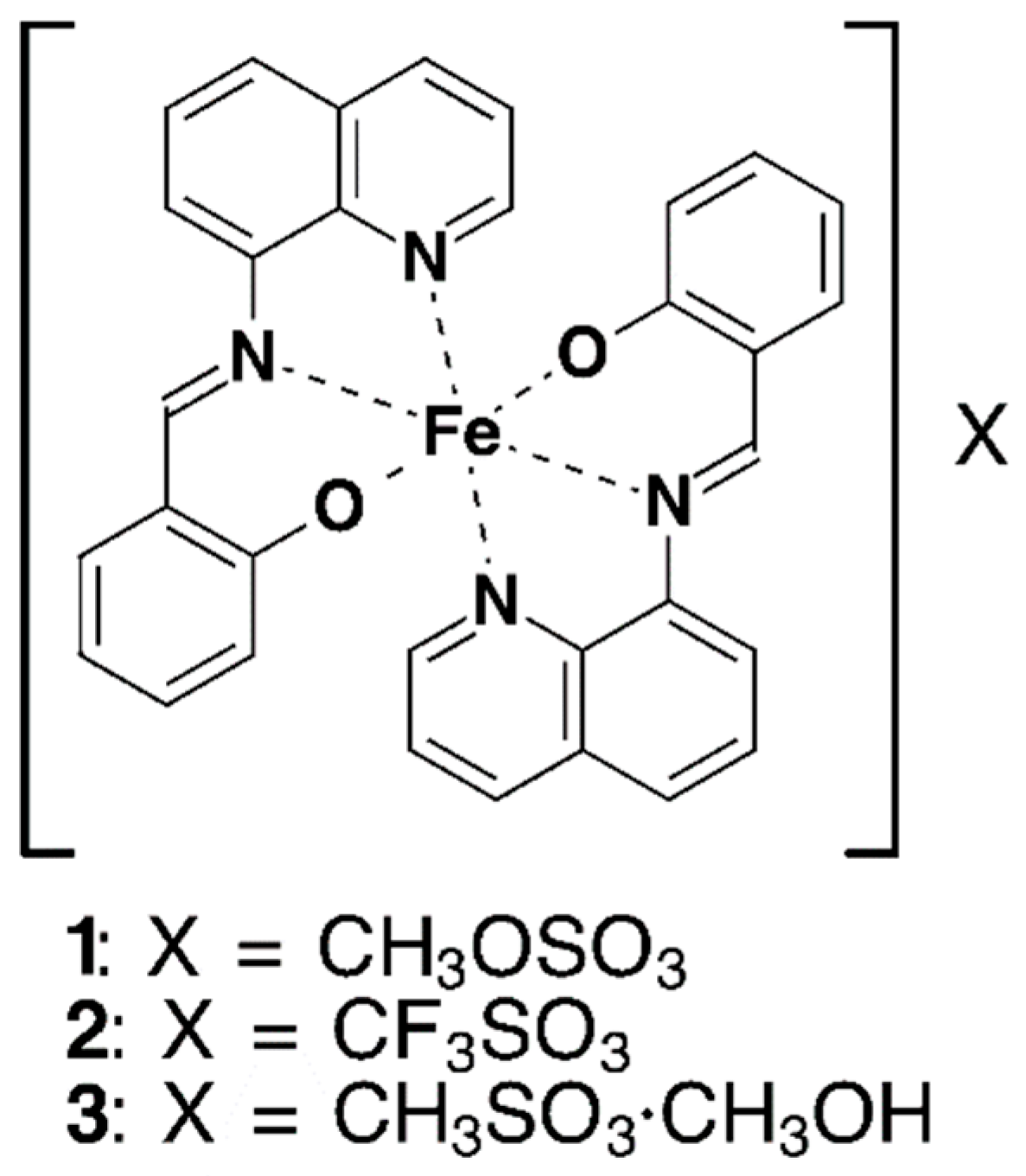
Abstract
New Fe(III) compounds from qsal ligand, [Fe(qsal)2](CH3OSO3) (1) and [Fe(qsal)2](CH3SO3)·CH3OH (3), along with known compound, [Fe(qsal)2](CF3SO3) (2), were obtained as large well-shaped crystals (Hqsal = N-(8-quinolyl)salicylaldimine). The compounds 1 and 2 were in the low-spin (LS) state at 300 K and exhibited a cooperative spin crossover (SCO) transition with a thermal hysteresis loop at higher temperatures, whereas 3 was in the high-spin (HS) state below 300 K. The optical conductivity spectra for 1 and 3 were calculated from the single-crystal reflection spectra, which were, to the best of our knowledge, the first optical conductivity spectra of SCO compounds. The absorption bands for the LS and HS [Fe(qsal)2] cations were assigned by time-dependent density functional theory calculations. The crystal structures of 1 and 2 consisted of a common one-dimensional (1D) array of the [Fe(qsal)2] cation, whereas that of 3 had an unusual 1D arrangement by π-stacking interactions which has never been reported. The crystal structures in the high-temperature phases for 1 and 2 indicate that large structural changes were triggered by the motion of counter anions. The comparison of the crystal structures of the known [Fe(qsal)2] compounds suggests the significant role of a large non-spherical counter-anion or solvate molecule for the total lattice energy gain in the crystal of a charged complex.
1. Introduction
Spin crossover (SCO) between a high-spin (HS) and low-spin (LS) state in a transition metal coordination compound is one of the molecular switching phenomena responsive to various external stimuli such as temperature, pressure, light, magnetic field, and chemicals. Significant attention has been attracted to SCO phenomena in the wide range of fields of chemical and physical sciences [1,2,3,4,5]. The SCO switches not only a spin-state but also color and coordination structure in a transition metal complex. Thus, the utilization of electronic and structural transformation accompanying SCO can lead to potential applications of display, memory, sensing, electronic, and mechanical devices.
A family of coordination compounds from qsal (Hqsal = N-(8-quinoyl)salicylaldimine), which is a π-extended tridentate Schiff base ligand, is known as one of well-studied SCO compounds. Dahl et al. first reported the synthesis, magnetic, and spectral properties of the qsal ligand and its transition metal coordination compounds in 1969 [6]. Dickinson et al. reported on the anomalous magnetic conversion of [Fe(qsal)2](NCS) in 1977 [7]. Successively the [Fe(qsal)2] derivatives with various counter-anions and solvate-molecules have been studied so far [8,9,10,11,12,13,14,15,16], leading to the appearance of a cooperative SCO transition with a very wide thermal hysteresis in [Fe(qsal)2](NCSe) [9] and giving a rare example of Fe(III) compounds exhibiting the light-induced excited spin trapping (LIESST) effect [11]. Moreover, substitution effects on the qsal ligand have been reported in recent years [17,18,19,20,21]. We focused on the [Fe(qsal)2] compounds showing a cooperative SCO transition probably due to strong π-stacking interactions and developed SCO conductors and magnets by combining the [Fe(qsal)2] cation with redox active functional anions [22,23]. A number of multifunctional materials from the [Fe(qsal)2] cation have been reported to date [24,25,26,27,28,29,30,31,32,33]. Recently, heteroleptic Fe(III) compounds [34,35,36,37] as well as Fe(II) compounds [38,39,40,41,42,43,44] from the qsal and its derivatives, were also reported.
As described above, the SCO compounds from qsal have attracted great deal attention, and some systems have been reported to undergo SCO phase transitions in a critical manner. However, the fundamental mechanism, namely how the spin-state changes in the individual iron sites interact with each other, has been virtually unknown. This is partly because it is often difficult to determine both the HS and LS structures due to deterioration in crystal through the SCO transition or the desorption of solvate molecules. In particular, the existence of solvate molecules in an SCO compound affords various undistinguishable effects on its SCO behavior as well as instability of the crystal. Therefore, non-solvate [Fe(qsal)2] compounds seem to be favorable for the elucidation of the SCO mechanism. Meanwhile, structure-characterized non-solvate [Fe(qsal)2] compounds were rare [11,13,15,24,33].
The optical spectra are useful to interpret the photoresponsive property of SCO compounds. Most absorption spectra for SCO compounds have been obtained by using a solution, KBr pellet, oil-dispersed powder, and cast film. Some diffuse reflectance spectra for powdered samples have also been reported. However, the sample preparations for these measurements often vary the spin-state and SCO behavior of the compound measured. To obtain an accurate optical spectrum of an SCO compound whose structure and spin-state is confirmed, a single-crystal optical spectrum may be most suitable. Although a single-crystal absorption spectrum is the most suitable for this purpose, it is not easy to record it because most Fe(III) compounds and LS Fe(II) compounds are intensely colored. A single-crystal reflection spectrum is the second choice. By the Kramers–Kronig analysis, it can be converted to its optical conductivity spectrum corresponding to the absorption spectrum. Meanwhile, the compound measured has a large area of a flat crystal surface without the occurrence of interference in the wide wavelength range from visible to near-infrared. Thus, it is also difficult to obtain a single-crystal reflection spectrum of an SCO compound.
Recently we found a new non-solvate compound [Fe(qsal)2](CH3OSO3) (1) as very large rhombic platelets with a flat crystal surface (Figure 1). As we tried to prepare the related compounds with similar size and shape anions, we obtained relatively large crystals of the known non-solvate compound [Fe(qsal)2](CF3SO3) (2) [12] and a new methanol-solvate compound [Fe(qsal)2](CH3SO3)·CH3OH (3). The compounds 1 and 2 were in the LS state, whereas compound 3 was in the HS state at room temperature. The compounds 1 and 2 showed a cooperative SCO transition with a thermal hysteresis above 350 K. Fortunately, we were successful to determine the crystal structures of the HS phases for 1 and 2 at 425 and 400 K, respectively. The structural comparison among the [Fe(qsal)2] compounds suggests the role of counter-anion or solvate molecule for the crystal of a charged complex. Moreover, single-crystal reflection spectra could be recorded for 1 and 3 in the wide wavelength range. The optical conductivity spectra from the single-crystal reflection spectra are, to the best our knowledge, first examples for SCO compounds. We report herein the preparation, crystal structures, and spectral characterizations for 1–3 and also discuss the mechanism of the cooperative SCO transitions in the [Fe(qsal)2] compounds.
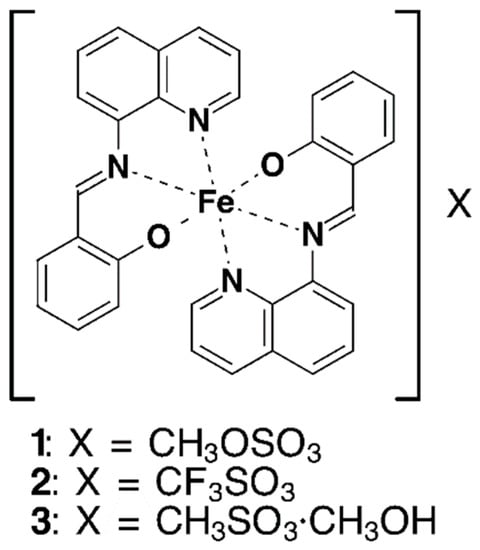
Figure 1.
Molecular structures of compounds 1–3.
2. Materials and Methods
2.1. Synthesis of Compounds 1–3
All chemicals were purchased and used without further purification. [Fe(qsal)2]Cl·1.5H2O was prepared according to the literature [7].
[Fe(qsal)2](CH3OSO3) (1): A saturated methanol solution of [Fe(qsal)2]Cl·1.5H2O was filtered by an Advantec 5B filter paper. Diffusion of an excess amount of (Bu3MeN)(CH3OSO3) to the filtered solution at room temperature gave 1 as large black rhombic platelets. Anal. Calcd. For C33H25N4O6FeS: C, 59.92; H, 3.81; N, 8.47%. Found: C, 59.85; H, 3.95; N, 8.18%.
[Fe(qsal)2](CF3SO3) (2): A saturated methanol solution of [Fe(qsal)2]Cl·1.5H2O was filtered by an Advantec 5B filter paper. Diffusion of an excess amount of Li(CF3SO3) to the filtered solution at room temperature gave 2 as black parallelogrammatic platelets. Anal. Calcd. For C33H22N4O5F3FeS: C, 56.67; H, 3.17; N, 8.01%. Found: C, 56.54; H, 3.21; N, 7.94%.
[Fe(qsal)2](CH3SO3)∙CH3OH (3): A saturated methanol solution of [Fe(qsal)2]Cl·1.5H2O was filtered by an Advantec 5B filter paper. Diffusion of an excess amount of (Bu4N)(CH3SO3) followed by cooling to 3 °C gave 3 as black rhombic platelets. Anal. Calcd. for C34H29N4O6FeS: C, 60.27; H, 4.31; N, 8.27%. Found: C, 60.00; H, 4.46; N, 8.13%.
2.2. Physical Measurements
Variable temperature direct current magnetic susceptibilities of polycrystalline samples were measured on a Quantum Design MPMS-XL magnetometer under a field of 0.5 T in the temperature range of 2−320 K. The oven option was used for the measurement in the temperature range of 300−450 K. The magnetic susceptibilities were corrected for diamagnetic contributions estimated by Pascal constants [45].
The Mössbauer spectra were recorded on a constant acceleration spectrometer with a source of 57Co/Rh in the transmission mode. The measurements at low temperature were performed with a closed-cycle helium refrigerator (Iwatani Co., Ltd., Japan). Velocity was calibrated by using an α-Fe standard. The obtained Mössbauer spectra were fitted with asymmetric Lorentzian doublets by the least squares fitting program (MossWinn).
The polarized reflection spectrum was recorded using an infrared microscope Spectratech IR-Plan combined with an FT-IR spectrometer Thermo Nicolet NEXUS 870 for 5000−12000 cm−1, and a multichannel visible spectrograph Atago Macs 320 for 11000−33000 cm−1. The spectrum was obtained from the developed plane of the thin plate crystal. The crystal orientation was adjusted so that the infrared reflectivity was maximized for the plane polarized light. The optical conductivity spectrum was calculated from the reflection spectrum by the Kramers–Kronig analysis.
2.3. Crystal Structure Determinations of 1–3
A crystal was mounted on a roll of 15 μm thick polyimide film by using the AralditeTM adhesive. A Nihon Thermal Engineering nitrogen gas flow temperature controller was used for the temperature variable measurements. All data were collected on a Bruker APEX II CCD area detector with monochromated Mo-Kα radiation generated by a Bruker Turbo X-ray Source coupled with Helios multilayer optics. All data collections were performed using the APEX2 crystallographic software package (Bruker AXS). The data were collected to a maximum 2θ value of 55.0°. A total of 720 oscillation images were collected. The APEX3 crystallographic software package (Bruker AXS) was used to determine the unit cell parameters. Data were integrated by using SAINT. Numerical absorption correction was applied by using SADABS. The structures at all temperatures were solved by direct methods and refined by full-matrix least-squares methods based on F2 by using the SHELXTL program. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were generated by calculation and refined using the riding model. CCDC 1891471-1891477 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
2.4. Density Functional Theory (DFT) Calculations
All theoretical calculations were performed using the Gaussian 09 program package [46]. The atomic coordinates for the LS and HS states of the [Fe(qsal)2] cation were taken from the single crystal structural data for 1 and 3, respectively. All geometry optimization and frequency calculations of the compounds were carried out at the B3LYP functional [47,48]. The Wachters-Hay basis set [49,50] for Fe atoms and the 6-31+G(d) basis set [51] for H, C, O, and N atoms were used. No imaginary frequencies were found in the optimized structures. Cartesian coordinates of the LS [Fe(qsal)2] and HS [Fe(qsal)2] cations calculated by the B3LYP level of theory are listed in Tables S1 and S2 in the supplementary materials. The transition energies of all electron transitions of the LS [Fe(qsal)2] and HS [Fe(qsal)2] cations were calculated by using the time-dependent DFT (TD-DFT) method [52] at the CAM-B3LYP level [53].
3. Results and Discussion
3.1. Synthesis of Compounds 1–3
Compounds 1–3 were prepared by the metathesis reaction between [Fe(qsal)2]Cl·1.5H2O and corresponding anion salts using the diffusion methods. Compound 1 was obtained as very large rhombic platelets (Figure 2a), whereas compounds 2 and 3 gave relatively large parallelogrammatic and rhombic platelets, respectively (Figure 2b,c). The compositions of 1 to 3 were confirmed by microanalyses and crystal analyses described below.

Figure 2.
Photographs of crystals for 1 (a), 2 (b), and 3 (c). The grid unit is 1 mm long.
3.2. Magnetic Susceptibility for 1–3
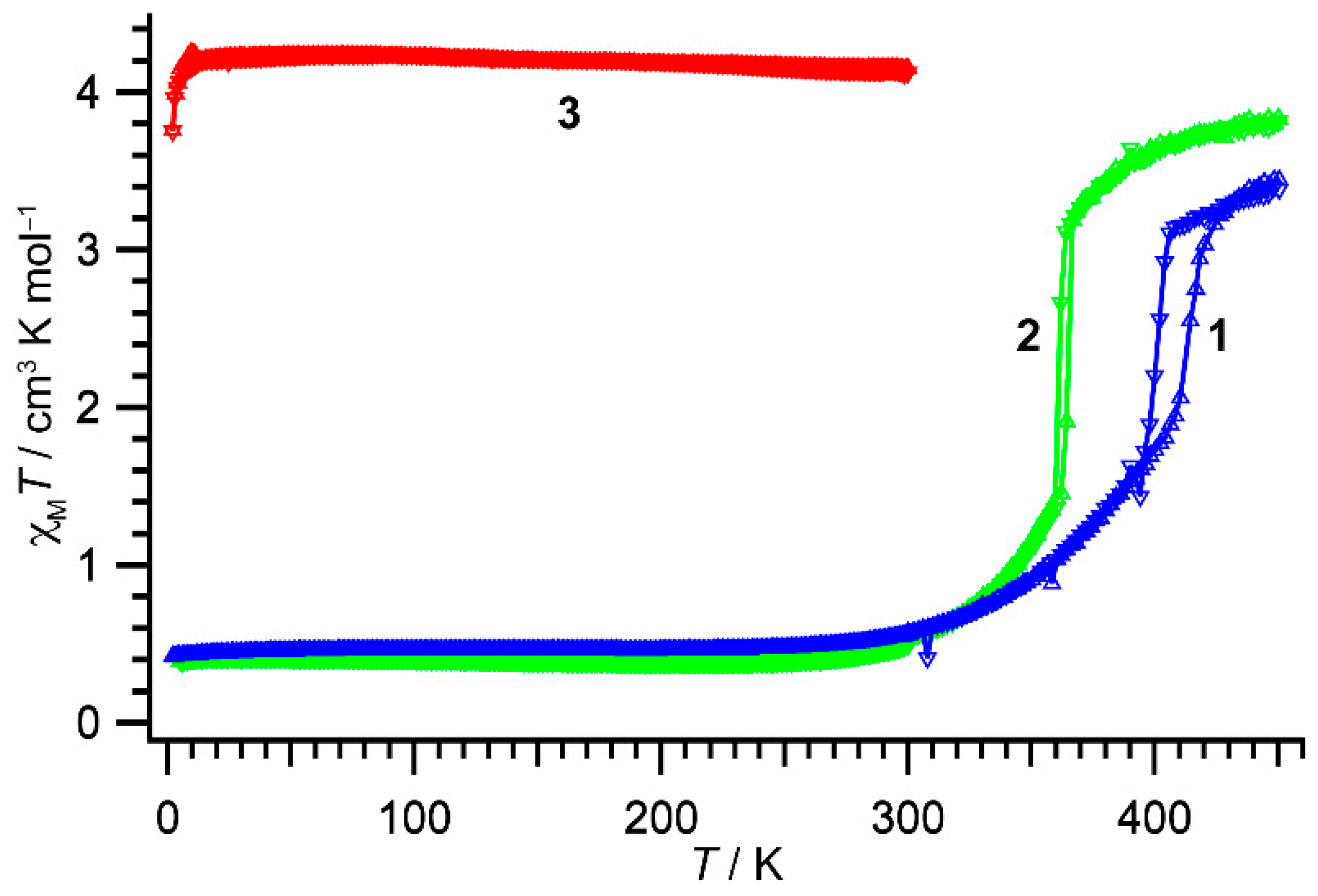
The temperature variations of magnetic susceptibility for compounds 1–3 are shown in Figure 3. The χMT value for compound 1 at 300 K was 0.56 cm3 K mol−1, suggesting 1 was almost in the LS state. Below 300 K, the χMT values were temperature-independent. The χMT value was 0.43 cm3 K mol−1 at 10 K. Meanwhile, on heating compound 1 above 300 K, the χMT values smoothly increased up to 410 K and then an abrupt change in χMT was observed (T1/2↑ = 414 K). After the steep transition, the χMT values still increased gradually again and reached 3.38 cm3 K mol−1 at 450 K, suggesting compound 1 exhibited an incomplete SCO transition. On successive lowering temperatures, a steep decrease in χMT was observed at 406 K (T1/2↓ = 402 K), and then the χMT curve traced that measured in the heating scan below 396 K. Thus, the compound 1 showed the cooperative SCO transition with a thermal hysteresis width of 12 K.
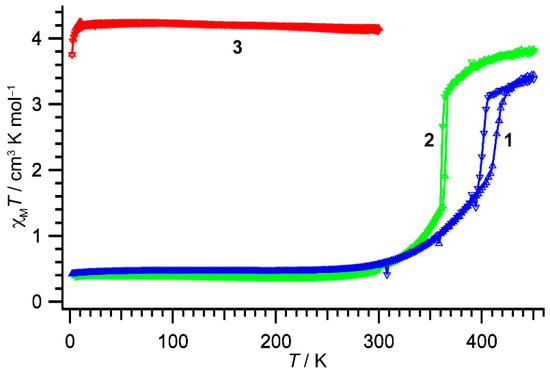
Figure 3.
The χMT vs. T products for 1 (blue triangles), 2 (green triangles), and 3 (red triangles). Triangles up and down indicate heating and cooling processes, respectively. Lines are used to guiding the eye.
The χMT value for compound 2 at 300 K was 0.54 cm3 K mol−1, suggesting 2 was also almost in the LS state. On heating the sample, the χMT values gradually rose, and an abrupt increase in χMT was observed at 362 K (T1/2↑ = 365 K). The χMT value reached to 3.84 cm3 K mol−1 at 450 K. On lowering temperatures, the χMT values traced those of the heating scan and then abruptly decreased at 364 K (T1/2↓ = 361 K), resulting in a cooperative transition with a thermal hysteresis width of 4 K.
The χMT value for compound 3 at 300 K was 4.15 cm3 K mol−1, suggesting 3 was almost in the HS state. On decreasing temperatures, the χMT values were almost constant in the temperature range of 2–300 K.
3.3. Mössbauer Spectroscopy for 1 and 3
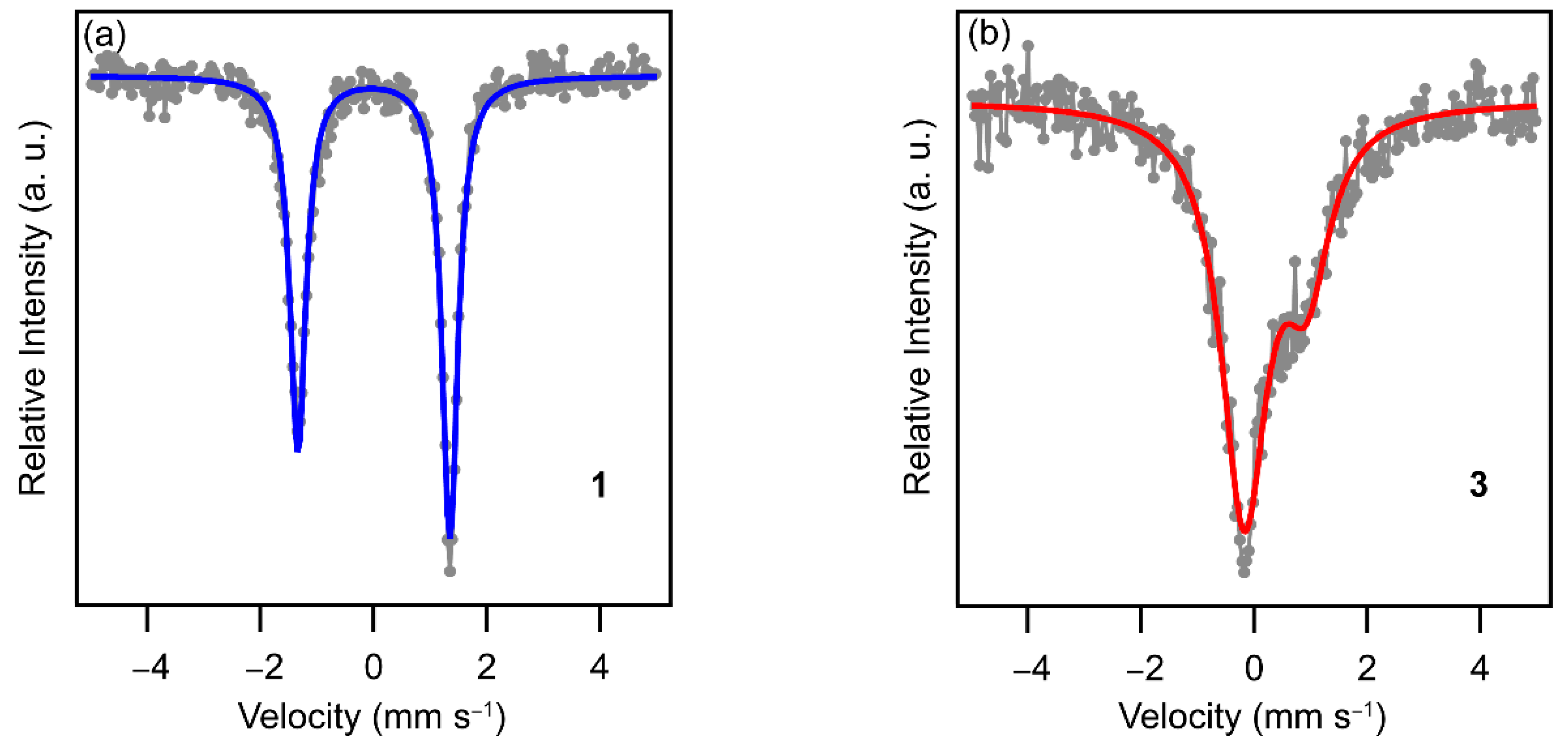
To confirm the spin-states of the Fe ion for compound 1 and 3 at 293 K, the Mössbauer spectra for 1 and 3 were recorded (Figure 4). The spectrum of 1 at 293 K consisted of only a sharp asymmetric doublet. The asymmetry of the spectrum may originate from the preferred orientation of the crystals. As compared with the Mössbauer parameters reported in the literature (Table 1), the spectrum of 1 can be ascribed to the LS spectrum, which is consistent with the χMT value of 1 at 293 K. On the other hand, the spectrum of 3 at 293 K consisted of a very broad asymmetric doublet. The isomer shift of 3 is similar to those of the simulated HS spectra reported in the literature. Note that the quadrupole splitting of 3 is the largest among those of the HS spectra in the related compounds and corresponds to that of the [Ni(nmt)2] compound having the significantly distorted coordination octahedron (mnt = maleonitriledithiolate) [33]. This suggests that the compound 3 might have a distorted coordination sphere due to some crystal packing effect.
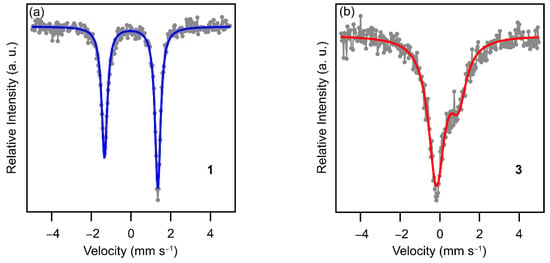
Figure 4.
Mössbauer spectra for 1 (a) and 3 (b) at 293 K. Gray circles are recorded data points. The blue and red curves are fitted LS and HS spectra, respectively.

Table 1.
Mössbauer parameters (mm s−1) for [Fe(qsal)2](X)·solv.
3.4. Optical Properties of the [Fe(qsal)2] Compounds
3.4.1. Reflection and Optical Conductivity Spectra of Single Crystals for 1 and 3
Since 1 and 3 have relatively large flat crystal surfaces, we tried to record reflection spectra at room temperature. The obtained reflection spectrum for 1 and 3 and their optical conductivity spectrum calculated by the Kramers–Kronig analysis are shown in Figure 5a,b, respectively.
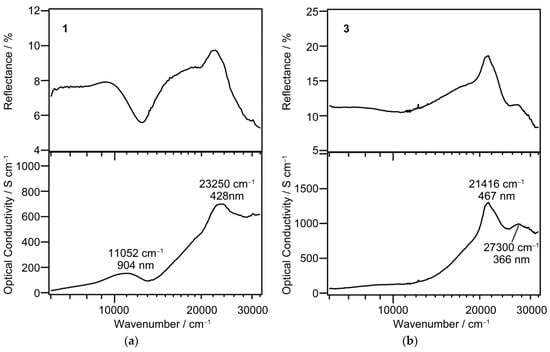
Figure 5.
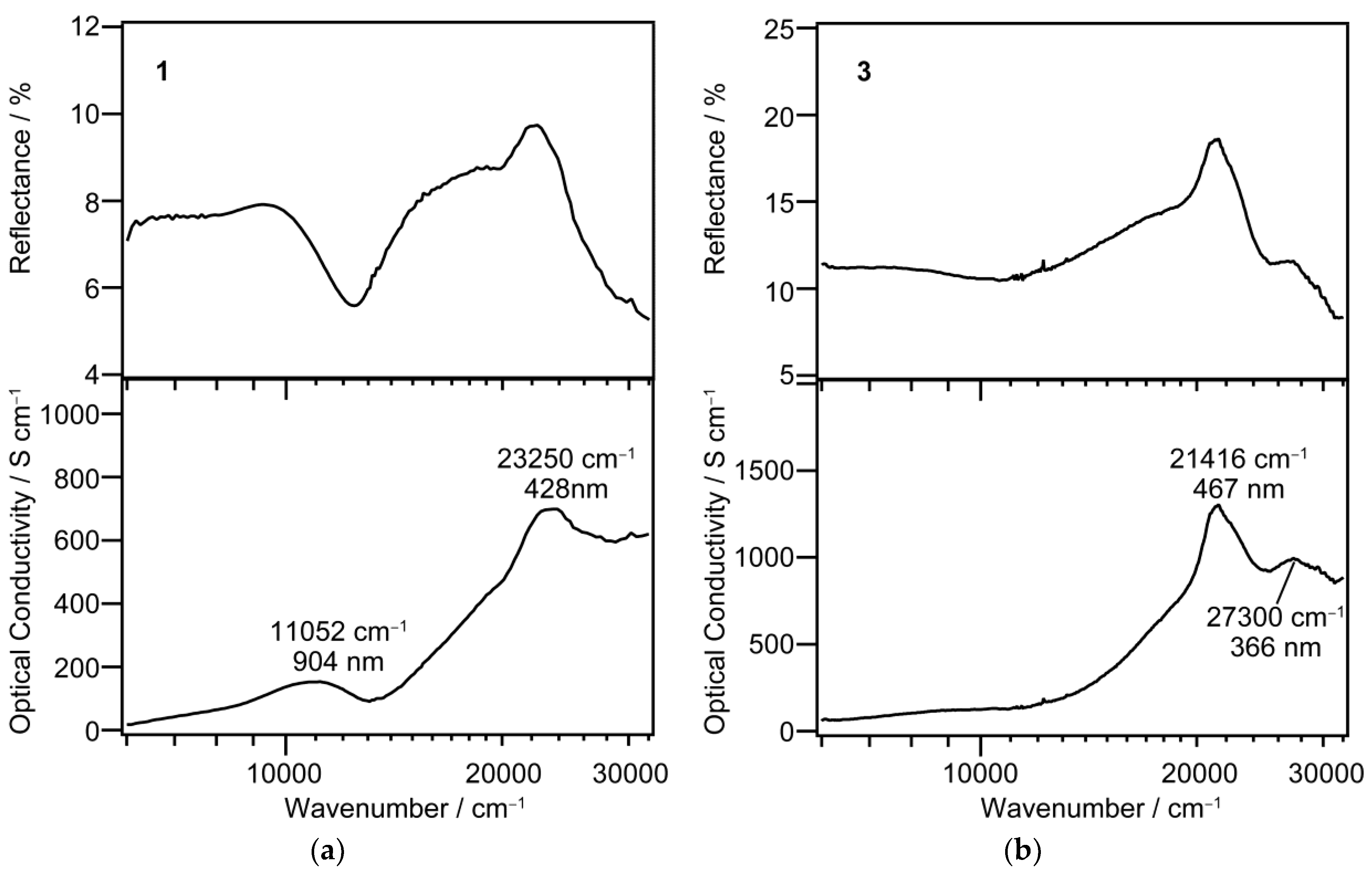
Single-crystal reflectance (upper) and optical conductivity (lower) spectra at room temperature for 1 (a) and 3 (b).
As is shown in the panel (a), the spectrum of the LS state in 1 displays the two clear absorption maxima: the strong visible absorption at 428 nm and the near-infrared medium band at 904 nm. On the other hand, the spectrum of the HS state in 3 does not show any clear absorption in the near-infrared region, whereas in the visible region there are two absorption bands at 467 and 366 nm.
Some examples of the light-induced excited spin-state trapping (LIESST) effect on the [Fe(qsal)2] compounds have ever been reported to date. The excitation wavelengths used for the LIESST effect were 808 [11] and 830 nm [22,23]. These wavelengths are in good agreement with the absorption band observed for 1, namely the LS [Fe(qsal)2] compound. Therefore, the LIESST scheme from the LS to HS states in the [Fe(qsal)2] compounds is evidenced.
3.4.2. Time-Dependent Density Functional Theory (TD-DFT) Calculations for the LS and HS [Fe(qsal)2] Cations
To provide an insight into the absorption bands for 1 and 3, the transition energies of all electron transitions in the LS [Fe(qsal)2] and HS [Fe(qsal)2] cations were calculated by using the time-dependent density functional theory (TD-DFT) method. The transition energies were strongly dependent on both the density functionals and [Fe(qsal)2] structures, and the most reasonable energies could be obtained by calculating the B3LYP-optimized structures at the CAM-B3LYP level. The transition wavelengths and assignments are summarized in Table 2.
Table 2.
Excitation wavelengths, oscillator strengths (f), and absorption assignments for the LS [Fe(qsal)2] and HS [Fe(qsal)2] cations.
The wavelengths calculated for the HS [Fe(qsal)2] cation were ascribed mainly to ligand-to-metal charge transfer (LMCT) transition and intra-ligand π-π* transition and were shorter than 656 nm, which was consistent with no remarkable absorption band observed in the near-infrared region for 3. The absorption bands observed at 467 and 366 nm for 3 were ascribed to π-π* and LMCT transitions, respectively. On the other hand, the wavelengths calculated for the LS [Fe(qsal)2] cation were ascribed mainly to d-d and LMCT transitions. Very weak absorption bands for the LS [Fe(qsal)2] cation were calculated in the near-infrared region. This is probably because these absorption bands would originate mainly from forbidden d-d transitions. The observed absorption bands at 904 and 428 nm for 1 can be ascribed to d-d and LMCT transitions, respectively.
3.5. Crystal Structures of 1–3
The variable temperature single crystal X-ray structural analyses for 1, 2, and 3 were performed using a Bruker AXS APEXII Ultra diffractometer. Crystallographic data are listed in Table 3 and Table 4. The crystal structures for 1 and 3 at 293 K belonged to a triclinic system with P, whereas the crystal structure of 2 at 293 K belonged to monoclinic P21/n. Each asymmetrical unit consisted of one [Fe(qsal)2] cation and one corresponding anion, and additionally one methanol molecule for 3. Fortunately, it was successful to determine the high-temperature phase structures of 1 and 2 at 400 and 425 K, respectively. The crystal space group for 2 maintained at 400 K, whereas that for 1 was changed into monoclinic P2/n. As associated with the crystal transformation, the crystallographically independent molecules in 1 at 425 K increased to three and a half of the [Fe(qsal)2] cations and CH3OSO3 anions. The independent [Fe(qsal)2] cations in 1 at 425 K are hereafter designated as A, B, C, and D (a half independent cation). The atomic numbers of the [Fe(qsal)2] cations A to D also add the notations of A to D to the corresponding atomic numbers.
Table 3.
Crystallographic data for 1.
Table 4.
Crystallographic data for 2 and 3.
3.5.1. Molecular Structure Description of the [Fe(qsal)2] Cation in 1–3
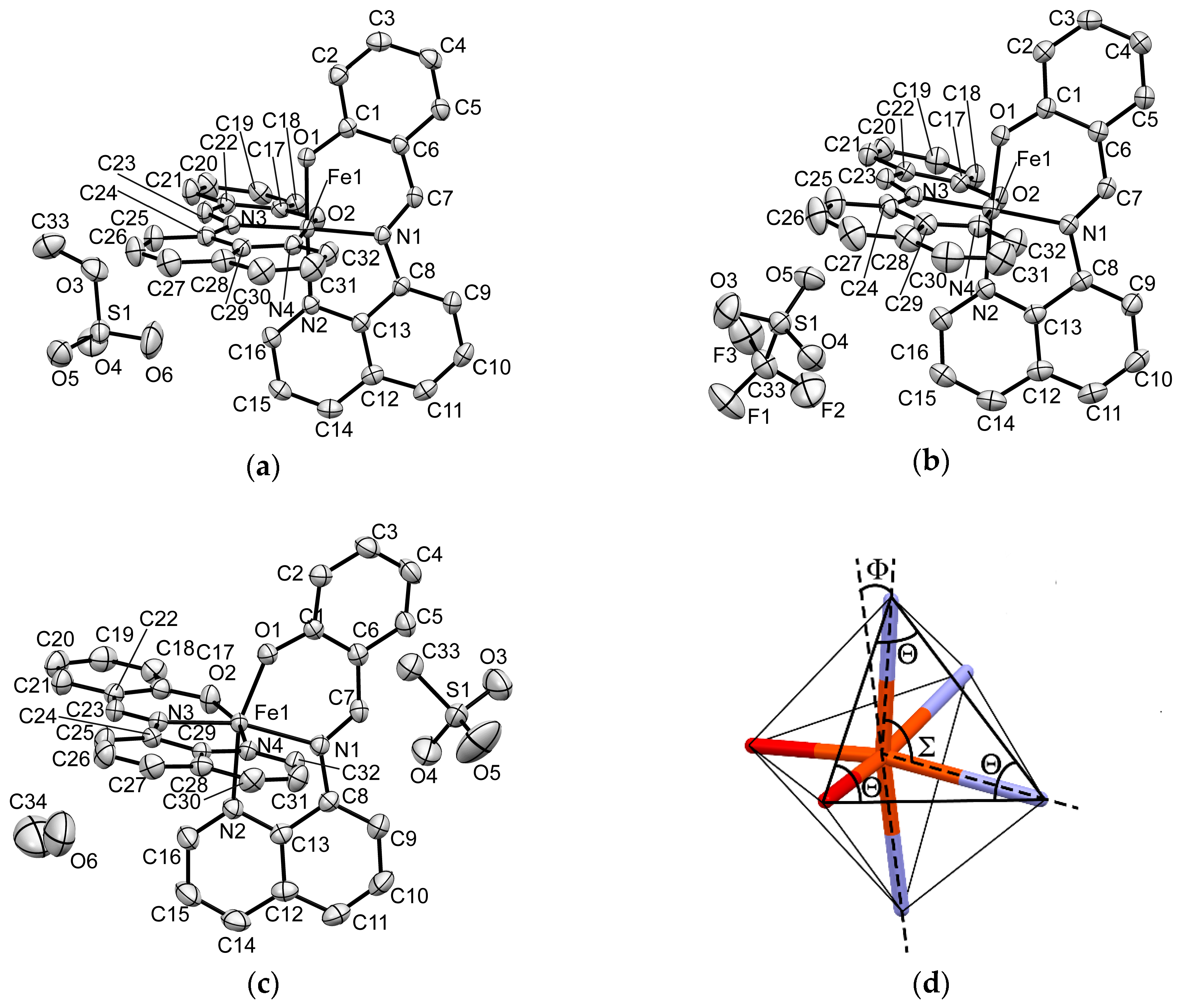
The π-ligand qsal anion was coordinated to a central Fe atom as a tridentate chelate ligand and thus two coordinated ligand molecules were arranged in an almost perpendicular manner (Figure 6a–c). The coordination bond lengths and distortion parameters (Σ, Θ, Φ, see Figure 6d) for 1, 2, and 3 along with those of the HS and LS [Fe(qsal)2] compounds confirmed by Mössbauer spectra are listed in Table 5. The distortion parameter Σ is the sum of the absolute differences in 12 coordination bite angles from 90°. The distortion parameter Θ is the sum of the absolute differences in 24 angles from 60° on 8 surface triangles of a coordination octahedron. Both Σ and Θ are zero if the coordination sphere is a regular octahedron. The distortion parameter Φ is the deviation of the angle between two coordination bonds from Fe to N atoms of two imine groups from 180°. All the distortion parameters increase their values on distorting the coordination octahedron.
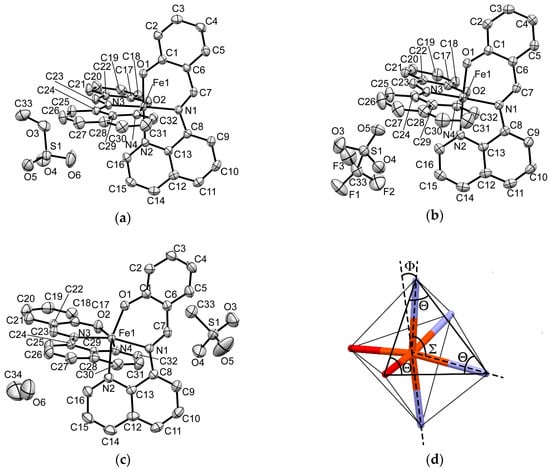
Figure 6.
ORTEP drawings of 50% probability with atomic numberings for the [Fe(qsal)2] cation, counter anion, and solvate molecule at 293 K for 1 (a); for 2 (b); for 3 (c). Hydrogen atoms are omitted for clarity; (d) The angles of a coordination octahedron concerning the distortion parameters.
Table 5.
Coordination bond lengths and distortion parameters for 1, 2, 3, and related compounds.
As compared with the coordination bond lengths and distortion parameters, the Fe–O and Fe–N bond lengths and distortion parameters for 1 and 2 at 293 K were quite similar to those of the LS NCSe compound, indicating that the [Fe(qsal)2] cations in 1 and 2 were in the LS state at 293 K. On the other hand, the coordination bond lengths and distortion parameters for 3 at 293 K were much larger than those in 1 and 2, whereas they were similar to those in the HS [Ni(dmit)2] compound (dmit = 4,5-dithiolato-1,3-dithiole-2-thione). Thus, the [Fe(qsal)2] cation in 3 was in the HS state at 293 K. These observations are in good agreement of the spin-states of 1–3 from the magnetic susceptibilities and Mössbauer spectra at 293 K. On increasing temperatures, the coordination bond lengths of 1 were gradually lengthened, which was consistent with the gradual increase in the χMT in 1. Note that the distortion parameters Σ in 1 decreased on increasing temperature to 400 K. This suggests that the distortion parameter Σ may not closely correlate with the spin-state of the [Fe(qsal)2] cation. At 425 K all the coordination bond lengths of three and a half independent [Fe(qsal)2] cations except the Fe-O bond lengths in cation A were longer than those in 1 at 400 K. Although the distortion parameters Σ of the three and a half cations were varied, all the distortion parameters Θ and Φ were larger than those at 400 K. As compared with the LS NCSe and HS [Ni(dmit)2] compounds, the cations A and D may contain certain LS fractions. Therefore, the SCO in 1 proved to be an incomplete cooperative transition accompanying a crystal structure phase transition. On the other hand, the coordination bond lengths and distortion parameters Θ and Φ in 2 at 400 K were a little smaller than those in the HS [Ni(dmit)2] compound. This indicates that the SCO transition in 2 was also an incomplete one.
The Fe−O, Fe−N(imine), and Fe−N(quinoline) bond lengths in the [Fe(qsal)2] compounds showing SCO conversions were varied from 1.869 to 1.914 Å, from 1.943 to 2.143 Å, and from 1.978 to 2.164 Å, respectively (Table 5). The Fe1−N2(quinoline) bond length of 2.172 Å in HS compound 3, the Fe1−N1(imine) bond length of 1.936 Å in the LS NCSe compound, and the Fe1−O1 and Fe1−N2 bond lengths of 1.9278 and 2.195 Å in the HS [Ni(mnt)2] compound were deviated from the above bond length range. Since the degrees of deviations were small, the occurrence of SCO cannot be judged only from the coordination bond lengths. As mentioned above, the parameters Σ may not be related to the spin-states of the [Fe(qsal)2] cation. On the other hand, the parameters Θ and Φ are probably reflected in their spin-states. In particular, the parameters Φ may be useful to judge the occurrence of SCO because the largest Φ values are observed in the HS [Ni(mnt)2] compound and HS compound 3. Halcrow reported that the similar deviations of Φ are related to the occurrence of SCO in the Fe(II) complexes from bpp ligands (bpp = 2,6-di(pyrazol-1-yl)pyridine) [54]. These findings indicate that the central donor atoms of tridentate ligands can greatly impact the ligand field splitting energies in their homoleptic complexes, which is in good agreement with the role of the azo-functional group in new anionic SCO Fe(III) complexes from azobisphenolate ligands [55].
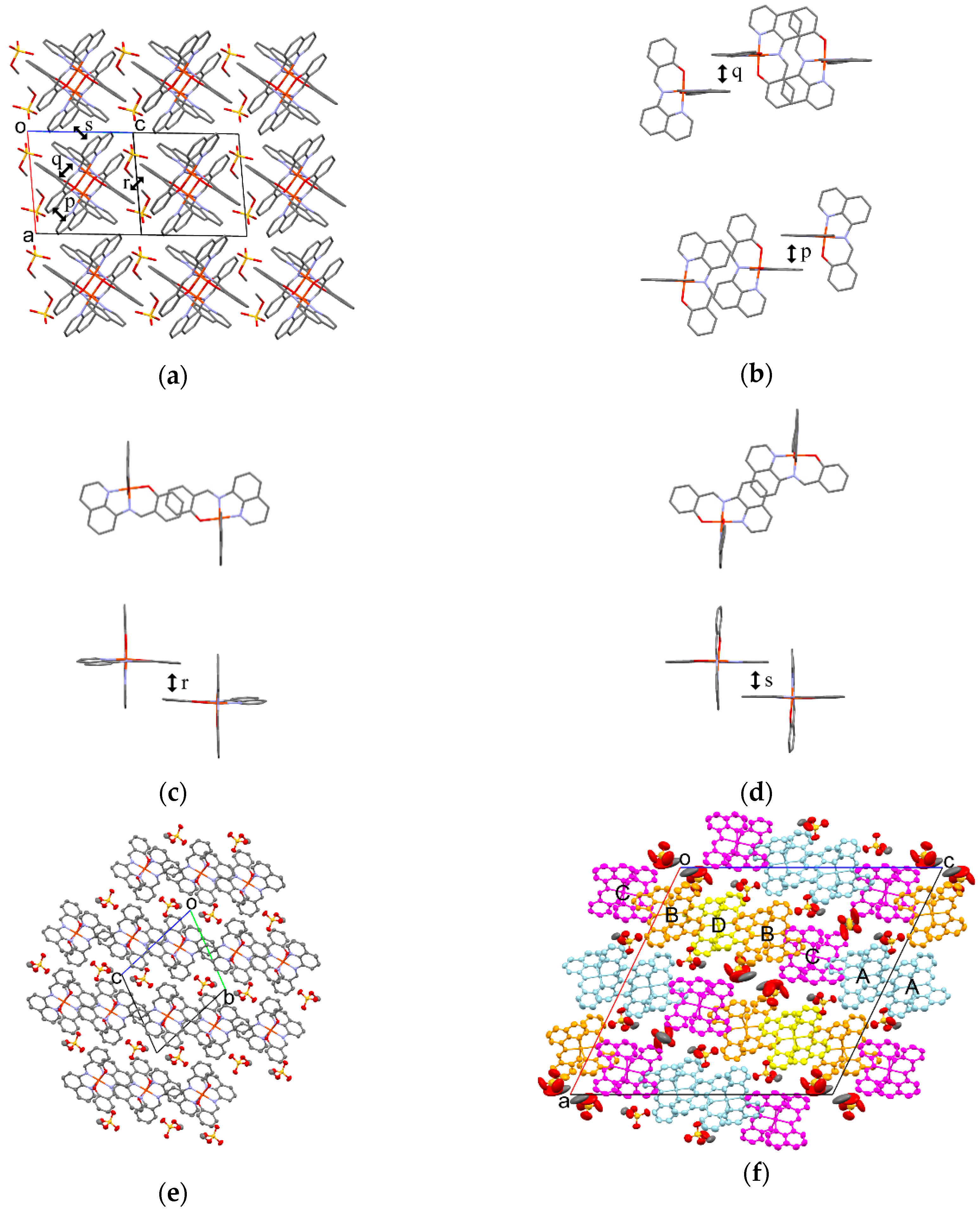
3.5.2. Molecular Arrangement of 1
The molecular arrangement of the [Fe(qsal)2] cations and CH3OSO3 anions in 1 along the b−c direction is shown in Figure 7a. Selected intermolecular distances in 1 are listed in Table 6. The phenyl ring (C1–C6) in the [Fe(qsal)2] cation was stacked with the quinolyl ring (C8–C16,N2) in the nearest neighboring [Fe(qsal)2] cation in a parallel-displaced manner, to form a π-stacking [Fe(qsal)2] dimer (p in Figure 7b). The π-stacking dimers were arranged along the b−c direction through short C···C contacts between the imine and quinolyl moieties (q in Figure 7b) and thus afforded an alternate one-dimensional (1D) [Fe(qsal)2] array. This type of 1D [Fe(qsal)2] arrangement was found in [Fe(qsal)2](NCX)·solv (X = S, Se) [9,10,11] and [Fe(qsal)2](I3) [13]. Moreover, a parallel-displaced π-stacking between the phenyl rings (C17–C22) (r in Figure 7c) and edge-shared π-stacking between the quinolyl rings (C8–C16,N2) (s in Figure 7d) were observed along the b+c and a directions, respectively. Therefore, the formation of a three-dimensional (3D) π-stacking interaction network is elucidated in 1. The CH3OSO3 anions were located in the cavity of the 3D π-stacking network of the [Fe(qsal)2] cation. All the edge oxygen atoms in the CH3OSO3 anion were contacted with the hydrogen atoms of four neighboring [Fe(qsal)2] cations.
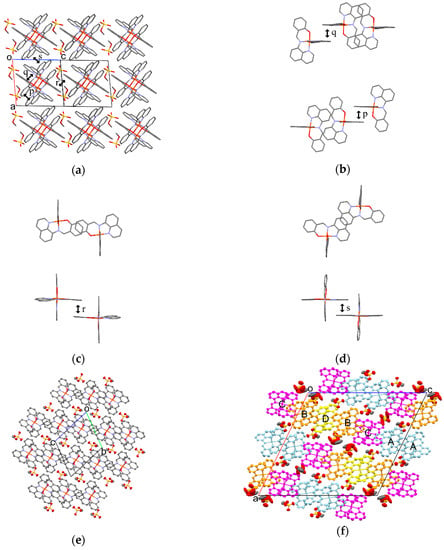
Figure 7.
(a) Crystal structure viewed along the b−c direction of 1 at 293 K; Top (upper) and side (bottom) views of π-stacking structures of the [Fe(qsal)2] cations for p (b), r (c), and s (d) at 293 K; ORTEP drawings of 1 with 50% probability at 293 K (e) and 425 K (f). The light blue, orange, magenta, and yellow [Fe(qsal)2] cations correspond to cations A, B, C, and D, respectively. Hydrogen atoms are omitted for clarity.
Table 6.
Selected intermolecular short distances (Å) at 293, 360, and 400 K in 1.
On increasing temperatures from 293 to 400 K, the intermolecular distances for 1 were gradually increased (Table 6), but the overlapping modes were not changed. On the other hand, the molecular arrangement of the [Fe(qsal)2] cations and CH3OSO3 anions at 425 K were dramatically changed, which can be easily recognized by comparison between Figure 7e,f. The periodic units of the 1D arrays of the [Fe(qsal)2] cation and CH3OSO3 anion were two and seven at 293 and 425 K, respectively. The incommensurate periodic units suggest that a large structural rearrangement would take place through the structural phase transition accompanying SCO. Within the 1D [Fe(qsal)2] array at 425 K, the π-overlaps similar to p shown in Figure 7b were found both between cations A and between cations B and D. Although the π-overlaps were deformed and twisted, several C···C contacts shorter than the sum of van der Waals (vdW) radii (C: 1.70 Å) [56] were observed between cations A and C, whereas there is only one short C···C contact between cations B and C. Thus, the 1D [Fe(qsal)2] array seemed to consist of C···A···A···C tetramer and D··B···D trimer at 425 K. Between the 1D [Fe(qsal)2] arrays, most π-overlaps similar to s shown in Figure 7d were lengthened in the range of 3.407−3.610 Å along the b axis, whereas one half of π-overlaps similar to r shown in Figure 7c disappeared and the remained π-overlaps were observed only between cations A and B along the a axis. Thus, the 3D π-stacking interaction network was rearranged and weakened by the deformation of the [Fe(qsal)2] array. Since the orientations of the CH3OSO3 anions were partly different from those below 400 K and their thermal ellipsoids were also much larger than those of the [Fe(qsal)2] cation at 425 K, the crystal structure phase transition in 1 seems to arise from the thermal motion of the CH3OSO3 anions.
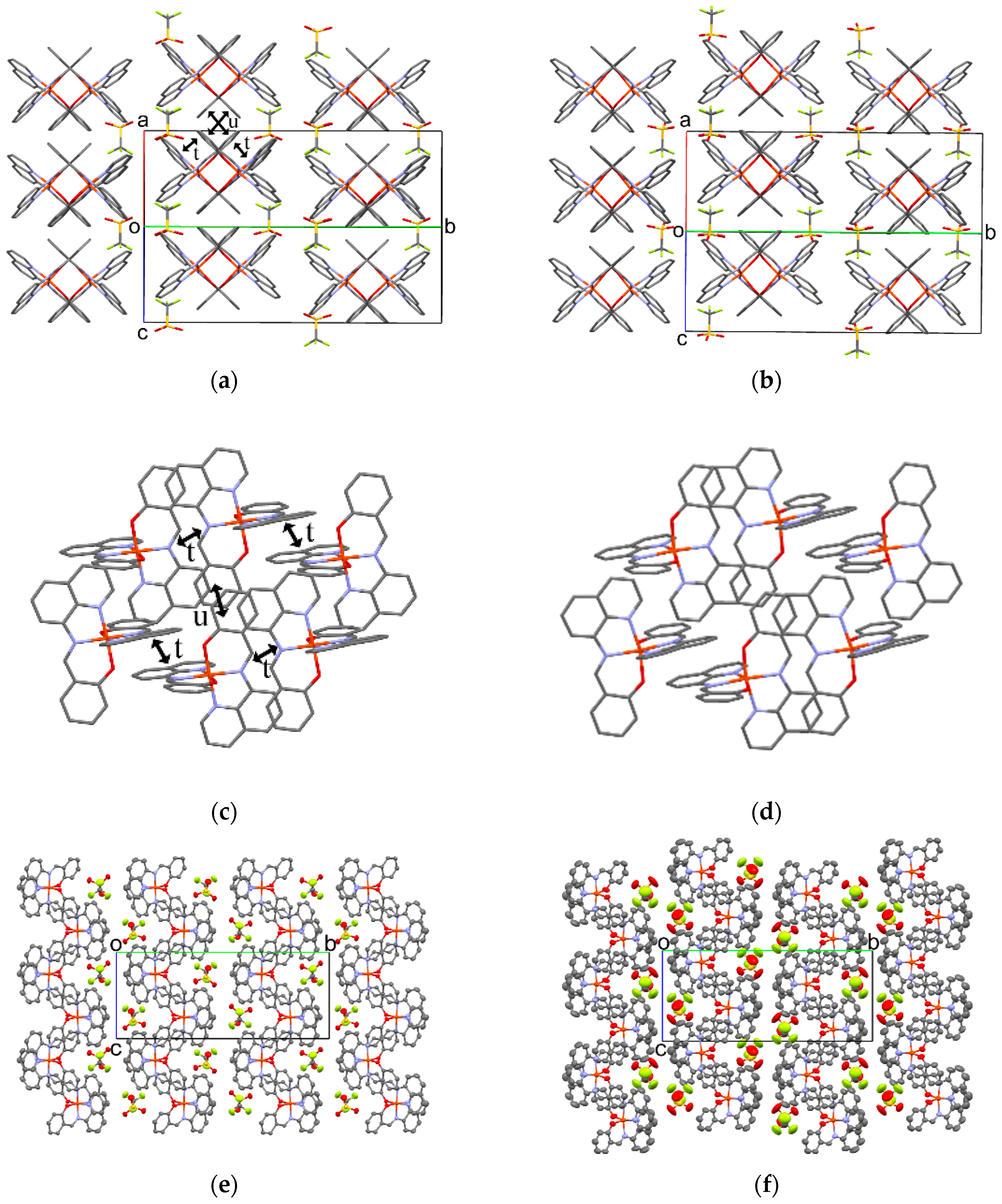
3.5.3. Molecular Arrangement of 2
The molecular arrangement of the [Fe(qsal)2] cations and CF3SO3 anions in 2 along the a+c direction at 293 and 400 K are shown in Figure 8a,b. Selected intermolecular distances are listed in Table 7. Similar to the π-stacking [Fe(qsal)2] dimer in 1, the qsal ligand of the [Fe(qsal)2] cation was stacked with that of the neighboring [Fe(qsal)2] cation in a head-to-tail manner (t in Figure 8c). On the other hand, the π-stacked [Fe(qsal)2] cations were related each other by a symmetry operation of n glide plane and thus gave a uniform 1D molecular array along the a+c direction. This type of the uniform 1D [Fe(qsal)2] arrangement was found in [Fe(qsal)2](NCS) [11] and [Fe(qsal)2](I) [15]. The π-overlaps between the 1D [Fe(qsal)2] arrays were found between the phenyl moieties along the a−c direction (u in Figure 8a,c). Thus, the [Fe(qsal)2] cations formed a two-dimensional (2D) regular network along the ac plane. Meanwhile, there was no remarkable short contact between the 2D [Fe(qsal)2] networks.
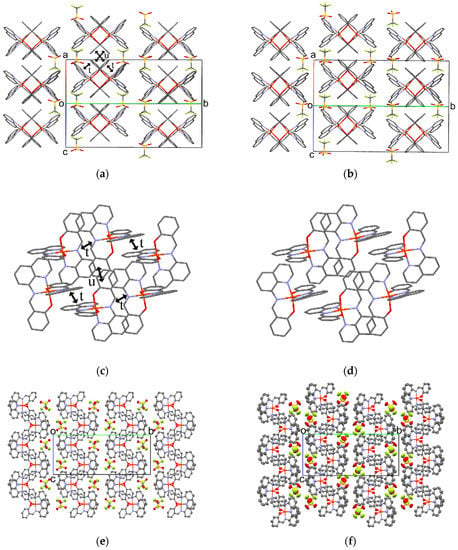
Figure 8.
Crystal structure viewed along the a+c direction of 2 at 293 K (a) and 400 K (b). Hydrogen atoms are omitted for clarity; π-stacking structures of the [Fe(qsal)2] cations along the ac plane at 293 K (c) and 400 K (d); ORTEP drawings of 2 with 50% probability at 293 K (e) and 400 K (f).
Table 7.
Selected intermolecular short distances (Å) in 2.
The CF3SO3 anions were located between the 2D [Fe(qsal)2] networks. The S-O bond lengths imply that the negative charge in the CF3SO3 anion may be delocalized at all oxygen atoms. The O5···C23(imine) and O4···C14(quinolyl) distances were found to be 3.078(3) and 3.179(3) Å, respectively. These suggest the existence of effective Coulomb interactions between the [Fe(qsal)2] cation and CF3SO3 anion.
The molecular arrangement of 2 at 400 K was very similar to that at 293 K. The difference in the 1D π-stacking arrays (Figure 8a,b) and 2D π-stacking network structure (Figure 8c,d) were hardly observed, but the 2D π-stacking layers glided alternately along the c axis (Figure 8e,f). Since the short O5···C23(imine) distance was shortened to be 3.039(6) Å at 400 K, this structure transition would be involved in the Coulomb interaction. The CF3SO3 anions in a 2D π-stacking layer were located near the quinolyl rings in the neighboring 2D layer at 293 K, whereas the CF3SO3 anions were shifted from the quinolyl rings in the neighboring 2D layer at 400 K. Moreover, the thermal ellipsoids of the CF3SO3 anion and quinolyl moiety were larger than those of the other molecular components at 400 K. These observations indicate that this structure transition in 2 may result from the thermal motion of the CF3SO3 anion, which is reminiscent of the structural transition in 1.
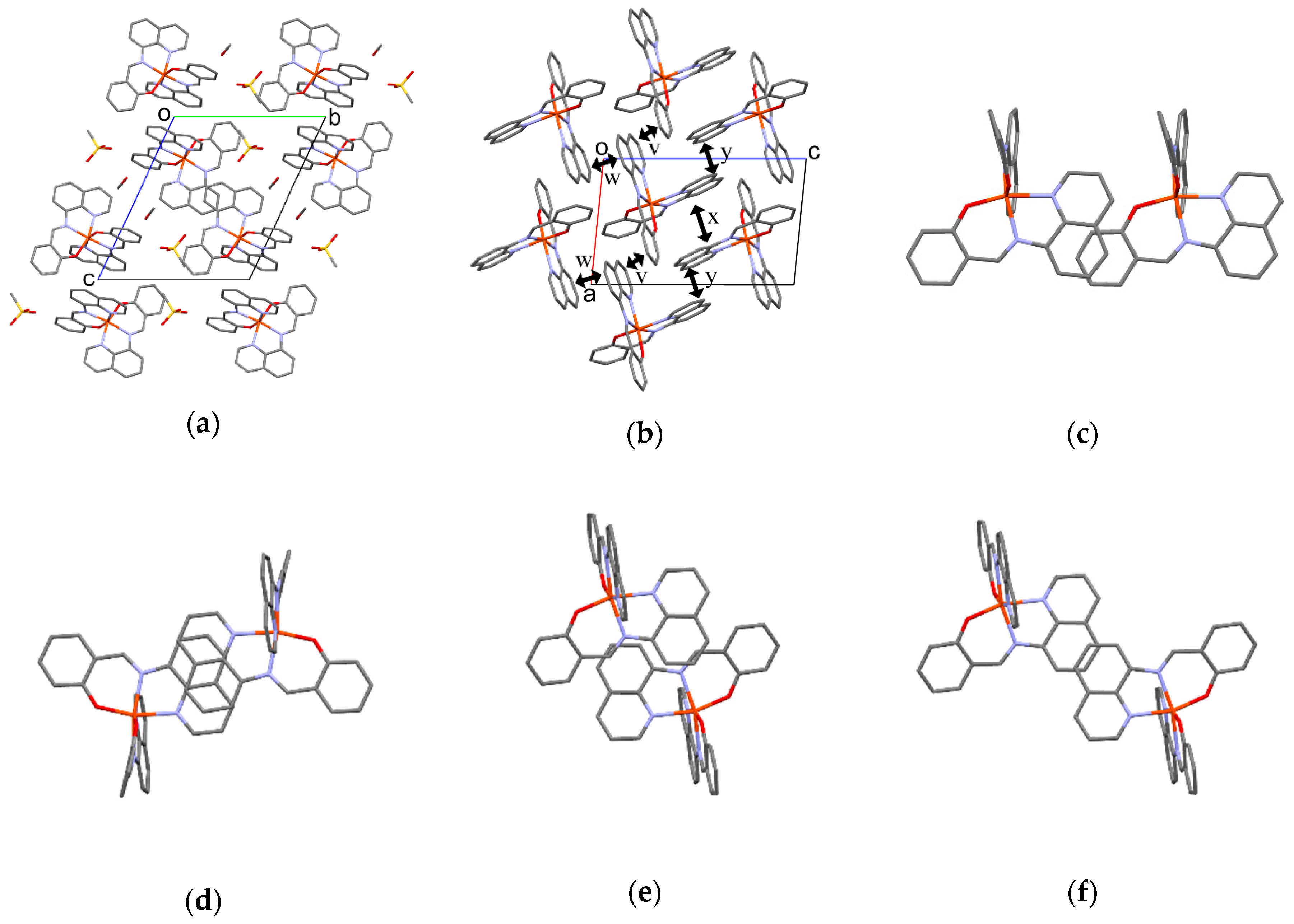
3.5.4. Molecular Arrangement of 3 at 293 K
The molecular arrangement of the [Fe(qsal)2] cations in 3 was quite different from those found in common [Fe(qsal)2] compounds (Figure 9a). Selected intermolecular distances are listed in Table 8. Relatively strong π-stacking interactions between the [Fe(qsal)2] cations afforded a 1D array along the a axis (v in Figure 9b,c). There were two kinds of π-overlaps between the 1D [Fe(qsal)2] arrays. One overlaps were between the quinolyl planes (w in Figure 9b,d), the other ones were between the quinolyl and imine moieties (x in Figure 9b,e). These overlaps were alternately arranged along the b+c direction. Meanwhile, π-overlaps y between the quinolyl planes shown in Figure 9b,f seem to be weak, because the shortest distance is 3.679(4) Å. The above-mentioned three π-stacking interactions gave a 2D network parallel to the b+c direction (Figure 9b). Since the intermolecular C3···C4 and C15···C20 distances between the 2D [Fe(qsal)2] networks were 3.620(4) and 3.627(5) Å, respectively, the [Fe(qsal)2] cations in 3 formed the 2D interaction network.
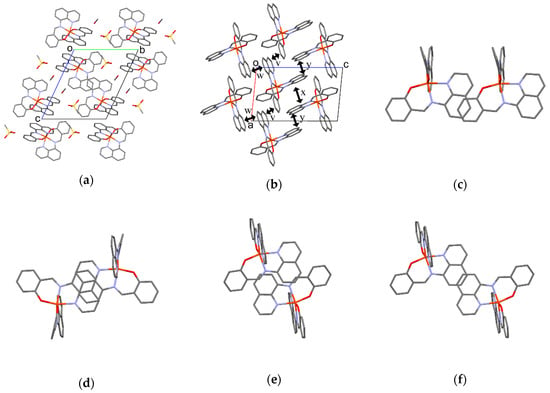
Figure 9.
(a) Crystal structure viewed along the a axis of 3 at 293 K; (b) Side view of the π-stacking structure of 3; Top views of the π-stacking structure for v (c), w (d), x (e), and y (f). Hydrogen atoms are omitted for clarity.
Table 8.
Selected intermolecular distances (Å) in 3.
The CH3SO3 anions and methanol molecules were located between the 2D [Fe(qsal)2] networks. The S-O bond lengths imply that the negative charge in the CH3SO3 anion may be localized mainly at O3 and O4 atoms. The short O4···C7 and O3···O6 distances were found to be 3.070(3) and 2.814(4) Å, respectively. The former suggests the existence of effective Coulomb interactions between the [Fe(qsal)2] cation and CH3SO3 anion, the latter indicates that of hydrogen bonding interactions between the CH3SO3 anion and methanol molecule. We can assume that both strong Coulomb interaction and hydrogen bonding interaction may induce the distortion of a coordination octahedron, leading to the HS state in the whole temperature range.
3.5.5. Correlation between the Crystal Structures and Magnetic Behaviors for the [Fe(qsal)2] Compounds
The magnetic behaviors and short cation···cation distances for the [Fe(qsal)2] compounds with nonplanar counter-anions whose crystal structures were deposited to date are summarized in Table 9. Interestingly, the [Fe(qsal)2] cation arrangements except compound 3 are quite similar to each other. All the [Fe(qsal)2] compounds except 3 consist of a 1D π-stacking [Fe(qsal)2] cation array shown in Figure 7b and Figure 8c. On the other hand, the arrangements between the 1D [Fe(qsal)2] cation arrays are slightly different, for example, between Figure 7a and Figure 8a. Basically, the counter-anions or solvate molecules may determine the molecular arrangement of the [Fe(qsal)2] cations.
Table 9.
Magnetic behaviors and selected intermolecular short C···C distances between the [Fe(qsal)2] cations for [Fe(qsal)2](X)·solv.
Let us consider the comparison with the most intriguing compounds [Fe(qsal)2](NCS) and [Fe(qsal)2](NCSe), both of which exhibited a cooperative SCO transition with a large thermal hysteresis loop. It is very difficult to quantitatively evaluate the strength of π-stacking interactions in various stacking manners. However, the number and shrinkage of intermolecular short distances between the atoms can estimate the strength of π-stacking interactions qualitatively. It should be noted that neither C···C nor C···N distance between the [Fe(qsal)2] cations is shorter than the sum of vdW radii (C: 1.70, N: 1.55 Å) [56] for [Fe(qsal)2](NCS) and [Fe(qsal)2](NCSe). The short C···C distances in [Fe(qsal)2](NCS) are 3.466, 3.527, and 3.593 Å along the 1D [Fe(qsal)2] array, whereas those in [Fe(qsal)2](NCSe) are 3.491, 3.533, and 3.545 Å along the 1D [Fe(qsal)2] array, and 3.538 Å between the 1D [Fe(qsal)2] arrays. On the contrary, as shown in Table 9, several C···C distances shorter than the sum of vdW radii (< 3.40 Å) along the 1D [Fe(qsal)2] array are found in the LS [Fe(qsal)2] compounds, and moreover, between the 1D [Fe(qsal)2] arrays for 1. As compared between [Fe(qsal)2](NCX) and [Fe(qsal)2](NCX)·solv (X = S, Se), the existence of solvate molecules should result in the expansion of intermolecular distances. However, the intermolecular [Fe(qsal)2] cation distances involved in π-stacking interactions in the solvate compounds are much shorter than those in the non-solvate compounds.
The literature [9] disclosed that [Fe(qsal)2](NCSe)·solv were transformed into [Fe(qsal)2](NCSe) by desolvation. Moreover, the time-dependence of the spin-states in [Fe(qsal)2](NCS) was reported in the literature [8], suggesting that the desolvation from [Fe(qsal)2](NCS)·solv to [Fe(qsal)2](NCS) took place at 286 K. Very recently, similar desolvation-induced crystal structure transformations in the charged Fe(II) and Fe(III) compounds were reported [57,58], which seemed to be driven by Coulomb interactions. In general, the Coulomb interaction is much stronger than other intermolecular interactions. Thus, the structure transformation through desolvation in charged complexes may be driven by the Coulomb energy gain, namely shrinking the distances between the cations and anions. Several C···N and C···S distances shorter than the sum of vdW radii are found between the [Fe(qsal)2] cation and NCS anion for [Fe(qsal)2](NCS), indicating the Coulomb interactions may operate the crystal structure transformation for [Fe(qsal)2](NCX)·solv.
One may ask why solvate molecules are included in a crystal lattice although they will reduce Coulomb energy gain. If the energy gain from other intermolecular interactions in a solvate compound is larger than the loss of Coulomb energy gain, the total energy gain can be larger than that in a non-solvate compound. The shorter intermolecular distances involved in π-stacking interactions in [Fe(qsal)2](NCX)·solv suggest that the strong π-stacking interactions may result from the [Fe(qsal)2] cation arrangement given by the inclusion of the solvate molecules. Therefore, we can assume that the solvate molecule in a solvate complex seems to play a significant role in the enhancement of the total lattice energy gain by various intermolecular interactions between its molecular components.
This idea is also applicable to 1, 2, and the I3 compound. Although the crystal structure from a charged non-solvate complex should be a typical ionic crystal which has a larger coordination number and shorter cation···anion distances, the crystal structures for 1, 2, and the I3 compound were not those of a typical ionic crystal but those similar to [Fe(qsal)2](NCS)·CH2Cl2 and [Fe(qsal)2](NCSe)·CH2Cl2. This suggests that the total lattice energy gain from the strong π-stacking interactions found in 1, 2, and the I3 compound may exceed the loss of Coulomb energy gain in the crystal structure of a typical ionic crystal. Consequently, we can discuss the role of large non-spherical counter-anions or solvate molecules for a crystal packing in a charged complex by means of the competition between Coulomb and intermolecular interactions. Further investigations on other charged complexes are needed for verification of the present finding.
Next, we will discuss the large thermal hysteresis loops of the magnetic susceptibility found in [Fe(qsal)2](NCS) and [Fe(qsal)2](NCSe). As shown in Table 9, more than two-step variations in the cooperative SCO transition are one of the characteristic points for 1, 2, [Fe(qsal)2](NCS) and [Fe(qsal)2](NCSe). Moreover, large thermal ellipsoids or disorder of counter-anions were observed in the crystal structures of their HS phases. This implies that the thermal motion of the counter-anion may play an important role in their HS crystal structures. The thermal variations in the crystal structures of 1 from 293 to 400 K (Table 6) revealed that strong intermolecular π-stacking interactions were retained despite the gradual SCO conversion. Recently, we found similar gradual SCO conversion in the charged Fe(II) compound having strong intermolecular interactions, in which the large difference in lattice enthalpy between the LS and HS states leads to a gradual SCO conversion despite the existence of strong intermolecular interactions [58]. Therefore, to undergo an abrupt SCO transition with a large hysteresis loop, it may be important to realize a small difference in lattice enthalpy between the LS and HS states by the choice of a counter-anion or solvate molecule.
4. Conclusions
New [Fe(qsal)2] compounds, [Fe(qsal)2](CH3OSO3) 1 and [Fe(qsal)2](CH3SO3)·CH3OH 3, along with the reported compound, [Fe(qsal)2](CF3SO3) 2, were prepared and characterized. The compounds 1 and 2 exhibited a cooperative SCO transition at higher temperatures. The optical conductivity spectrum from the single-crystal reflection spectrum of 1 revealed that the photo-excitation band for the LIESST effect on the LS [Fe(qsal)2] compounds is attributed to d-d transition. The successful crystal structure determinations of 1 and 2 in the high-temperature phase reveal that large structural changes were triggered by the motion of counter anions. The structural comparison among the [Fe(qsal)2] compounds determined to date suggests that the counter-anions and solvate molecules play a significant role in the total lattice energy gain in a charged complex. The present finding may lead to elucidation of the role of counter-anions and solvate molecules for controlling SCO transition behaviors. To do this end, the quantitative evaluation of lattice enthalpy for each spin-state in SCO compounds may be required. Since the [Fe(qsal)2] derivatives are suitable candidates for this purpose, we are now investigating a family of the [Fe(qsal)2] derivatives.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/9/2/81/s1, Table S1: Cartesian coordinates of the LS [Fe(qsal)2] cation, Table S2: Cartesian coordinates of the HS [Fe(qsal)2] cation.
Author Contributions
Conceptualization, K.T.; Methodology, K.T.; Validation, K.T.; Formal analysis, K.T., K.Y., T.Y., and Y.S.; Investigation, K.T., K.Y., T.Y, Y.E., Y.S., K.Y., and H.M.; Resources, K.T. and H.M.; Data curation, K.T., K.Y., T.Y., and Y.S.; Writing—original draft preparation, K.T.; Writing—review and editing, K.Y., T.Y., Y.E., Y.S., K.Y., and H.M.; Visualization, K.T.; Supervision, K.T.; Project administration, K.T.; Funding acquisition, K.T. and H.M.
Funding
This research was funded partly by a Grant-in-Aid for Scientific Research on the Priority Area of Molecular Conductors (no. 18028024) and on Innovative Areas of Molecular Degrees of Freedom (no. 20110007), and a Grant-in-Aid for Young Scientists (B) (no. 19750107) and for Scientific Research (C) (no. 25410068) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Acknowledgments
K.T. is grateful to T. Ishikawa at Tokyo Institute of Technology for discussing the optical properties of the Fe complexes. K.T. also thanks to A. Sakamoto at the University of Tokyo, and Y. Furuie at Kobe University for performing the elemental analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gütlich, P.; Goodwin, H.A. (Eds.) Spin Crossover in Transition Metal Compounds I−III; Springer: Berlin/Heidelberg, Germany, 2004; ISBN 3-540-40394-9, 3-540-40396-5 and 3-540-40395-7. [Google Scholar]
- Halcrow, M.A. (Ed.) Spin-Crossover Materials; John Wiley & Sons, Ltd.: Oxford, UK, 2013; ISBN 978-1-119-99867-9. [Google Scholar]
- Takahashi, K. (Ed.) Spin-Crossover Complexes; MDPI: Basel, Switzerland, 2018; ISBN 978-3-03842-825-1. [Google Scholar]
- Bousseksou, A.; Molnár, G.; Salmon, L.; Nicolazzi, W. Molecular spin crossover phenomenon: Recent achievements and prospects. Chem. Soc. Rev. 2011, 40, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Gütlich, P.; Gaspar, A.B.; Garcia, Y. Spin state switching in iron coordination compounds. Beilstein J. Org. Chem. 2013, 9, 342–391. [Google Scholar] [CrossRef] [PubMed]
- Dahl, B.M.; Dahl, O. Studies of Chelates with Heterocyclic Ligands IV. Transition Metal Complexes with N-(8-quinolyl)-salicylaldimine. Acta Chem. Scand. 1969, 23, 1503–1513. [Google Scholar] [CrossRef]
- Dickinson, R.C.; Baker, W.A.; Collins, R.L. The Magnetic Properties of bis[N-(8-quinolyl)-salicylaldimine]halogenoiron(III)·X hydrate, Fe(8-QS)2X·xH2O: A Reexamination. J. Inorg. Nucl. Chem. 1977, 39, 1531–1533. [Google Scholar] [CrossRef]
- Oshio, H.; Kitazaki, K.; Mishiro, J.; Kato, N.; Maeda, Y.; Takashima, Y. New Spin-crossover Iron(III) Complexes with Large Hysteresis Effects and Time Dependence of their Magnetism. J. Chem. Soc. Dalton Trans. 1987, 1341–1347. [Google Scholar] [CrossRef]
- Hayami, S.; Gu, Z.; Yoshiki, H.; Fujishima, A.; Sato, O. Iron(III) Spin-Crossover Compounds with a Wide Apparent Thermal Hysteresis around Room Temperature. J. Am. Chem. Soc. 2001, 123, 11644–11650. [Google Scholar] [CrossRef] [PubMed]
- Hayami, S.; Kawahara, T.; Juhasz, G.; Kawamura, K.; Uehashi, K.; Sato, O.; Maeda, Y. Iron(III) Spin Transition Compound with a Large Thermal Hysteresis. J. Radioanal. Nucl. Chem. 2003, 255, 443–447. [Google Scholar] [CrossRef]
- Hayami, S.; Hiki, K.; Kawahara, T.; Maeda, Y.; Urakami, D.; Inoue, K.; Ohama, M.; Kawata, S.; Sato, O. Photo-Induced Spin Transition of Iron(III) Compounds with π-π Intermolecular Interactions. Chem. A Eur. J. 2009, 15, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, T.A.; Ovchinnikov, I.V.; Garipov, R.R.; Ivanova, G.I. Spin Crossover [Fe(qsal)2]X (X = Cl, SCN, CF3SO3) Complexes: EPR and DFT Study. Appl. Magn. Reson. 2011, 40, 1–10. [Google Scholar] [CrossRef]
- Takahashi, K.; Sato, T.; Mori, H.; Tajima, H.; Sato, O. Correlation between the Magnetic Behaviors and Dimensionality of Intermolecular Interactions in Fe(III) Spin Crossover Compounds. Phys. B Condens. Matter 2010, 405, S65–S68. [Google Scholar] [CrossRef]
- Takahashi, K.; Sato, T.; Mori, H.; Tajima, H.; Einaga, Y.; Sato, O. Cooperative Spin Transition and Thermally Quenched High-Spin State in New Polymorph of [Fe(qsal)2]I3. Hyperfine Interact. 2012, 206, 1–5. [Google Scholar] [CrossRef]
- Djukic, B.; Jenkins, H.A.; Seda, T.; Lemaire, M.T. Structural and Magnetic Properties of Homoleptic Iron(III) Complexes Containing N-(8-Quinolyl)-Salicylaldimine [Fe(Qsal)2]+X− {X = I or (Qsal)FeCl3}. Transit. Metal Chem. 2013, 38, 207–212. [Google Scholar] [CrossRef]
- Tsukiashi, A.; Nakaya, M.; Kobayashi, F.; Ohtani, R.; Nakamura, M.; Harrowfield, J.M.; Kim, Y.; Hayami, S. Intermolecular Interaction Tuning of Spin-Crossover Iron(III) Complexes with Aromatic Counteranions. Inorg. Chem. 2018, 57, 2834–2842. [Google Scholar] [CrossRef] [PubMed]
- Harding, D.J.; Phonsri, W.; Harding, P.; Gass, I.A.; Murray, K.S.; Moubaraki, B.; Cashion, J.D.; Liu, L.; Telfer, S.G. Abrupt Spin Crossover in an Iron(III) Quinolylsalicylaldimine Complex: Structural Insights and Solvent Effects. Chem. Commun. 2013, 49, 6340–6342. [Google Scholar] [CrossRef] [PubMed]
- Harding, D.J.; Sertphon, D.; Harding, P.; Murray, K.S.; Moubaraki, B.; Cashion, J.D.; Adams, H. FeIII Quinolylsalicylaldimine Complexes: A Rare Mixed-Spin-State Complex and Abrupt Spin Crossover. Chem. A Eur. J. 2013, 19, 1082–1090. [Google Scholar] [CrossRef]
- Harding, D.J.; Phonsri, W.; Harding, P.; Murray, K.S.; Moubaraki, B.; Jameson, G.N.L. Abrupt Two-Step and Symmetry Breaking Spin Crossover in an Iron(III) Complex: An Exceptionally Wide [LS-HS] Plateau. Dalton Trans. 2014, 44, 15079–15082. [Google Scholar] [CrossRef] [PubMed]
- Phonsri, W.; Harding, D.J.; Harding, P.; Murray, K.S.; Moubaraki, B.; Gass, I.A.; Cashion, J.D.; Jameson, G.N.L.; Adams, H. Stepped Spin Crossover in Fe(III) Halogen Substituted Quinolylsalicylaldimine Complexes. Dalton Trans. 2014, 43, 17509–17518. [Google Scholar] [CrossRef] [PubMed]
- Phonsri, W.; Harding, P.; Liu, L.; Telfer, S.G.; Murray, K.S.; Moubaraki, B.; Ross, T.M.; Jameson, G.N.L.; Harding, D.J. Solvent Modified Spin Crossover in an Iron(III) Complex: Phase Changes and an Exceptionally Wide Hysteresis. Chem. Sci. 2017, 8, 3949–3959. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Cui, H.; Kobayashi, H.; Einaga, Y.; Sato, O. The Light-Induced Excited Spin State Trapping Effect on Ni(dmit)2 Salt with an Fe(III) Spin-Crossover Cation: [Fe(qsal)2][Ni(dmit)2]·2CH3CN. Chem. Lett. 2005, 34, 1240–1241. [Google Scholar] [CrossRef]
- Takahashi, K.; Cui, H.-B.; Okano, Y.; Kobayashi, H.; Einaga, Y.; Sato, O. Electrical Conductivity Modulation Coupled to a High-Spin-Low-Spin Conversion in the Molecular System [FeIII(qsal)2][Ni(dmit)2]3·CH3CN·H2O. Inorg. Chem. 2006, 45, 5739–5741. [Google Scholar] [CrossRef]
- Faulmann, C.; Dorbes, S.; Lampert, S.; Jacob, K.; Garreau de Bonneval, B.; Molnár, G.; Bousseksou, A.; Real, J.A.; Valade, L. Crystal Structure, Magnetic Properties and Mössbauer Studies of [Fe(qsal)2][Ni(dmit)2]. Inorg. Chim. Acta 2007, 360, 3870–3878. [Google Scholar] [CrossRef]
- Takahashi, K.; Cui, H.-B.; Okano, Y.; Kobayashi, H.; Mori, H.; Tajima, H.; Einaga, Y.; Sato, O. Evidence of the Chemical Uniaxial Strain Effect on Electrical Conductivity in the Spin-Crossover Conducting Molecular System: [FeIII(qnal)2][Pd(dmit)2]5·Acetone. J. Am. Chem. Soc. 2008, 130, 6688–6689. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Mori, H.; Kobayashi, H.; Sato, O. Mechanism of Reversible Spin Transition with a Thermal Hysteresis Loop in [FeIII(qsal)2][Ni(dmise)2]·2CH3CN: Selenium Analogue of the Precursor of an Fe(III) Spin-Crossover Molecular Conducting System. Polyhedron 2009, 28, 1776–1781. [Google Scholar] [CrossRef]
- Neves, A.I.S.; Dias, J.C.; Vieira, B.J.C.; Santos, I.C.; Castelo Branco, M.B.; Pereira, L.C.J.; Waerenborgh, J.C.; Almeida, M.; Belo, D.; da Gama, V.A. New Hybrid Material Exhibiting Room Temperature Spin-Crossover and Ferromagnetic Cluster-Glass Behavior. CrystEngComm 2009, 11, 2160–2168. [Google Scholar] [CrossRef]
- Faulmann, C.; Chahine, J.; Valade, L.; Chastanet, G.; Létard, J.F.; De Caro, D. Photomagnetic Studies of Spin-Crossover-and Photochromic-Based Complexes. Eur. J. Inorg. Chem. 2013, 5, 1058–1067. [Google Scholar] [CrossRef]
- Togo, T.; Amolegbe, S.A.; Yamaguchi, R.; Kuroda-Sowa, T.; Nakaya, M.; Shimayama, K.; Nakamura, M.; Hayami, S. Crystal Structure and Spin-Crossover Behavior of Iron(III) Complex with Nitroprusside. Chem. Lett. 2013, 42, 1542–1544. [Google Scholar] [CrossRef]
- Fukuroi, K.; Takahashi, K.; Mochida, T.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y.; Mori, H. Synergistic Spin Transition between Spin Crossover and Spin-Peierls-like Singlet Formation in the Halogen-Bonded Molecular Hybrid System: [Fe(Iqsal)2][Ni(dmit)2]⋅CH3CN⋅H2O. Angew. Chem. Int. Ed. 2014, 53, 1983–1986. [Google Scholar] [CrossRef]
- Vieira, B.J.C.; Dias, J.C.; Santos, I.C.; Pereira, L.C.J.; da Gama, V.; Waerenborgh, J.C. Thermal Hysteresis in a Spin-Crossover FeIII Quinolylsalicylaldimine Complex, FeIII(5-Br-qsal)2Ni(dmit)2·solv: Solvent Effects. Inorg. Chem. 2015, 54, 1354–1362. [Google Scholar] [CrossRef]
- Vieira, B.J.C.; Coutinho, J.T.; Dias, J.C.; Nunes, J.C.; Santos, I.C.; Pereira, L.C.J.; Da Gama, V.; Waerenborgh, J.C. Crystal Structure and Spin Crossover Behavior of the [Fe(5-Cl-Qsal)2][Ni(Dmit)2]·2CH3CN Complex. Polyhedron 2015, 85, 643–651. [Google Scholar] [CrossRef]
- Takahashi, K.; Sakurai, T.; Zhang, W.-M.; Okubo, S.; Ohta, H.; Yamamoto, T.; Einaga, Y.; Mori, H. Spin-Singlet Transition in the Magnetic Hybrid Compound from a Spin-Crossover Fe(III) Cation and π-Radical Anion. Inorganics 2017, 5, 54. [Google Scholar] [CrossRef]
- Phonsri, W.; Davies, C.G.; Jameson, G.N.L.; Moubaraki, B.; Murray, K.S. Spin Crossover, Polymorphism and Porosity to Liquid Solvent in Heteroleptic Iron(III) {Quinolylsalicylaldimine/Thiosemicarbazone-Salicylaldimine} Complexes. Chem. A Eur. J. 2016, 22, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Phonsri, W.; Macedo, D.; Moubaraki, B.; Cashion, J.; Murray, K. Heteroleptic Iron(III) Spin Crossover Complexes; Ligand Substitution Effects. Magnetochemistry 2016, 2, 3. [Google Scholar] [CrossRef]
- Murata, S.; Takahashi, K.; Mochida, T.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y. Cooperative Spin-Crossover Transition from Three-Dimensional Purely π-Stacking Interactions in a Neutral Heteroleptic Azobisphenolate FeIII Complex with a N3O3 Coordination Sphere. Dalton Trans. 2017, 46, 5786–5789. [Google Scholar] [CrossRef] [PubMed]
- Phonsri, W.; Macedo, D.S.; Davies, C.G.; Jameson, G.N.L.; Moubaraki, B.; Murray, K.S. Heteroleptic Iron(III) Schiff Base Spin Crossover Complexes: Halogen Substitution, Solvent Loss and Crystallite Size Effects. Dalton Trans. 2017, 46, 7020–7029. [Google Scholar] [CrossRef] [PubMed]
- Kuroda-Sowa, T.; Yu, Z.; Senzaki, Y.; Sugimoto, K.; Maekawa, M.; Munakata, M.; Hayami, S.; Maeda, Y. Abrupt Spin Transitions and LIESST Effects Observed in FeII Spin-Crossover Complexes with Extended π-Conjugated Schiff-Base Ligands Having N4O2 Donor Sets. Chem. Lett. 2008, 37, 1216–1217. [Google Scholar] [CrossRef]
- Yu, Z.; Kuroda-Sowa, T.; Kume, H.; Okubo, T.; Maekawa, M.; Munakata, M. Effects of Metal Doping on the Spin-Crossover Properties of an Iron(II) Complex with Extended π-Conjugated Schiff-Base Ligand Having an N4O2 Donor Set. Bull. Chem. Soc. Jpn. 2009, 82, 333–337. [Google Scholar] [CrossRef]
- Kuroda-Sowa, T.; Kimura, K.; Kawasaki, J.; Okubo, T.; Maekawa, M. Effects of Weak Interactions on Spin Crossover Properties of Iron(II) Complexes with Extended π-Conjugated Schiff-Base Ligands. Polyhedron 2011, 30, 3189–3192. [Google Scholar] [CrossRef]
- Iasco, O.; Rivière, E.; Guillot, R.; Buron-Le Cointe, M.; Meunier, J.-F.; Bousseksou, A.; Boillot, M.-L. FeII(pap-5NO2)2 and FeII(qsal-5NO2)2 Schiff-Base Spin-Crossover Complexes: A Rare Example with Photomagnetism and Room-Temperature Bistability. Inorg. Chem. 2015, 54, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Phonsri, W.; Macedo, D.S.; Vignesh, K.R.; Rajaraman, G.; Davies, C.G.; Jameson, G.N.L.; Moubaraki, B.; Ward, J.S.; Kruger, P.E.; Chastanet, G.; et al. Halogen Substitution Effects on N2O Schiff Base Ligands in Unprecedented Abrupt FeII Spin Crossover Complexes. Chem. A Eur. J. 2017, 23, 7052–7065. [Google Scholar] [CrossRef] [PubMed]
- Kuroda-Sowa, T.; Isobe, R.; Yamao, N.; Fukumasu, T.; Okubo, T.; Maekawa, M. Variety of Spin Transition Temperatures of Iron(II) Spin Crossover Complexes with Halogen Substituted Schiff-Base Ligands, HqsalX(X = F, Cl, Br, and I). Polyhedron 2017, 136, 74–78. [Google Scholar] [CrossRef]
- Atzori, M.; Poggini, L.; Squillantini, L.; Cortigiani, B.; Gonidec, M.; Bencok, P.; Sessoli, R.; Mannini, M. Thermal and Light-Induced Spin Transition in a Nanometric Film of a New High-Vacuum Processable Spin Crossover Complex. J. Mater. Chem. C 2018, 6, 8885–8889. [Google Scholar] [CrossRef]
- König, E. Landolt-Börnstein Neue Serie Gruppe II, Vol. 2; Hellwege, K.-H., Hellwege, A.M., Eds.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1966; pp. 16–18. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Wachters, A.J.H. Gaussian Basis Set for Molecular Wavefunctions Containing Third-Row Atoms. J. Chem. Phys. 1970, 52, 1033–1036. [Google Scholar] [CrossRef]
- Hay, P.J. Gaussian Basis Sets for Molecular Calculations. The Representation of 3d Orbitals in Transition-Metal Atoms. J. Chem. Phys. 1977, 66, 4377–4384. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Halcrow, M.A. Iron(II) Complexes of 2,6-di(pyrazol-1-yl)pyridines—A Versatile System for Spin-Crossover Research. Coord. Chem. Rev. 2009, 253, 2493–2514. [Google Scholar] [CrossRef]
- Takahashi, K.; Kawamukai, K.; Okai, M.; Mochida, T.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y.; Shiota, Y.; Yoshizawa, K. A new family of anionic FeIII spin crossover complexes featuring a weak-field N2O4 coordination octahedron. Chem. Eur. J. 2016, 22, 1253–1257. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Murata, S.; Takahashi, K.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y.; Shiota, Y.; Yoshizawa, K. The Role of Coulomb Interactions for Spin Crossover Behaviors and Crystal Structural Transformation in Novel Anionic Fe(III) Complexes from a π-Extended ONO Ligand. Crystals 2016, 6, 49. [Google Scholar] [CrossRef]
- Takahashi, K.; Okai, M.; Mochida, T.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y.; Shiota, Y.; Yoshizawa, K.; Konaka, H.; et al. Contribution of Coulomb Interactions to a Two-Step Crystal Structure Phase Transformation Coupled with a Significant Change in Spin Crossover Behavior for a Series of Charged FeII Complexes from 2,6-bis(2-methylthiazol-4-yl)pyridine. Inorg. Chem. 2018, 57, 1277–1287. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).