Acta Cryst. A 2008, 64, 404–418. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | ||||
| Chemical formula | CHNaO | |||
| 82.03 | ||||
| Crystal system | orthorhombic | orthorhombic | tetragonal | orthorhombic |
| space group | (more details in [5]) | |||
| Temperature (K) | 298 K | 100 K | ||
| a (Å) | 17.850 (15) | 5.951 (2) | 4.1560 (15) | 5.9136 (5) |
| b (Å) | 9.982 (7) | 20.213 (7) | 4.1560 (15) | 10.0457 (8) |
| c (Å) | 6.068 (4) | 5.902 (3) | 10.053 (7) | 5.8572 (4) |
| V (Å) | 1081.2 (14) | 709.9 (5) | 173.64 (17) | 347.96 (4) |
| Z | 12 | 8 | 2 | 4 |
| Radiation type | Mo K | |||
| wavelength Å | 0.7107 | |||
| (mm) | 0.247 | 0.237 | ||
| Crystal size (mm) | 0.6 × 0.3 × 0.05 | several crystals | 0.12 × 0.45 × 0.54 | |
| Data Collection | ||||
| Diffractometer | n. a. | Huber Type 512 with APEX I | ||
| Absorption correction | n. a. | Empirical [10] | ||
| T, T | n. a. | 0.5594, 0.6507 | 0.5303, 0.6499 | |
| No. of meas. refl. | 1426 | 1046 | 15,026 | 93,856 |
| No. of indep. refl. | n. a. | 342 | 840 | |
| No of obs. [] refl. | 556 | 427 | 306 | 703 |
| R | 0.046 | 0.042 | ||
| ( (Å) | 0.88 | 0.83 | ||
| Refinement | ||||
| H-atom treatment | H-atoms not present in CIF | -parameters constr. and refined | ||
| No. of reflections | 556 | 427 | 306 | 703 |
| IAM | ||||
| , , S | 0.0610, n. a., n. a. | 0.0740, n. a., n. a. | 0.0322, 0.0887, 2.0288 | 0.0304, 0.0849, 1.6601 |
| , (e Å) | n. a. | 0.867, −0.307 | 0.453, −0.198 | |
| Invariom | ||||
| , , S | n. a. | 0.0270, 0.0777, 1.7752 | 0.287, 0.0811, 1.5854 | |
| , (e Å) | n. a. | 0.482,−0.340 | 0.227, −0.193 | |
| Atom Name | Invariom Assigned | Local-Atomic Site Symmetry | Model Compound |
|---|---|---|---|
| Na | Na | none | Sodium cation |
| O(3) | O1.5c[1.5o1c] | m | acetic acid anion |
| C(1) | C1.5o1.5o1c | m | acetic acid anion |
| C(2) | C1c1h1h1h | ethane | |
| H(2A,B,C) | H1c[1c1h1h] | 6 | ethane |
| Form I | Form II | Form III | Form IV | |||
|---|---|---|---|---|---|---|
| IAM | IAM | Invariom | IAM | Invariom | IAM | |
| R(F) | 6.10% | 7.40% | 2.70% | 3.22% | 2.87% | 3.04% |
| wR(F) | — | — | 4.35% | 4.91% | 4.26% | 4.49% |
| S | — | — | 1.78 | 2.03 | 1.59 | 1.66 |
| No. of parameters | — | — | 17 | 31 | ||
| H-atom positions | not present | constrained to parent site | ||||
| H-atom displacements | — | refined | ||||
| Average DMSDA | — | — | 11.5 × 10 Å | 13.5 × 10 Å | 4.5 × 10 Å | 9.5 × 10 Å |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dittrich, B.; Bergmann, J.; Roloff, P.; Reiss, G.J. New Polymorphs of the Phase-Change Material Sodium Acetate. Crystals 2018, 8, 213. https://doi.org/10.3390/cryst8050213
Dittrich B, Bergmann J, Roloff P, Reiss GJ. New Polymorphs of the Phase-Change Material Sodium Acetate. Crystals. 2018; 8(5):213. https://doi.org/10.3390/cryst8050213
Chicago/Turabian StyleDittrich, Birger, Justin Bergmann, Peter Roloff, and Guido J. Reiss. 2018. "New Polymorphs of the Phase-Change Material Sodium Acetate" Crystals 8, no. 5: 213. https://doi.org/10.3390/cryst8050213
APA StyleDittrich, B., Bergmann, J., Roloff, P., & Reiss, G. J. (2018). New Polymorphs of the Phase-Change Material Sodium Acetate. Crystals, 8(5), 213. https://doi.org/10.3390/cryst8050213