TMP-SSurface: A Deep Learning-Based Predictor for Surface Accessibility of Transmembrane Protein Residues

 ,
,
Abstract
1. Introduction
2. Results and Discussion
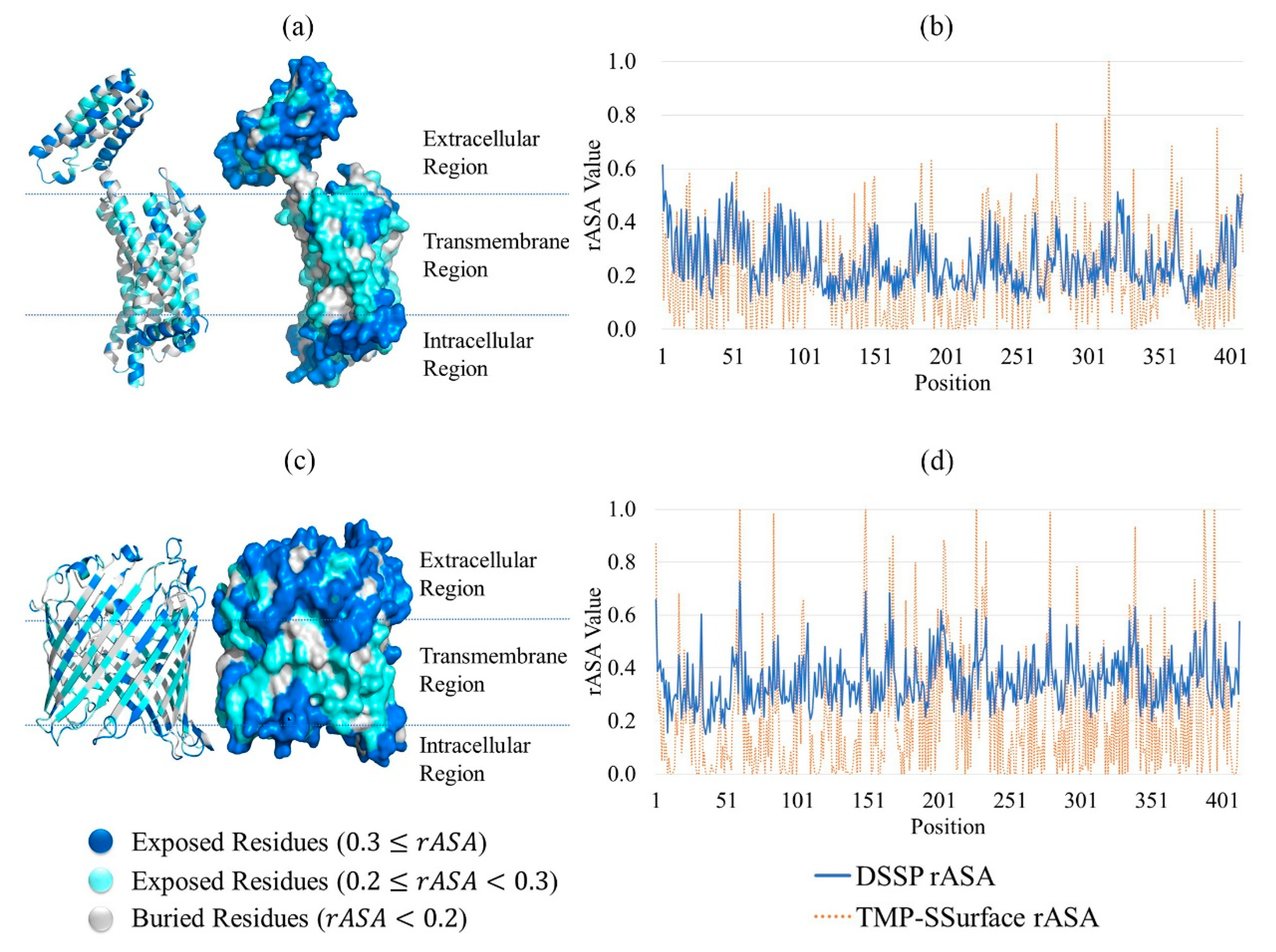
2.1. Feature Analysis
2.2. Effect of Window Size
2.3. Hyper-Parameter Tuning
2.4. Ablation Study
2.5. Comparison with Previous Predictors
2.6. Short Sequence Test
2.7. TMP Type Test
2.8. Case Study
3. Materials and Methods
3.1. Benchmark Datasets
3.2. Calculation of rASA
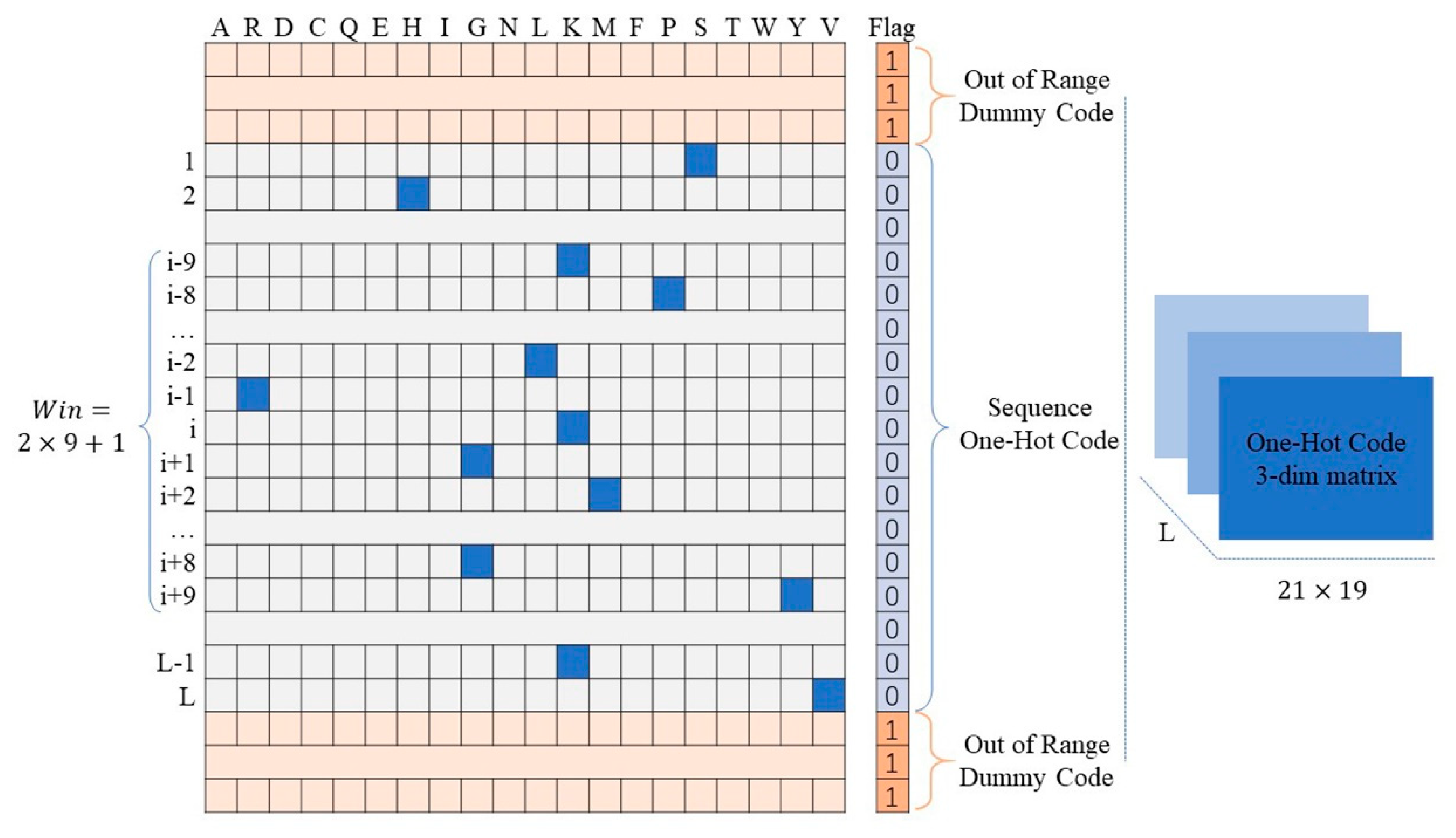
3.3. Encoding of Protein Fragments
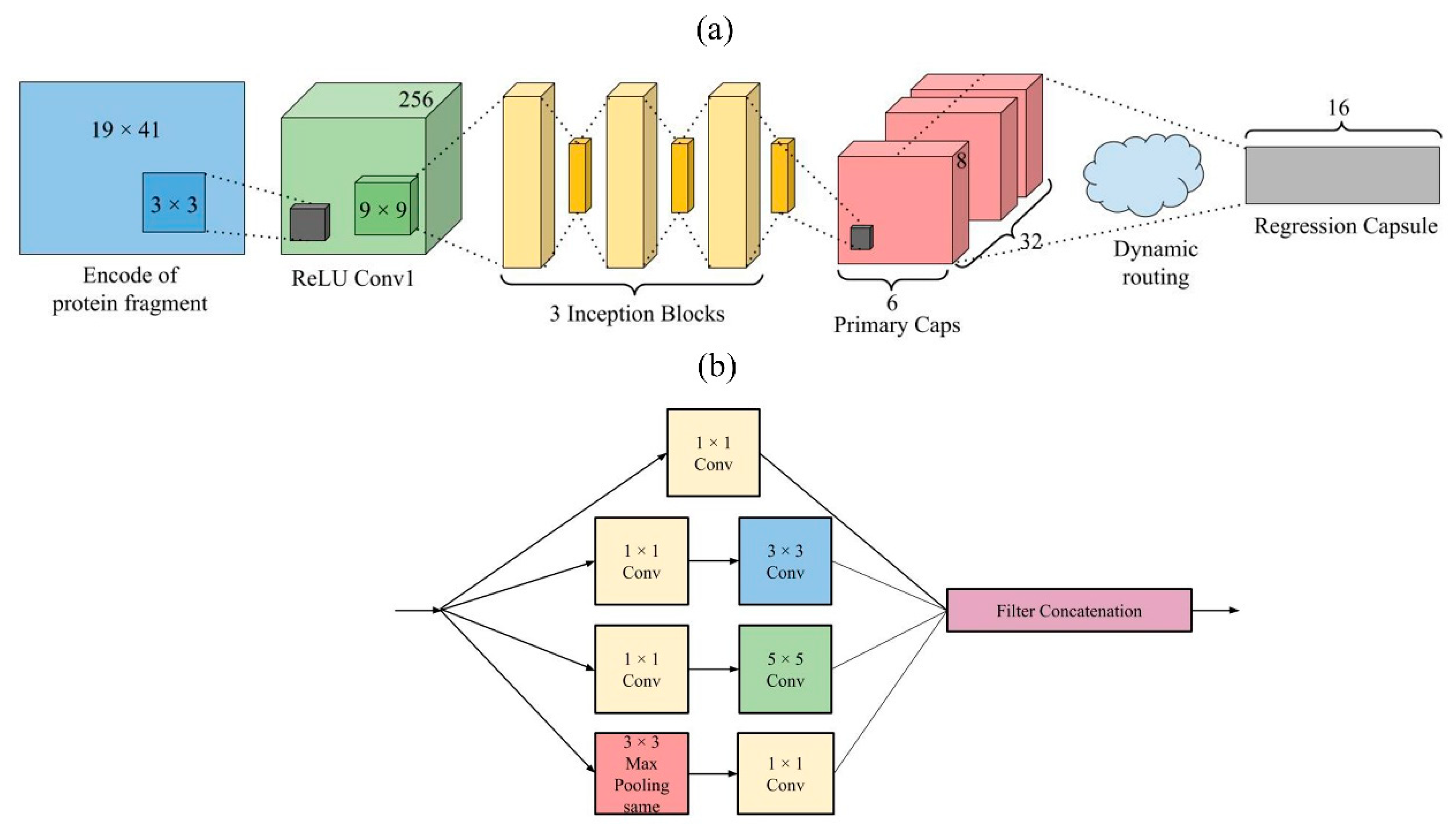
3.4. Model Design
3.5. From Capsule Length to rASA
3.6. Performance Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Puder, S.; Fischer, T.; Mierke, C.T. The transmembrane protein fibrocystin/polyductin regulates cell mechanics and cell motility. Phys. Biol. 2019, 16, 066006. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Cohen, E.B.; Edwards, A.P.B.; Xavier-Ferrucio, J.; Bugge, K.; Federman, R.S.; Absher, D.; Myers, R.M.; Kragelund, B.B.; Krause, D.S.; et al. Transmembrane Protein Aptamer Induces Cooperative Signaling by the EPO Receptor and the Cytokine Receptor beta-Common Subunit. Iscience 2019, 17, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Oguro, A.; Imaoka, S. Thioredoxin-related transmembrane protein 2 (TMX2) regulates the Ran protein gradient and importin-beta-dependent nuclear cargo transport. Sci. Rep. 2019, 9, 15296. [Google Scholar] [CrossRef] [PubMed]
- Rafi, S.K.; Fernandez-Jaen, A.; Alvarez, S.; Nadeau, O.W.; Butler, M.G. High Functioning Autism with Missense Mutations in Synaptotagmin-Like Protein 4 (SYTL4) and Transmembrane Protein 187 (TMEM187) Genes: SYTL4- Protein Modeling, Protein-Protein Interaction, Expression Profiling and MicroRNA Studies. Int. J. Mol. Sci. 2019, 20, 3358. [Google Scholar] [CrossRef] [PubMed]
- Weihong, C.; Bin, C.; Jianfeng, Y. Transmembrane protein 126B protects against high fat diet (HFD)-induced renal injury by suppressing dyslipidemia via inhibition of ROS. Biochem. Biophys. Res. Commun. 2019, 509, 40–47. [Google Scholar] [CrossRef]
- Tanabe, Y.; Taira, T.; Shimotake, A.; Inoue, T.; Awaya, T.; Kato, T.; Kuzuya, A.; Ikeda, A.; Takahashi, R. An adult female with proline-rich transmembrane protein 2 related paroxysmal disorders manifesting paroxysmal kinesigenic choreoathetosis and epileptic seizures. Rinsho Shinkeigaku 2019, 59, 144–148. [Google Scholar] [CrossRef][Green Version]
- Moon, Y.H.; Lim, W.; Jeong, B.C. Transmembrane protein 64 modulates prostate tumor progression by regulating Wnt3a secretion. Oncol. Lett. 2019, 18, 283–290. [Google Scholar] [CrossRef]
- Tao, D.; Liang, J.; Pan, Y.; Zhou, Y.; Feng, Y.; Zhang, L.; Xu, J.; Wang, H.; He, P.; Yao, J.; et al. In Vitro and In Vivo Study on the Effect of Lysosome-associated Protein Transmembrane 4 Beta on the Progression of Breast Cancer. J. Breast Cancer. 2019, 22, 375–386. [Google Scholar] [CrossRef]
- Yan, J.; Jiang, Y.; Lu, J.; Wu, J.; Zhang, M. Inhibiting of Proliferation, Migration, and Invasion in Lung Cancer Induced by Silencing Interferon-Induced Transmembrane Protein 1 (IFITM1). Biomed. Res. Int. 2019, 2019, 9085435. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef]
- Lu, C.; Liu, Z.; Zhang, E.; He, F.; Ma, Z.; Wang, H. MPLs-Pred: Predicting Membrane Protein-Ligand Binding Sites Using Hybrid Sequence-Based Features and Ligand-Specific Models. Int. J. Mol. Sci. 2019, 20, 3120. [Google Scholar] [CrossRef] [PubMed]
- Tarafder, S.; Toukir Ahmed, M.; Iqbal, S.; Tamjidul Hoque, M.; Sohel Rahman, M. RBSURFpred: Modeling protein accessible surface area in real and binary space using regularized and optimized regression. J. Biol. 2018, 441, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chai, H. Recent In Silico Research in High-Throughput Drug Discovery and Molecular Biochemistry. Curr. Top. Med. Chem. 2019, 19, 103–104. [Google Scholar] [CrossRef] [PubMed]
- Beuming, T.; Weinstein, H. A knowledge-based scale for the analysis and prediction of buried and exposed faces of transmembrane domain proteins. Bioinformatics 2004, 20, 1822–1835. [Google Scholar] [CrossRef][Green Version]
- Park, Y.; Hayat, S.; Helms, V. Prediction of the burial status of transmembrane residues of helical membrane proteins. BMC Bioinform. 2007, 8, 302. [Google Scholar] [CrossRef]
- Wang, C.; Li, S.; Xi, L.; Liu, H.; Yao, X. Accurate prediction of the burial status of transmembrane residues of alpha-helix membrane protein by incorporating the structural and physicochemical features. Amino Acids 2011, 40, 991–1002. [Google Scholar] [CrossRef]
- Lai, J.S.; Cheng, C.W.; Lo, A.; Sung, T.Y.; Hsu, W.L. Lipid exposure prediction enhances the inference of rotational angles of transmembrane helices. BMC Bioinform. 2013, 14, 304. [Google Scholar] [CrossRef]
- Yuan, Z.; Zhang, F.; Davis, M.J.; Boden, M.; Teasdale, R.D. Predicting the solvent accessibility of transmembrane residues from protein sequence. J. Proteome Res. 2006, 5, 1063–1070. [Google Scholar] [CrossRef]
- Illergard, K.; Callegari, S.; Elofsson, A. MPRAP: An accessibility predictor for a-helical transmembrane proteins that performs well inside and outside the membrane. BMC Bioinform. 2010, 11, 333. [Google Scholar] [CrossRef]
- Wang, C.; Xi, L.; Li, S.; Liu, H.; Yao, X. A sequence-based computational model for the prediction of the solvent accessible surface area for alpha-helix and beta-barrel transmembrane residues. J. Comput. Chem. 2012, 33, 11–17. [Google Scholar] [CrossRef]
- Xiao, F.; Shen, H.B. Prediction Enhancement of Residue Real-Value Relative Accessible Surface Area in Transmembrane Helical Proteins by Solving the Output Preference Problem of Machine Learning-Based Predictors. J. Chem. Inf. Model. 2015, 55, 2464–2474. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Yang, J.; Xiao, F.; Yang, Y.; Shen, H.B. MemBrain: An Easy-to-Use Online Webserver for Transmembrane Protein Structure Prediction. Nano Micro Lett. 2018, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Tang, J.; Zou, Q. Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 2017, 384, 135–144. [Google Scholar] [CrossRef]
- Wei, L.; Liao, M.; Gao, X.; Zou, Q. An Improved Protein Structural Classes Prediction Method by Incorporating Both Sequence and Structure Information. IEEE Trans. Nanobiosci. 2015, 14, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.J.; Feng, C.Q.; Lai, H.Y.; Chen, W.; Lin, H. Predicting protein structural classes for low-similarity sequences by evaluating different features. Knowl. Based Syst. 2019, 163, 787–793. [Google Scholar] [CrossRef]
- Yang, W.; Zhu, X.J.; Huang, J.; Ding, H.; Lin, H. A brief survey of machine learning methods in protein sub-Golgi localization. Curr. Bioinform. 2019, 14, 234–240. [Google Scholar] [CrossRef]
- Tan, J.X.; Li, S.H.; Zhang, Z.M.; Chen, C.X.; Chen, W.; Tang, H.; Lin, H. Identification of hormone binding proteins based on machine learning methods. Math. Biosci. Eng. 2019, 16, 2466–2480. [Google Scholar] [CrossRef]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene Subsequence Embedding for Prediction of Mammalian N6-Methyladenosine Sites from mRNA. RNA 2019, 25, 205–218. [Google Scholar] [CrossRef]
- Lv, Z.B.; Ao, C.Y.; Zou, Q. Protein Function Prediction: From Traditional Classifier to Deep Learning. Proteomics 2019, 19, 2. [Google Scholar] [CrossRef]
- Peng, L.; Peng, M.M.; Liao, B.; Huang, G.H.; Li, W.B.; Xie, D.F. The Advances and Challenges of Deep Learning Application in Biological Big Data Processing. Curr. Bioinform. 2018, 13, 352–359. [Google Scholar] [CrossRef]
- Fang, C.; Shang, Y.; Xu, D. Improving Protein Gamma-Turn Prediction Using Inception Capsule Networks. Sci. Rep. 2018, 8, 15741. [Google Scholar] [CrossRef] [PubMed]
- Kozma, D.; Simon, I.; Tusnady, G.E. PDBTM: Protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res. 2013, 41, D524–D529. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of delta-opioid receptor signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Mihel, J.; Sikic, M.; Tomic, S.; Jeren, B.; Vlahovicek, K. PSAIA-protein structure and interaction analyzer. BMC Struct. Biol. 2008, 8, 21. [Google Scholar] [CrossRef]
- Lee, B.; Richards, F.M. The interpretation of protein structures: Estimation of static accessibility. J Mol Biol 1971, 55, 379–400. [Google Scholar] [CrossRef]
- Tien, M.Z.; Meyer, A.G.; Sydykova, D.K.; Spielman, S.J.; Wilke, C.O. Maximum allowed solvent accessibilites of residues in proteins. PLoS ONE 2013, 8, e80635. [Google Scholar] [CrossRef]
- Rose, G.D.; Geselowitz, A.R.; Lesser, G.J.; Lee, R.H.; Zehfus, M.H. Hydrophobicity of amino acid residues in globular proteins. Science 1985, 229, 834–838. [Google Scholar] [CrossRef]
- Miller, S.; Janin, J.; Lesk, A.M.; Chothia, C. Interior and surface of monomeric proteins. J. Mol. Biol. 1987, 196, 641–656. [Google Scholar] [CrossRef]
- Sun, P.; Ju, H.; Liu, Z.; Ning, Q.; Zhang, J.; Zhao, X.; Huang, Y.; Ma, Z.; Li, Y. Bioinformatics resources and tools for conformational B-cell epitope prediction. Comput. Math. Methods Med. 2013, 2013, 943636. [Google Scholar] [CrossRef]
- He, F.; Wang, R.; Li, J.; Bao, L.; Xu, D.; Zhao, X. Large-scale prediction of protein ubiquitination sites using a multimodal deep architecture. BMC Syst. Biol. 2018, 12, 109. [Google Scholar] [CrossRef]
- Ding, H.; Li, D. Identification of mitochondrial proteins of malaria parasite using analysis of variance. Amino Acids 2015, 47, 329–333. [Google Scholar] [CrossRef]
- Ding, H.; Deng, E.Z.; Yuan, L.F.; Liu, L.; Lin, H.; Chen, W.; Chou, K.C. iCTX-type: A sequence-based predictor for identifying the types of conotoxins in targeting ion channels. Biomed. Res. Int. 2014, 2014, 286419. [Google Scholar] [CrossRef]
- Jeong, J.C.; Lin, X.; Chen, X.W. On position-specific scoring matrix for protein function prediction. IEEE ACM Trans. Comput. Biol. Bioinform. 2011, 8, 308–315. [Google Scholar] [CrossRef]
- Zeng, B.; Honigschmid, P.; Frishman, D. Residue co-evolution helps predict interaction sites in alpha-helical membrane proteins. J. Struct. Biol. 2019, 206, 156–169. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Ma, Z. In silico Prediction of Human Secretory Proteins in Plasma Based on Discrete Firefly Optimization and Application to Cancer Biomarkers Identification. Front. Genet. 2019, 10, 542. [Google Scholar] [CrossRef]
- Zangooei, M.H.; Jalili, S. Protein secondary structure prediction using DWKF based on SVR-NSGAII. Neurocomputing 2012, 94, 87–101. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Sabour, S.; Frosst, N.; Hinton, G.E. Dynamic Routing Between Capsules. Adv. Neural Inf. Process. Syst. 2017, 30, 3856–3866. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Method | Year | Samples | Algorithm | TMP Type | Seq Region | Measure |
|---|---|---|---|---|---|---|
| ProperTM [14] | 2004 | 59 | knowledge | α-TMP | TM region | Burial state |
| ASAP [18] | 2006 | 73 | SVR | all TMP | TM region | ASA |
| TMX [15] | 2007 | 43 | SVC | α-TMP | TM region | Burial state |
| MPRAP [19] | 2010 | 80 | SVR | α-TMP | full sequence | rASA |
| Yao et al. (2011) [16] | 2011 | 53 | SVM | α-TMP | TM region | Burial state |
| Yao et al. (2012) [20] | 2012 | 122 | RF | all TMP | TM region | ASA |
| TMexpoSVR [17] | 2013 | 110 | SVR | α-TMP | TM region | rASA |
| TMexpoSVC [17] | 2013 | 110 | SVC | α-TMP | TM region | Burial state |
| MenBrain-Rasa [21,22] | 2015 | 80 | SVR | α-TMP | full sequence | rASA |
| Feature | CC | MAE |
|---|---|---|
| One-hot | 0.417 | 0.203 |
| PSSM | 0.387 | 0.206 |
| One-hot + PSSM | 0.577 | 0.158 |
| Window Size | CC |
|---|---|
| 13 | 0.534 |
| 15 | 0.551 |
| 17 | 0.576 |
| 19 | 0.581 |
| 21 | 0.578 |
| 23 | 0.565 |
| Num of Inception Blocks | No. of Parameters | CC | MAE |
|---|---|---|---|
| 1 | 3,790,671 | 0.506 | 0.203 |
| 2 | 6,617,295 | 0.537 | 0.170 |
| 3 | 12,614,607 | 0.579 | 0.157 |
| 4 | 25,798,927 | 0.568 | 0.164 |
| 5 | 58,045,711 | 0.577 | 0.158 |
| Num of Dynamic Routings | CC | MAE |
|---|---|---|
| 1 | 0.558 | 0.167 |
| 2 | 0.568 | 0.164 |
| 3 | 0.577 | 0.158 |
| 4 | 0.573 | 0.160 |
| 5 | 0.575 | 0.160 |
| 6 | 0.569 | 0.164 |
| Model | CC | MAE |
|---|---|---|
| CNN | 0.163 | 0.191 |
| Inception | 0.415 | 0.167 |
| CapsuleNet | 0.503 | 0.151 |
| Without inception | 0.504 | 0.150 |
| Without CapsuleNet | 0.422 | 0.166 |
| TMP-SSurface | 0.584 | 0.144 |
| Predictor | CC | MAE | Failure | Time Cost (min) |
|---|---|---|---|---|
| MPRAP | 0.397 | 0.176 | 9 | 6.5 |
| MemBrain-Rasa | 0.545 | 0.153 | 7 | 23.7 |
| TMP-Ssurface | 0.584 | 0.144 | 0 | 4.7 |
| Sequenc Length | Sequence Number | CC | MAE |
|---|---|---|---|
| Less than 30 | 89 | 0.533 | 0.224 |
| Testing dataset (30–5000) | 50 | 0.584 | 0.144 |
| TMP Types | Protein Number | CC | MAE |
|---|---|---|---|
| α-helical TMPs | 45 | 0.597 | 0.139 |
| β-barrel TMPs | 5 | 0.511 | 0.151 |
| all-TMP | 50 | 0.584 | 0.144 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.; Liu, Z.; Kan, B.; Gong, Y.; Ma, Z.; Wang, H. TMP-SSurface: A Deep Learning-Based Predictor for Surface Accessibility of Transmembrane Protein Residues. Crystals 2019, 9, 640. https://doi.org/10.3390/cryst9120640
Lu C, Liu Z, Kan B, Gong Y, Ma Z, Wang H. TMP-SSurface: A Deep Learning-Based Predictor for Surface Accessibility of Transmembrane Protein Residues. Crystals. 2019; 9(12):640. https://doi.org/10.3390/cryst9120640
Chicago/Turabian StyleLu, Chang, Zhe Liu, Bowen Kan, Yingli Gong, Zhiqiang Ma, and Han Wang. 2019. "TMP-SSurface: A Deep Learning-Based Predictor for Surface Accessibility of Transmembrane Protein Residues" Crystals 9, no. 12: 640. https://doi.org/10.3390/cryst9120640
APA StyleLu, C., Liu, Z., Kan, B., Gong, Y., Ma, Z., & Wang, H. (2019). TMP-SSurface: A Deep Learning-Based Predictor for Surface Accessibility of Transmembrane Protein Residues. Crystals, 9(12), 640. https://doi.org/10.3390/cryst9120640