New Directions in Metal Phosphonate and Phosphinate Chemistry

 , ,
, ,  , , ,
, , ,  , , and
, , and
Abstract
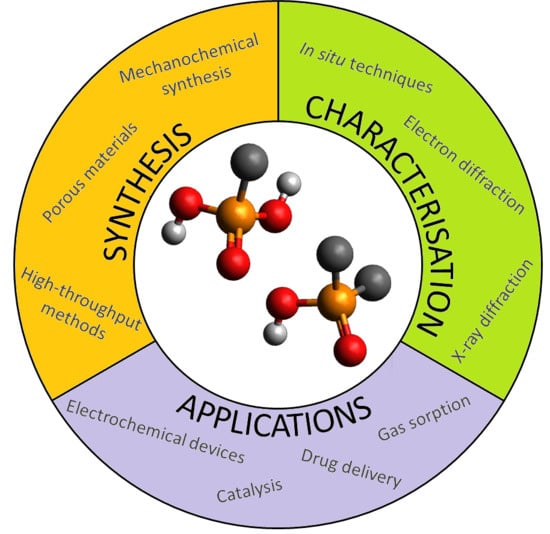
:
1. Introduction
2. Historical Landmarks
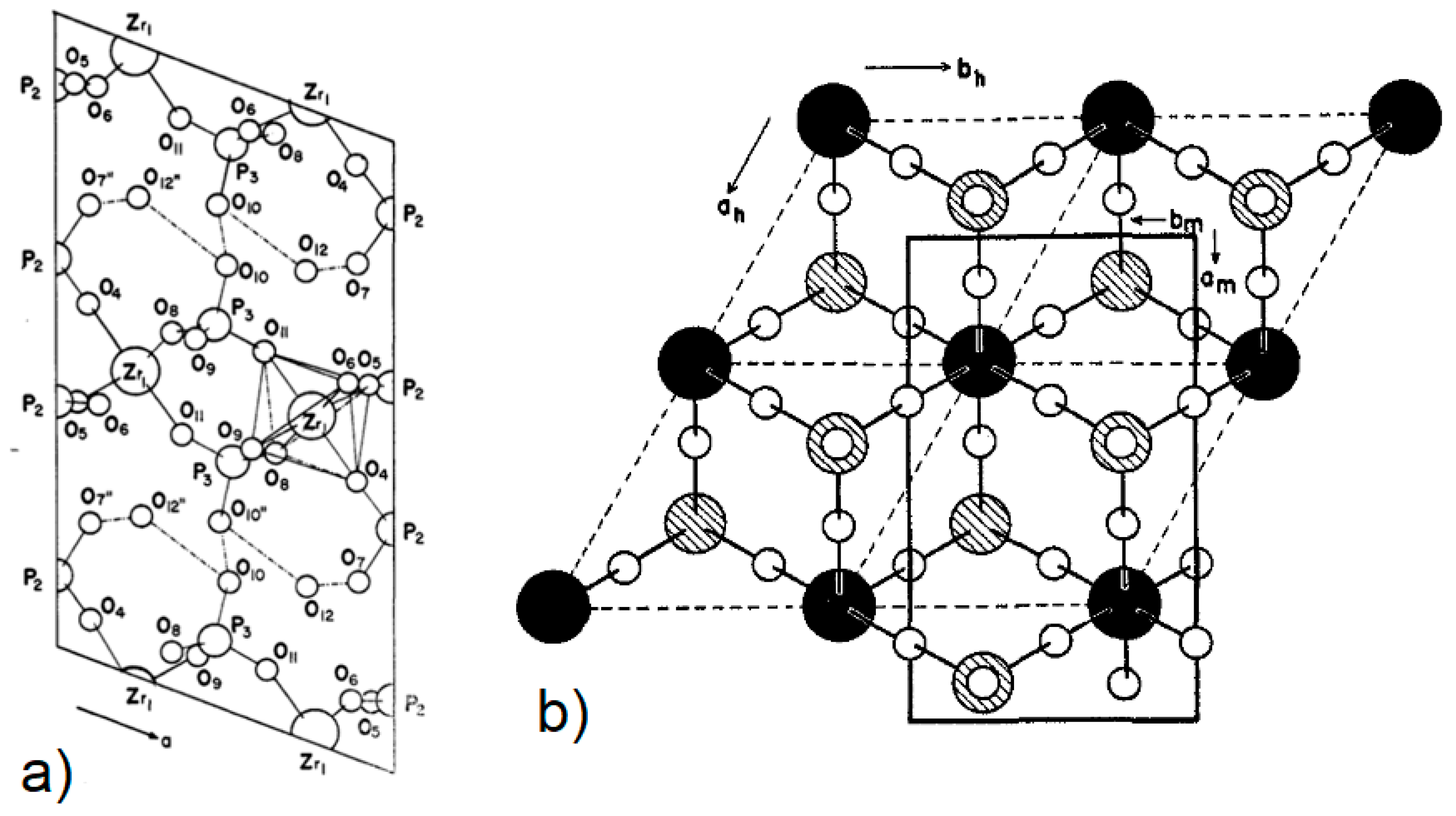
3. Synthesis and Characterisation of Metal Phosphonates
3.1. High-Throughput Methods
3.2. Mechanochemical Synthesis
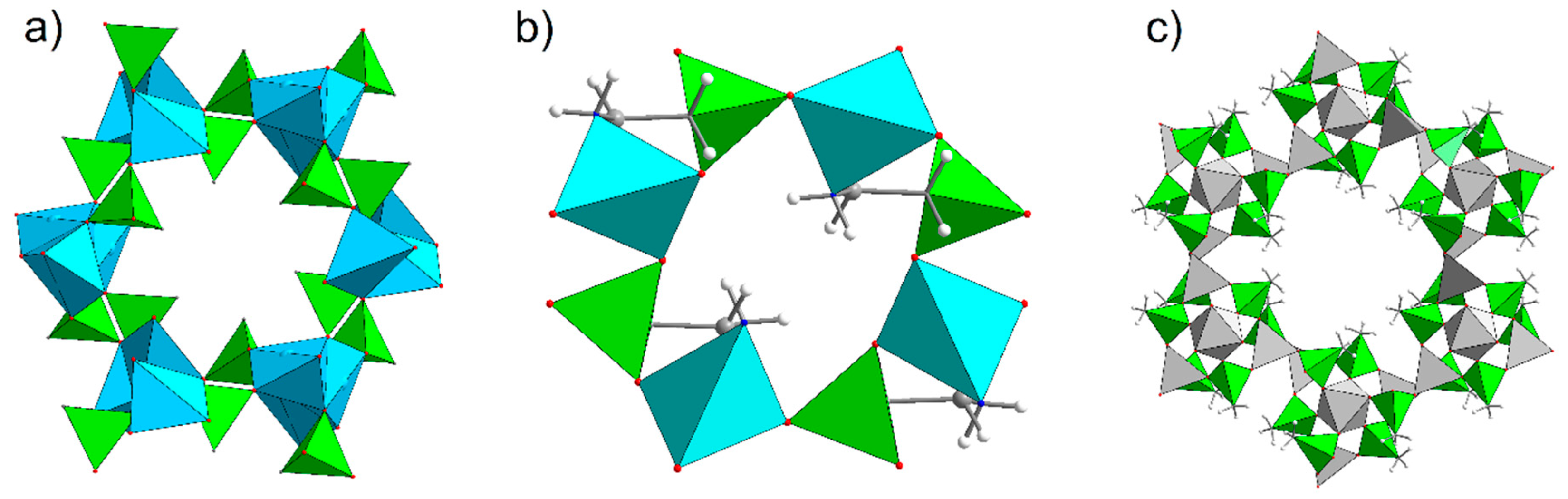
3.3. Porous Materials
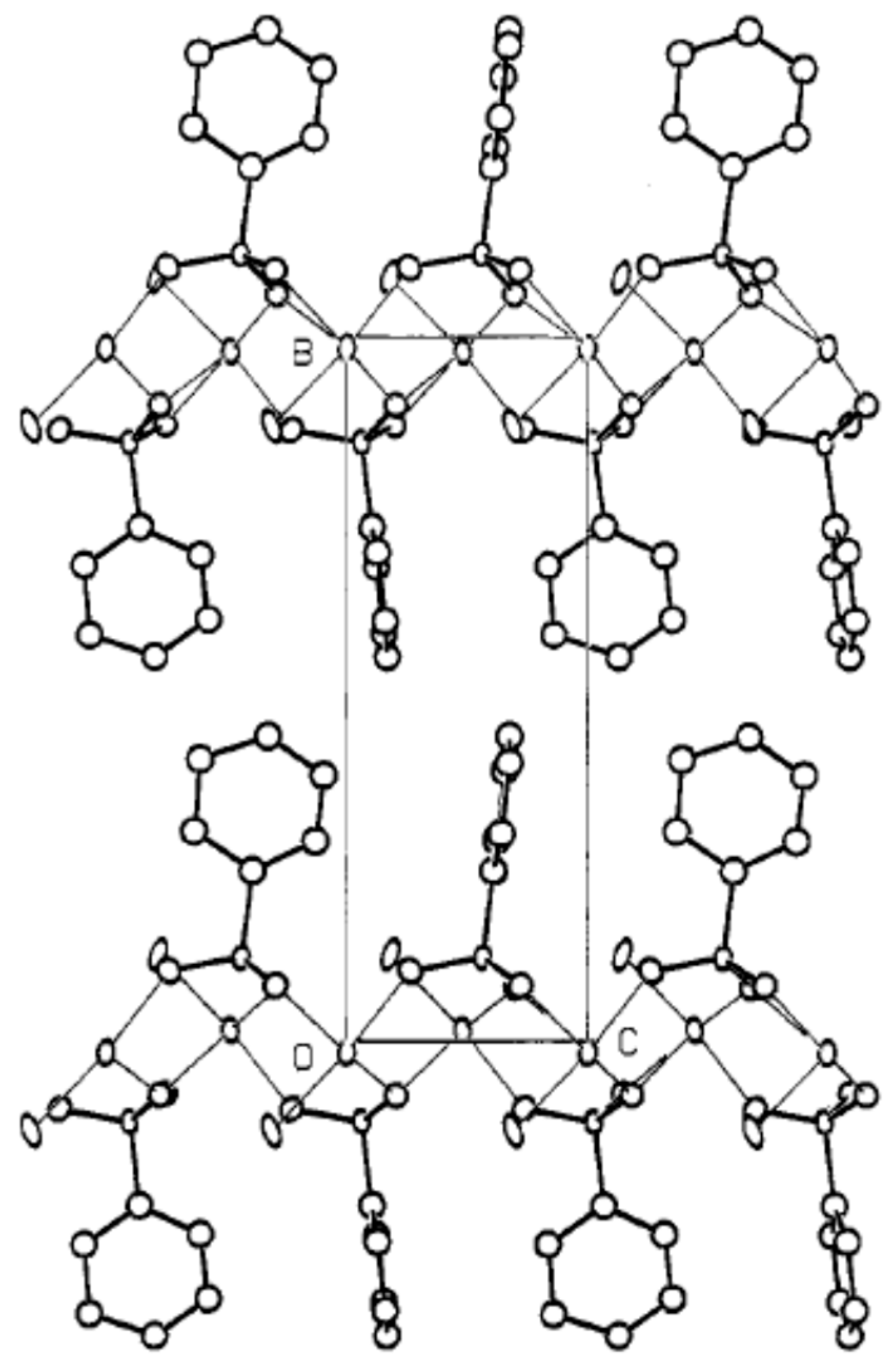
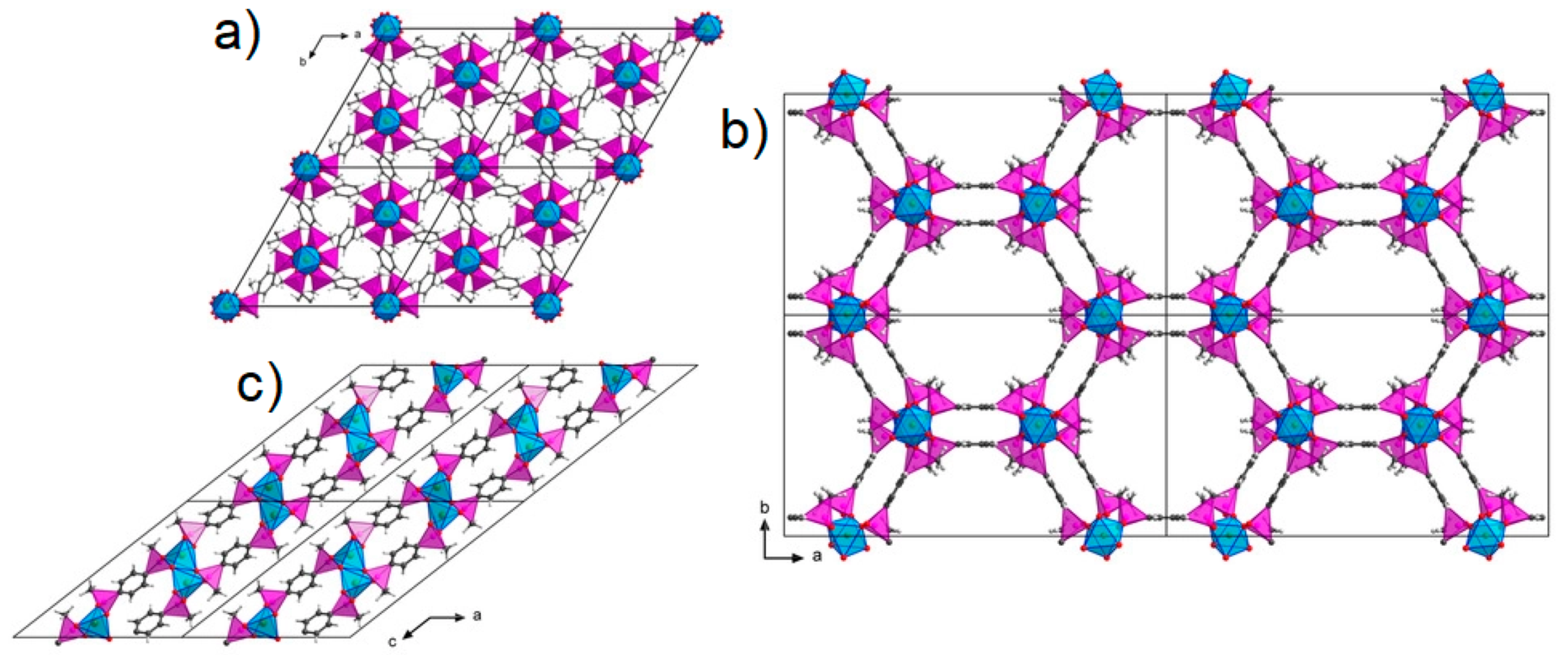
3.4. Phosphinic Acid-Based Materials
3.5. Structure Determination by Electron Diffraction
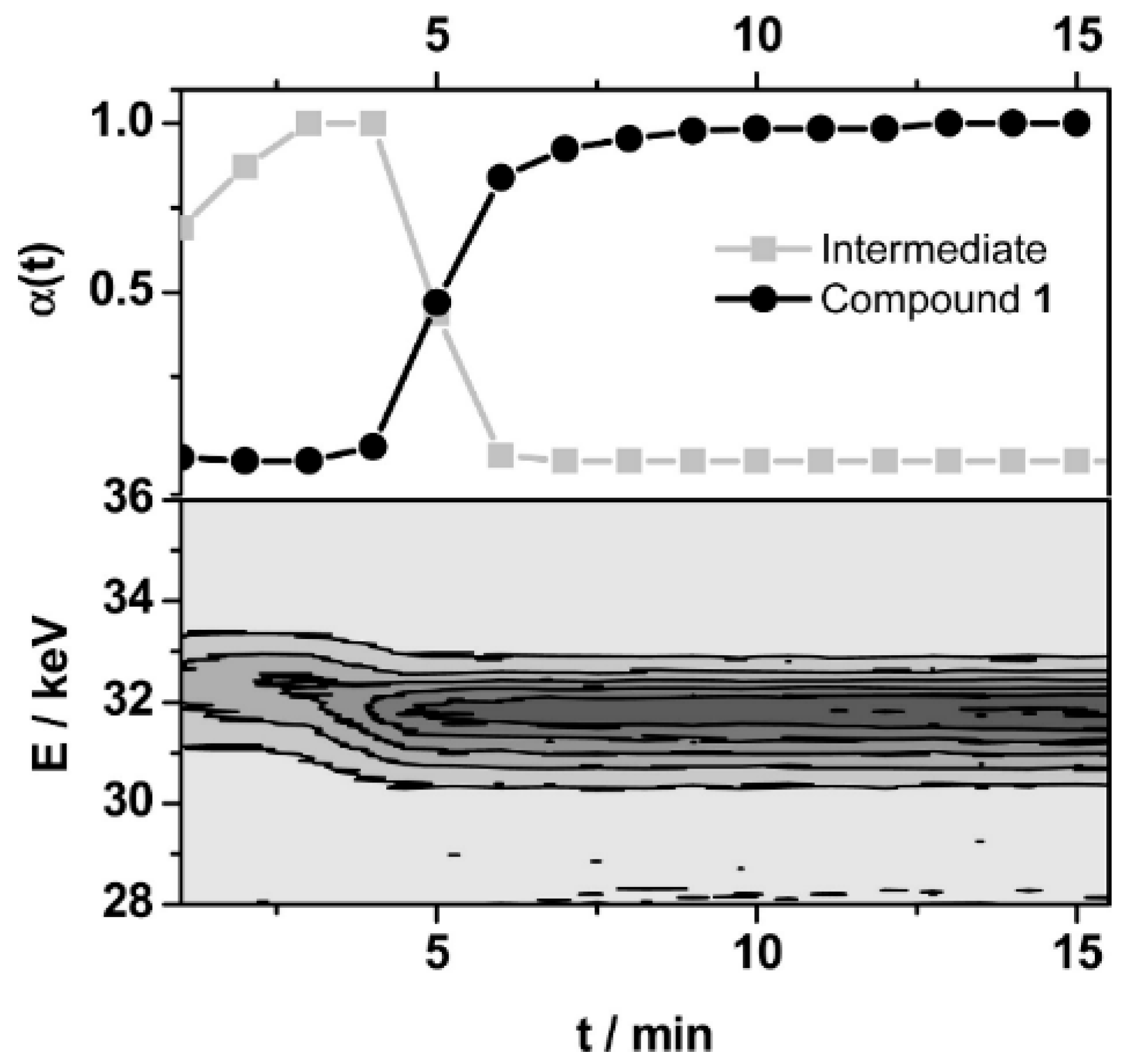
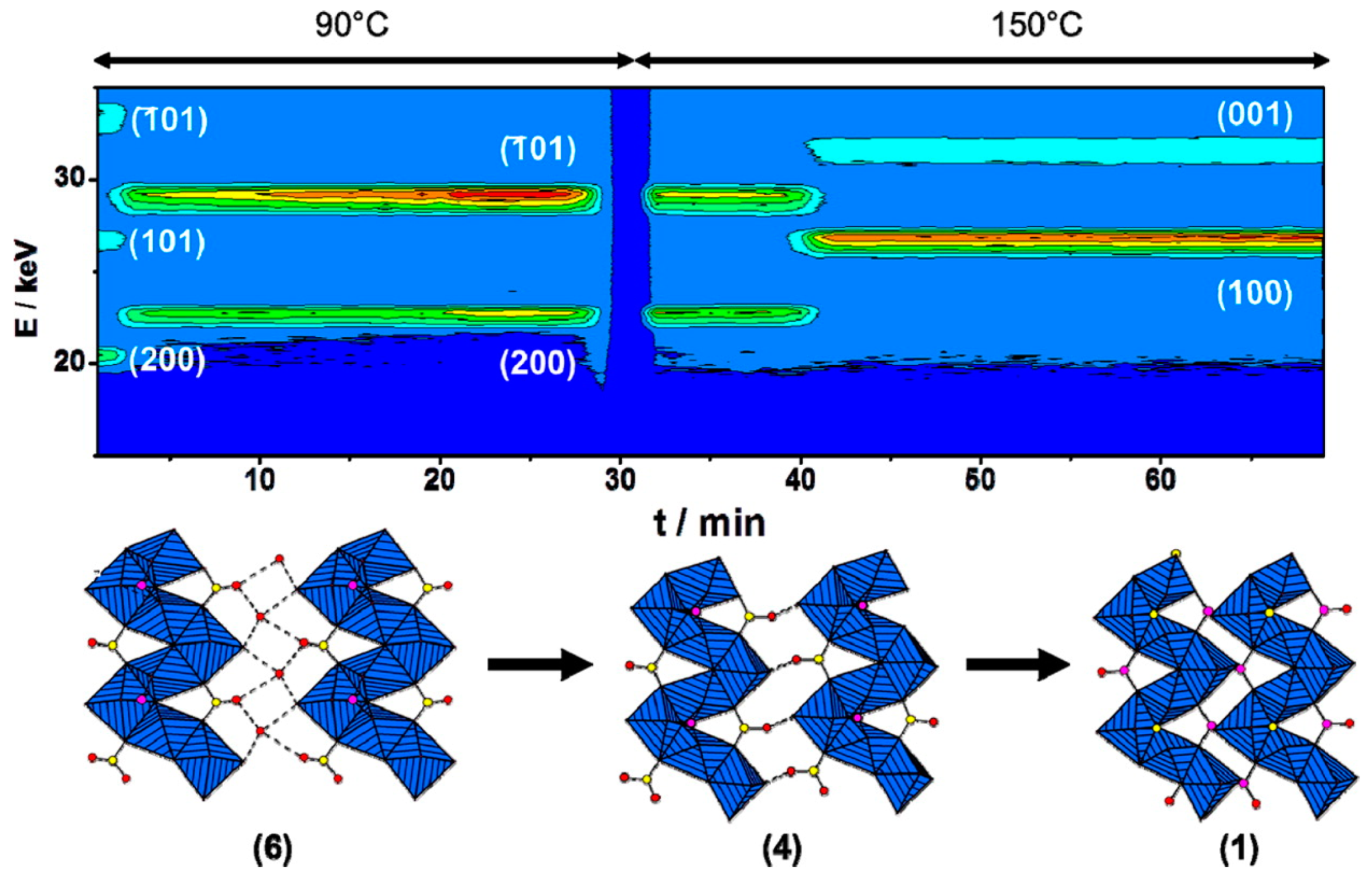
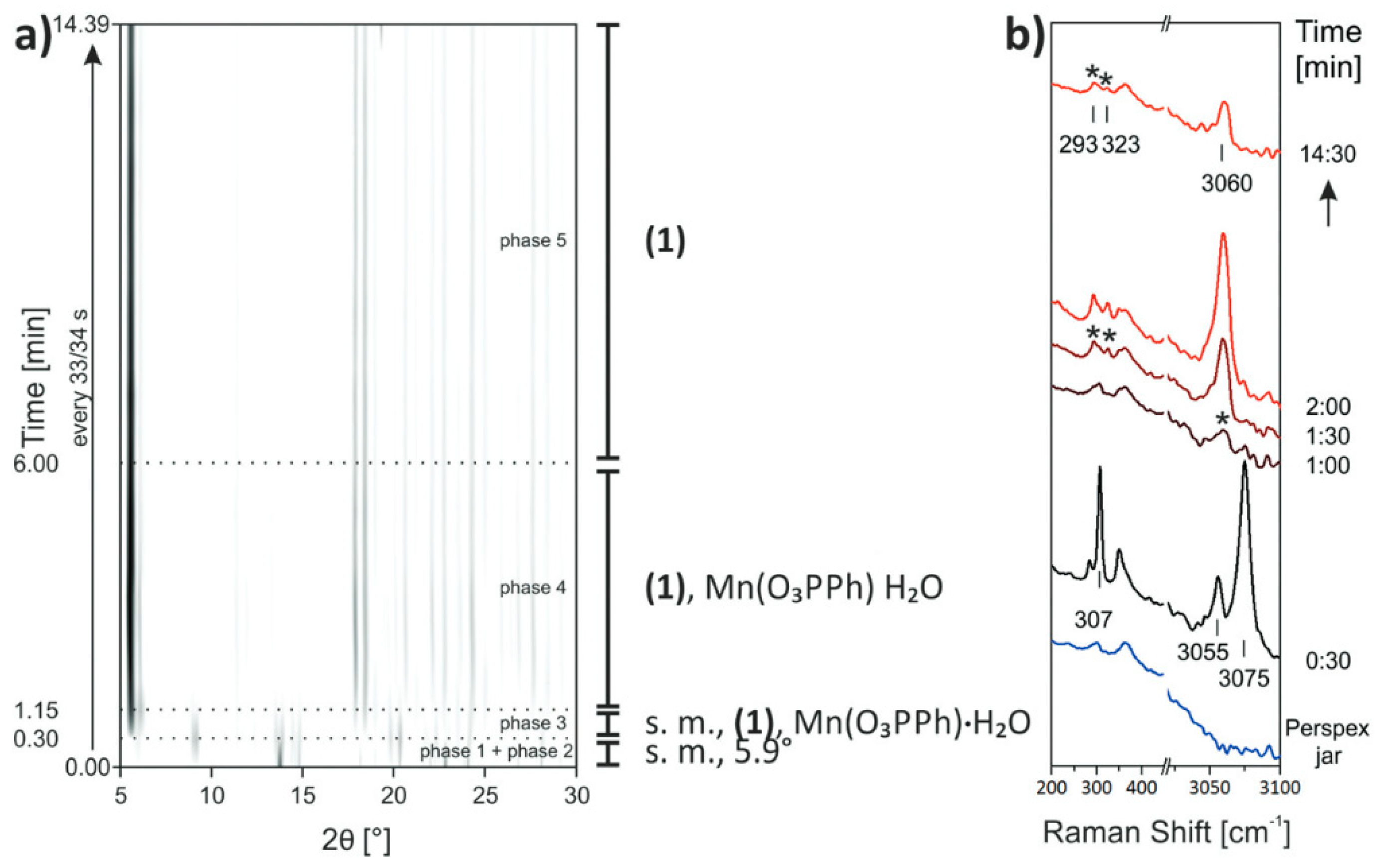
3.6. In Situ Characterisation
4. Applications of Metal Phosphonates
4.1. Catalysis
4.2. Gas Sorption and Separation
4.3. Electrochemical Devices
4.3.1. Solid-State Proton Conductors for Fuel Cells
4.3.2. Electrodes for Rechargeable Batteries
4.4. Drug Delivery
5. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Clearfield, A.; Demadis, K. (Eds.) Metal Phosphonate Chemistry; Royal Society of Chemistry: Cambridge, UK, 2011; ISBN 978-1-84973-356-4. [Google Scholar]
- Clearfield, A. Layered Phosphates, Phosphites and Phosphonates of Groups 4 and 14 Metals. Comments Inorg. Chem. 1990, 10, 89–128. [Google Scholar] [CrossRef]
- Cao, G.; Mallouk, T.E. Shape-Selective Intercalation Reactions of Layered Zinc and Cobalt Phosphonates. Inorg. Chem. 1991, 30, 1434–1438. [Google Scholar] [CrossRef]
- Zhang, Y.; Clearfield, A. Synthesis, Crystal Structures, and Coordination Intercalation Behavior of Two Copper Phosphonates. Inorg. Chem. 1992, 25, 2821–2826. [Google Scholar] [CrossRef]
- Zhang, B.; Poojary, D.M.; Clearfield, A.; Peng, G. Synthesis, Characterization, and Amine Intercalation Behavior of Zirconium N-(Phosphonomethyl)iminodiacetic Acid Layered Compounds. Chem. Mater. 2002, 8, 1333–1340. [Google Scholar] [CrossRef]
- Bao, S.S.; Shimizu, G.K.H.; Zheng, L.M. Proton conductive metal phosphonate frameworks. Coord. Chem. Rev. 2019, 378, 577–594. [Google Scholar] [CrossRef]
- Curini, M.; Rosati, O.; Costantino, U. Heterogeneous Catalysis in Liquid Phase Organic Synthesis, Promoted by Layered Zirconium Phosphates and Phosphonates. Curr. Org. Chem. 2005, 8, 591–606. [Google Scholar] [CrossRef]
- Zhu, Y.P.; Ma, T.Y.; Liu, Y.L.; Ren, T.Z.; Yuan, Z.Y. Metal phosphonate hybrid materials: From densely layered to hierarchically nanoporous structures. Inorg. Chem. Front. 2014, 1, 360–383. [Google Scholar] [CrossRef]
- Goura, J.; Chandrasekhar, V. Molecular Metal Phosphonates. Chem. Rev. 2015, 115, 6854–6965. [Google Scholar] [CrossRef]
- Vivani, R.; Alberti, G.; Costantino, F.; Nocchetti, M. New advances in zirconium phosphate and phosphonate chemistry: Structural archetypes. Microporous Mesoporous Mater. 2008, 107, 58–70. [Google Scholar] [CrossRef]
- Bao, S.S.; Zheng, L.M. Magnetic materials based on 3D metal phosphonates. Coord. Chem. Rev. 2016, 319, 63–85. [Google Scholar] [CrossRef]
- Shimizu, G.K.H.; Vaidhyanathan, R.; Taylor, J.M. Phosphonate and sulfonate metal organic frameworks. Chem. Soc. Rev. 2009, 38, 1430–1449. [Google Scholar] [CrossRef]
- Gagnon, K.J.; Perry, H.P.; Clearfield, A. Conventional and unconventional metal-organic frameworks based on phosphonate ligands: MOFs and UMOFs. Chem. Rev. 2012, 112, 1034–1054. [Google Scholar] [CrossRef]
- Yücesan, G.; Zorlu, Y.; Stricker, M.; Beckmann, J. Metal-organic solids derived from arylphosphonic acids. Coord. Chem. Rev. 2018, 369, 105–122. [Google Scholar] [CrossRef]
- Clearfield, A.; David Smith, G. The Crystallography and Structure of α-zirconium bis(monohydrogen orthophosphate) monohydrate. Inorg. Chem. 1969, 8, 431–436. [Google Scholar] [CrossRef]
- Alberti, G.; Costantino, U.; Allulli, S.; Tomassini, N. Crystalline Zr(R-PO3)2and Zr(R-OPO3)2compounds (R = organic radical). A new class of materials having layered structure of the zirconium phosphate type. J. Inorg. Nucl. Chem. 1978, 40, 1113–1117. [Google Scholar] [CrossRef]
- Alberti, G.; Casciola, M.; Costantino, U.; Vivani, R. Layered and pillared metal(IV) phosphates and phosphonates. Adv. Mater. 1996, 8, 291–303. [Google Scholar] [CrossRef]
- Vivani, R.; Costantino, F.; Taddei, M. Zirconium Phosphonates. In Metal Phosphonate Chemistry; Clearfield, A., Demadis, K.D., Eds.; Royal Society of Chemistry: Cambridge, UK, 2011; pp. 45–86. [Google Scholar]
- Cunningham, D.; Hennelly, P.J.D.; Deeney, T. Divalent metal phenylphosphonates and phenylarsonates. Inorg. Chim. Acta 1979, 37, 95–102. [Google Scholar] [CrossRef]
- Cao, G.; Lee, H.; Lynch, V.M.; Mallouk, T.E. Structural Studies of some New Lamellar Magnesium, Manganese and Calcium Phosphonates. Solid State Ion. 1988, 26, 63–69. [Google Scholar] [CrossRef]
- Cao, G.; Lee, H.; Lynch, V.M.; Mallouk, T.E. Synthesis and Structural Characterization of a Homologous Series of Divalent-Metal Phosphonates, MII(O3PR)·H2O and MII(HO3PR)2. Inorg. Chem. 1988, 27, 2781–2785. [Google Scholar] [CrossRef]
- Martin, K.J.; Squattrito, P.J.; Clearfield, A. The crystal and molecular structure of Zinc Phenylphosphonate. Carbohydr. Res. 1989, 194, 7–9. [Google Scholar] [CrossRef]
- Poojary, M.D.; Hu, H.L.; Campbell, F.L.; Clearfield, A. Determination of crystal structures from limited powder data sets: Crystal structure of zirconium phenylphosphonate. Acta Crystallogr. Sect. B 1993, 49, 996–1001. [Google Scholar] [CrossRef]
- Maeda, K.; Kiyozumi, Y.; Mizukami, F. Synthesis of the First Microporous Aluminum Phosphonate with Organic Groups Covalently Bonded to the Skeleton. Angew. Chem. Int. Ed. 1994, 33, 2335–2337. [Google Scholar] [CrossRef]
- Le Bideau, J.; Payen, C.; Palvadeau, P.; Bujoli, B. Preparation, Structure, and Magnetic Properties of Copper(II) Phosphonates. β-CuII(CH3PO3), an Original Three-Dimensional Structure with a Channel-Type Arrangement. Inorg. Chem. 1994, 33, 4885–4890. [Google Scholar] [CrossRef]
- Maeda, K.; Akimoto, J.; Kiyozumi, Y.; Mizukami, F. AlMepO-α: A Novel Open--Framework Aluminum Methylphosphonate with Organo-Lined Unidimensional Channels. Angew. Chem. Int. Ed. 1995, 34, 1199–1201. [Google Scholar] [CrossRef]
- Drumel, S.; Janvier, P.; Deniaud, D.; Bujoli, B. Synthesis and crystal structure of Zn(O3PC2H4NH2), the first functionalized zeolite-like phosphonate. J. Chem. Soc. Chem. Commun. 1995, 1051–1052. [Google Scholar] [CrossRef]
- Maeda, K.; Akimoto, J.; Kiyozumi, Y.; Mizukami, F. Structure of aluminium methylphosphonate, AlMepO-β, with unidimensional channels formed from ladder-like organic-inorganic polymer chains. J. Chem. Soc. Chem. Commun. 1995, 1033–1034. [Google Scholar] [CrossRef]
- Maeda, K. Metal phosphonate open-framework materials. Microporous Mesoporous Mater. 2004, 73, 47–55. [Google Scholar] [CrossRef]
- Lohse, D.L.; Sevov, S.C. Co2(O3P-CH2-PO3)·H2O: A Novel Microporous Diphosphonate with an Inorganic Framework and Hydrocarbon-Lined Hydrophobic Channels. Angew. Chem. Int. Ed. 1997, 36, 1619–1621. [Google Scholar] [CrossRef]
- Hix, G.B.; Kariuki, B.M.; Kitchin, S.; Tremayne, M. Synthesis and structural characterization of Zn(O3PCH2OH), a new microporous zinc phosphonate. Inorg. Chem. 2001, 40, 1477–1481. [Google Scholar] [CrossRef]
- Mason, M.R. Molecular Phosphates, Phosphonates, Phosphinates, and Arsonates of the Group 13 Elements. J. Clust. Sci. 1998, 9, 1–23. [Google Scholar] [CrossRef]
- Walawalkar, M.G.; Roesky, H.W.; Murugavel, R. Molecular phosphonate cages: Model compounds and starting materials for phosphate materials. Acc. Chem. Res. 1999, 32, 117–126. [Google Scholar] [CrossRef]
- Gopal, K.; Ali, S.; Winpenny, R.E.P. Structural Studies of Paramagnetic Molecular Phosphonates. In Metal Phosphonate Chemistry; Royal Society of Chemistry: Cambridge, UK, 2011; pp. 364–419. [Google Scholar]
- Chandrasekhar, V.; Senapati, T.; Dey, A.; Hossain, S. Molecular transition-metal phosphonates. Dalton Trans. 2011, 40, 5394–5418. [Google Scholar] [CrossRef] [PubMed]
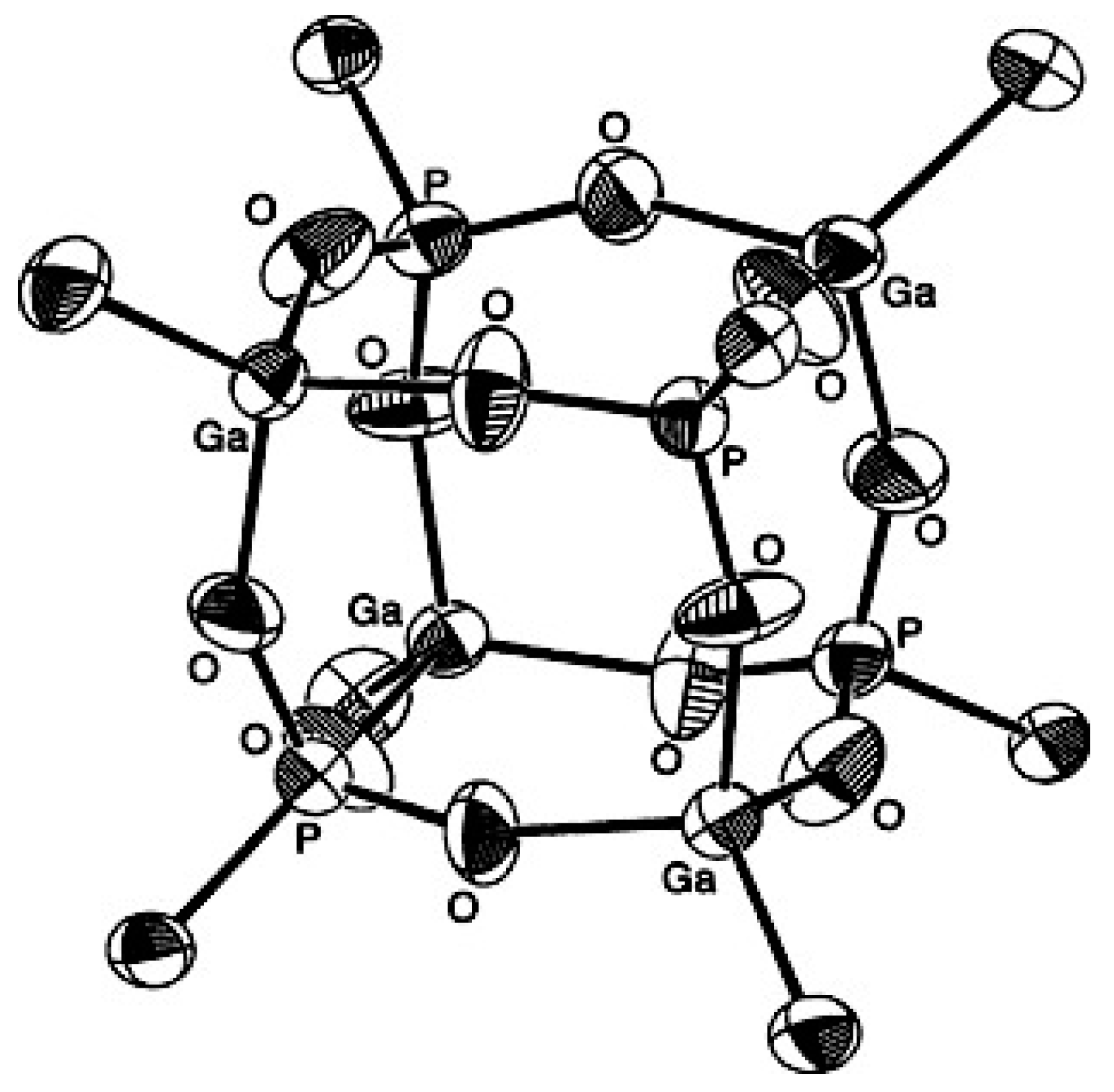
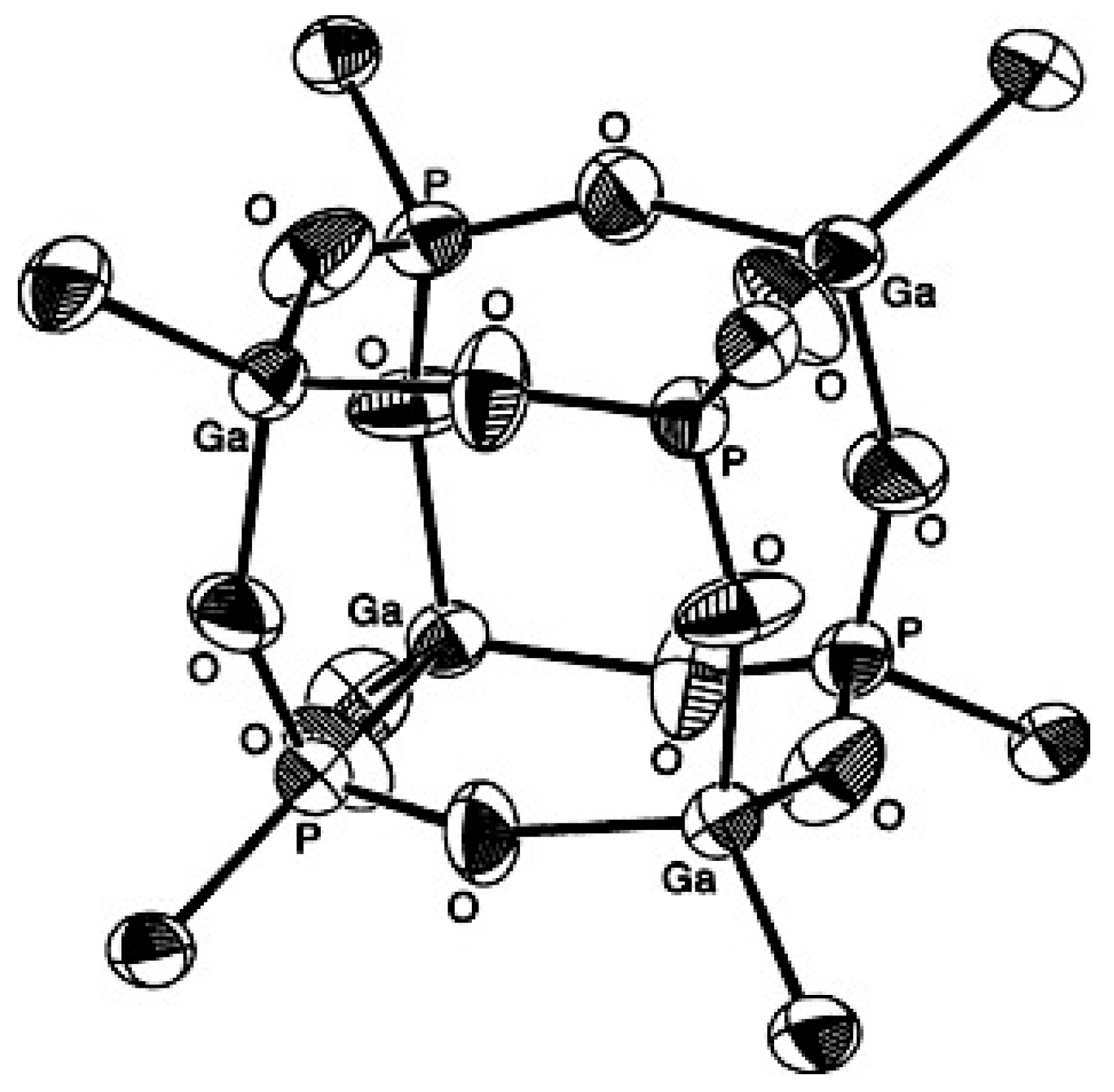
- Mason, M.R.; Mashuta, M.S.; Richardson, J.F. Cyclic and Cubic Organophosphonates of Gallium and Their Relationship to Structural Motifs in Gallophosphate Molecular Sieves. Angew. Chem. Int. Ed. 1997, 36, 239–241. [Google Scholar] [CrossRef]
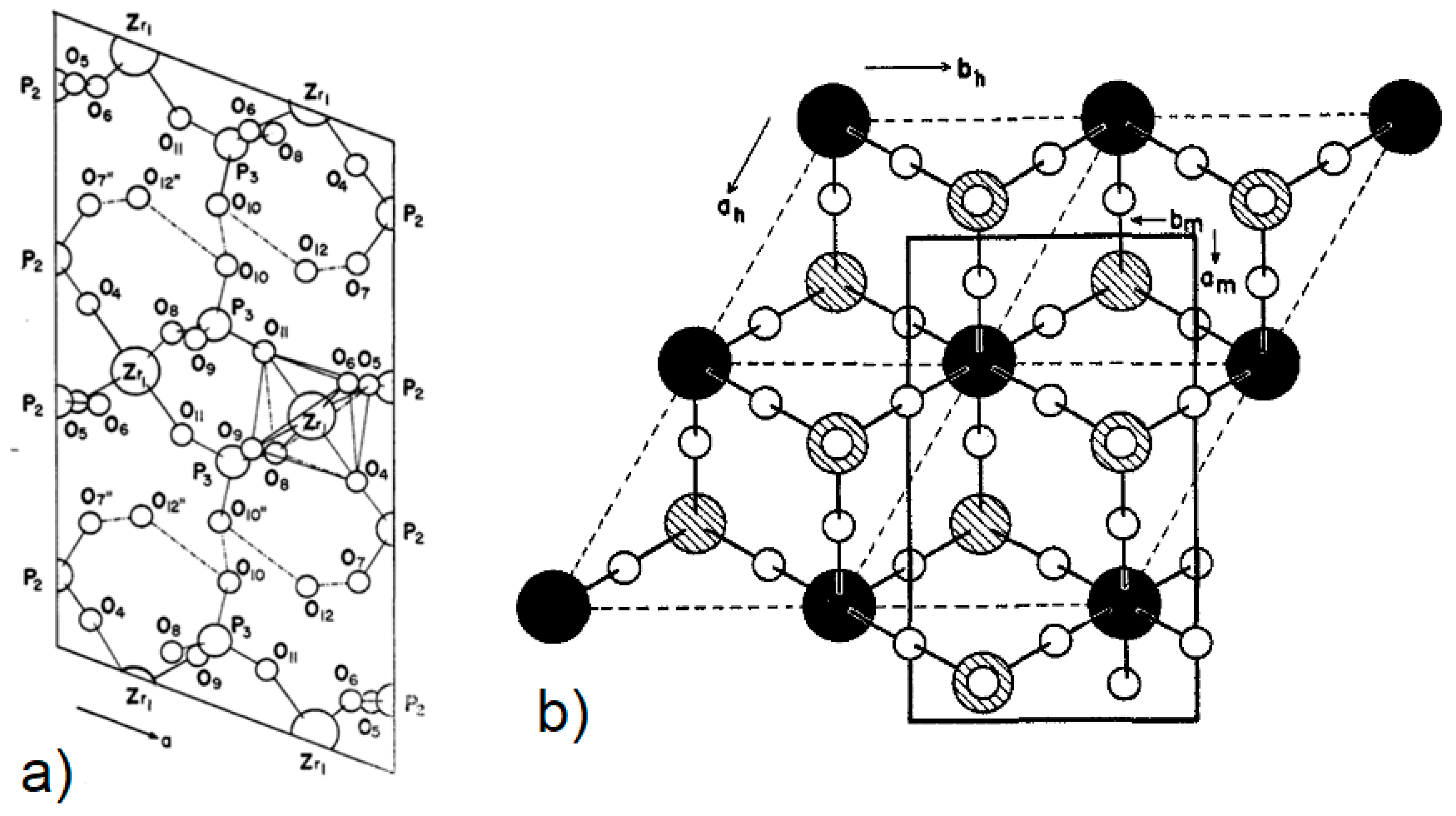
- Clearfield, A. The Early History and Growth of Metal Phosphonate Chemistry. In Metal Phosphonate Chemistry; Royal Society of Chemistry: Cambridge, UK, 2011; pp. 1–44. [Google Scholar]
- Stock, N. High-throughput investigations employing solvothermal syntheses. Microporous Mesoporous Mater. 2010, 129, 287–295. [Google Scholar] [CrossRef]
- Bauer, S.; Stock, N. Implementation of a temperature-gradient reactor system for high-throughput investigation of phosphonate-based inorganic-organic hybrid compounds. Angew. Chem. Int. Ed. 2007, 46, 6857–6860. [Google Scholar] [CrossRef] [PubMed]
- Stock, N.; Bein, T. High-Throughput Synthesis of Phosphonate-Based Inorganic-Organic Hybrid Compounds under Hydrothermal Conditions. Angew. Chem. Int. Ed. 2004, 43, 749–752. [Google Scholar] [CrossRef]
- Bauer, S.; Bein, T.; Stock, N. High-throughput investigation and characterization of cobalt carboxy phosphonates. Inorg. Chem. 2005, 44, 5882–5889. [Google Scholar] [CrossRef]
- Forster, P.M.; Stock, N.; Cheetham, A.K. A high-throughput investigation of the role of pH, temperature, concentration, and time on the synthesis of hybrid inorganic-organic materials. Angew. Chem. Int. Ed. 2005, 44, 7608–7611. [Google Scholar] [CrossRef]
- Stock, N.; Bein, T. High-throughput investigation of metal carboxyarylphosphonate hybrid compounds. J. Mater. Chem. 2005, 15, 1384–1391. [Google Scholar] [CrossRef]
- Maniam, P.; Stock, N. High-throughput Methods for the Systematic Investigation of Metal Phosphonate Synthesis Fields. In Metal Phosphonate Chemistry; Royal Society of Chemistry: Cambridge, UK, 2011; pp. 87–106. [Google Scholar]
- Hermer, N.; Stock, N. The new triazine-based porous copper phosphonate [Cu3(PPT)(H2O)3]·10H2O. Dalton Trans. 2015, 44, 3720–3723. [Google Scholar] [CrossRef]
- Rhauderwiek, T.; Wolkersdörfer, K.; Øien-Ødegaard, S.; Lillerud, K.P.; Wark, M.; Stock, N. Crystalline and permanently porous porphyrin-based metal tetraphosphonates. Chem. Commun. 2018, 54, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Rhauderwiek, T.; Stock, N.; Bueken, B.; Döblinger, M.; Wuttke, S.; Zhao, H.; Reinsch, H.; Hirschle, P.; De Vos, D.; Kolb, U. Highly stable and porous porphyrin-based zirconium and hafnium phosphonates—Electron crystallography as an important tool for structure elucidation. Chem. Sci. 2018, 9, 5467–5478. [Google Scholar] [CrossRef]
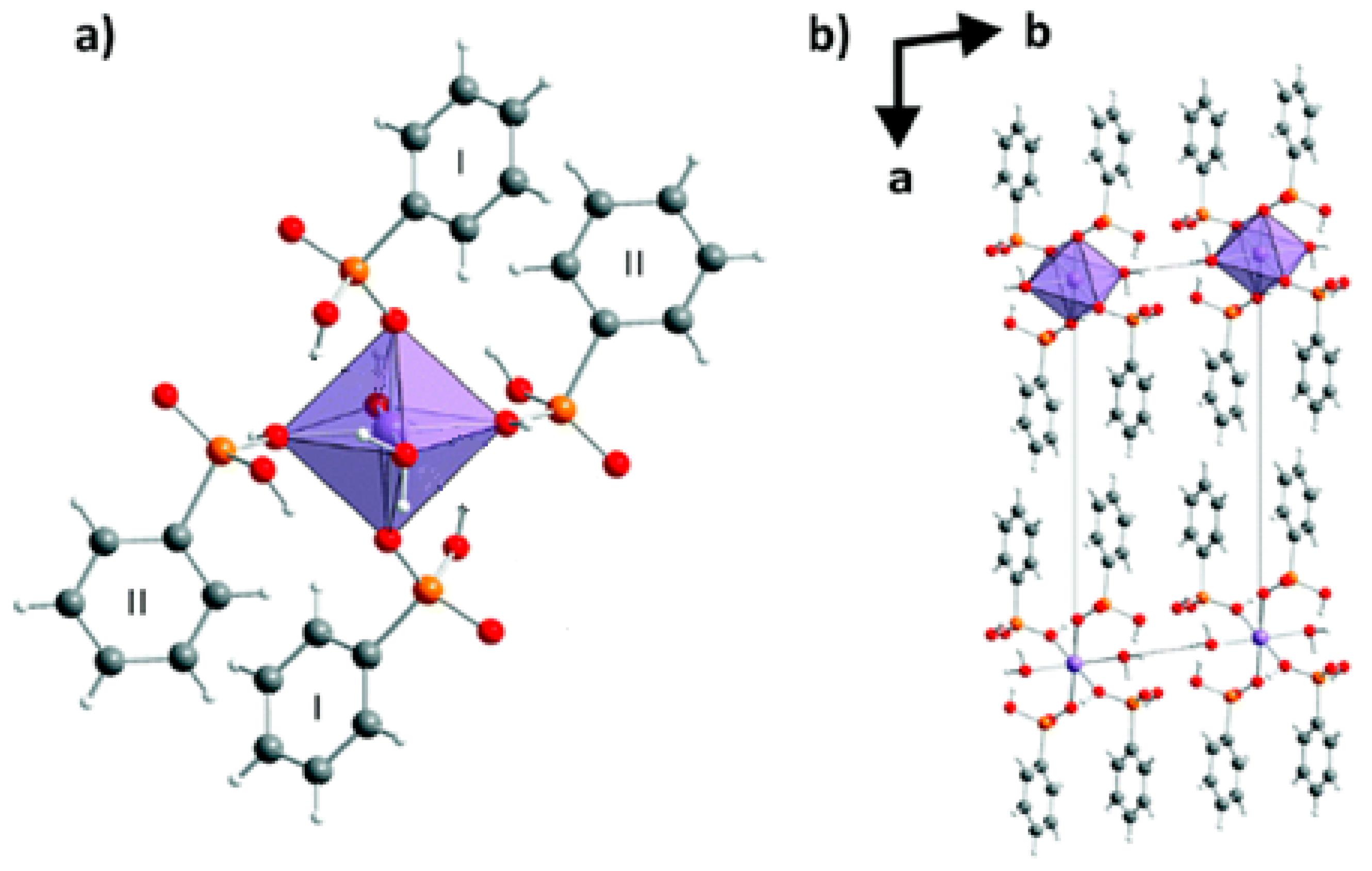
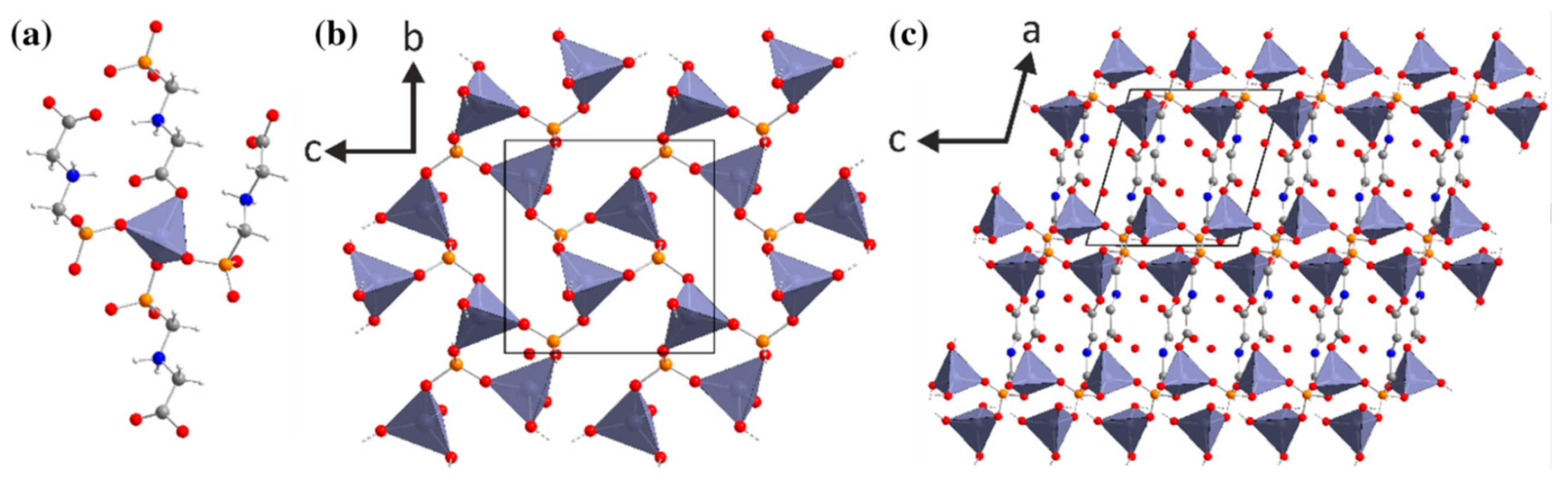
- Stock, N.; Rauscher, M.; Bein, T. Inorganic-organic hybrid compounds: Hydrothermal synthesis and characterization of a new three-dimensional metal tetraphosphonate Mn[(HO3PCH2)N(H)(CH2)4(H)N(CH2PO3H)2]. J. Solid State Chem. 2004, 177, 642–647. [Google Scholar] [CrossRef]
- Hermer, N.; Reinsch, H.; Mayer, P.; Stock, N. Synthesis and characterisation of the porous zinc phosphonate [Zn2(H2PPB)(H2O)2]·xH2O. CrystEngComm 2016, 18, 8147–8150. [Google Scholar] [CrossRef]
- Feyand, M.; Seidler, C.F.; Deiter, C.; Rothkirch, A.; Lieb, A.; Wark, M.; Stock, N. High-throughput microwave-assisted discovery of new metal phosphonates. Dalton Trans. 2013, 42, 8761–8770. [Google Scholar] [CrossRef]
- Schilling, L.H.; Stock, N. High-throughput ultrasonic synthesis and in situ crystallisation investigation of metal phosphonocarboxylates. Dalton Trans. 2014, 43, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, V.; Tkacova, K. Mechanochemistry of Solids: Past, Present, and Prospects. J. Mater. Synth. Process. 2000, 8, 121–122. [Google Scholar] [CrossRef]
- Takacs, L. The historical development of mechanochemistry. Chem. Soc. Rev. 2013, 42, 7649–7659. [Google Scholar] [CrossRef] [PubMed]
- Bruckmann, A.; Krebs, A.; Bolm, C. Organocatalytic reactions: Effects of ball milling, microwave and ultrasound irradiation. Green Chem. 2008, 10, 1131–1141. [Google Scholar] [CrossRef]
- Howard, J.L.; Cao, Q.; Browne, D.L. Mechanochemistry as an emerging tool for molecular synthesis: What can it offer? Chem. Sci. 2018, 9, 3080–3094. [Google Scholar] [CrossRef]
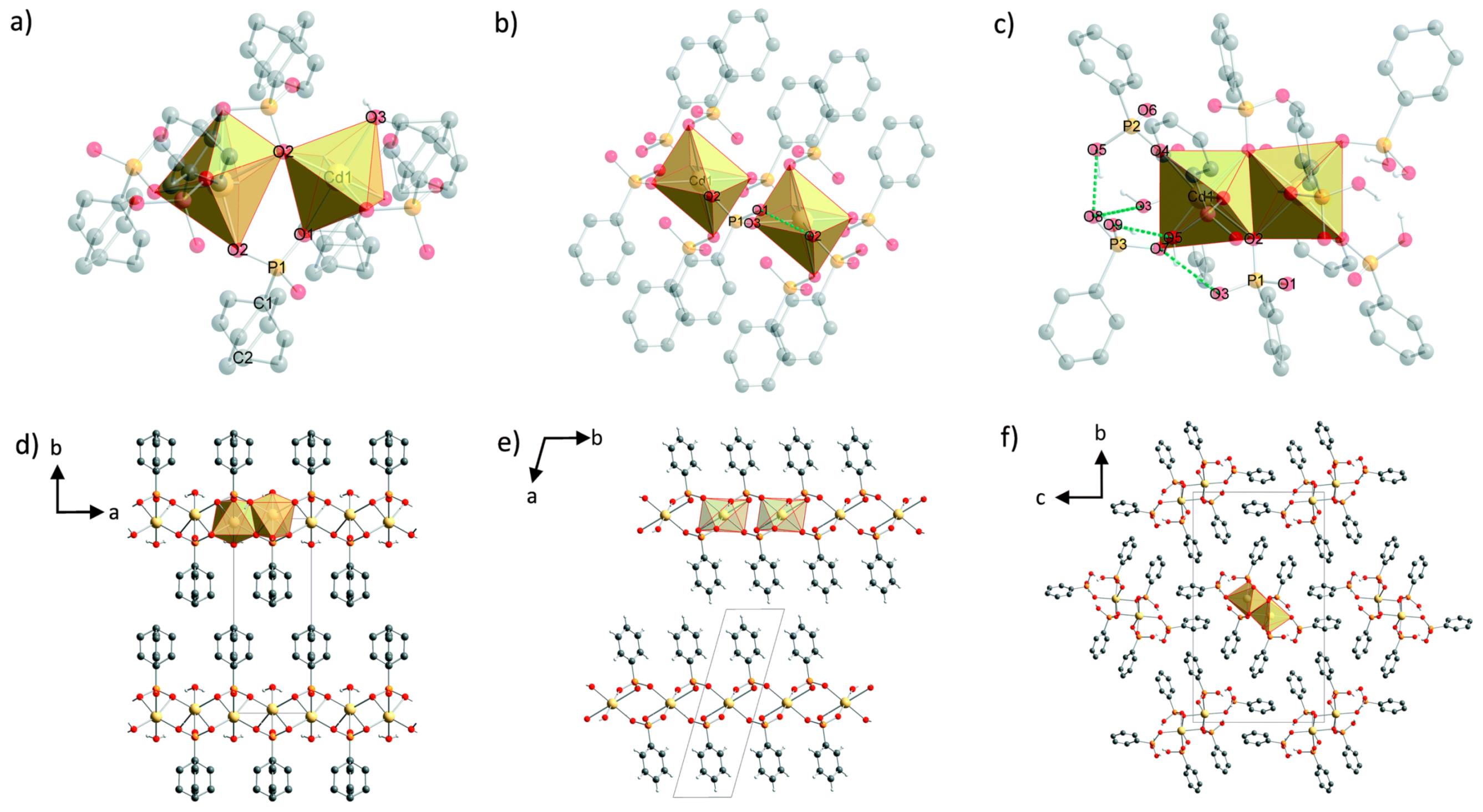
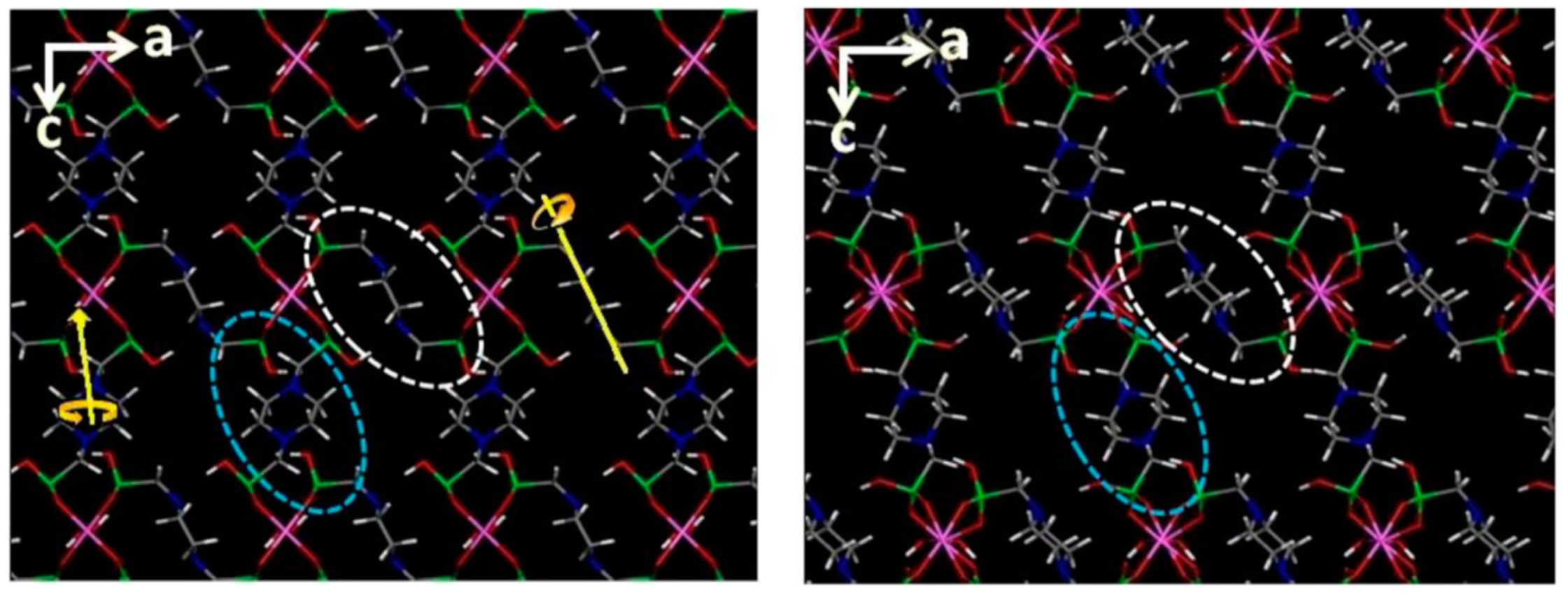
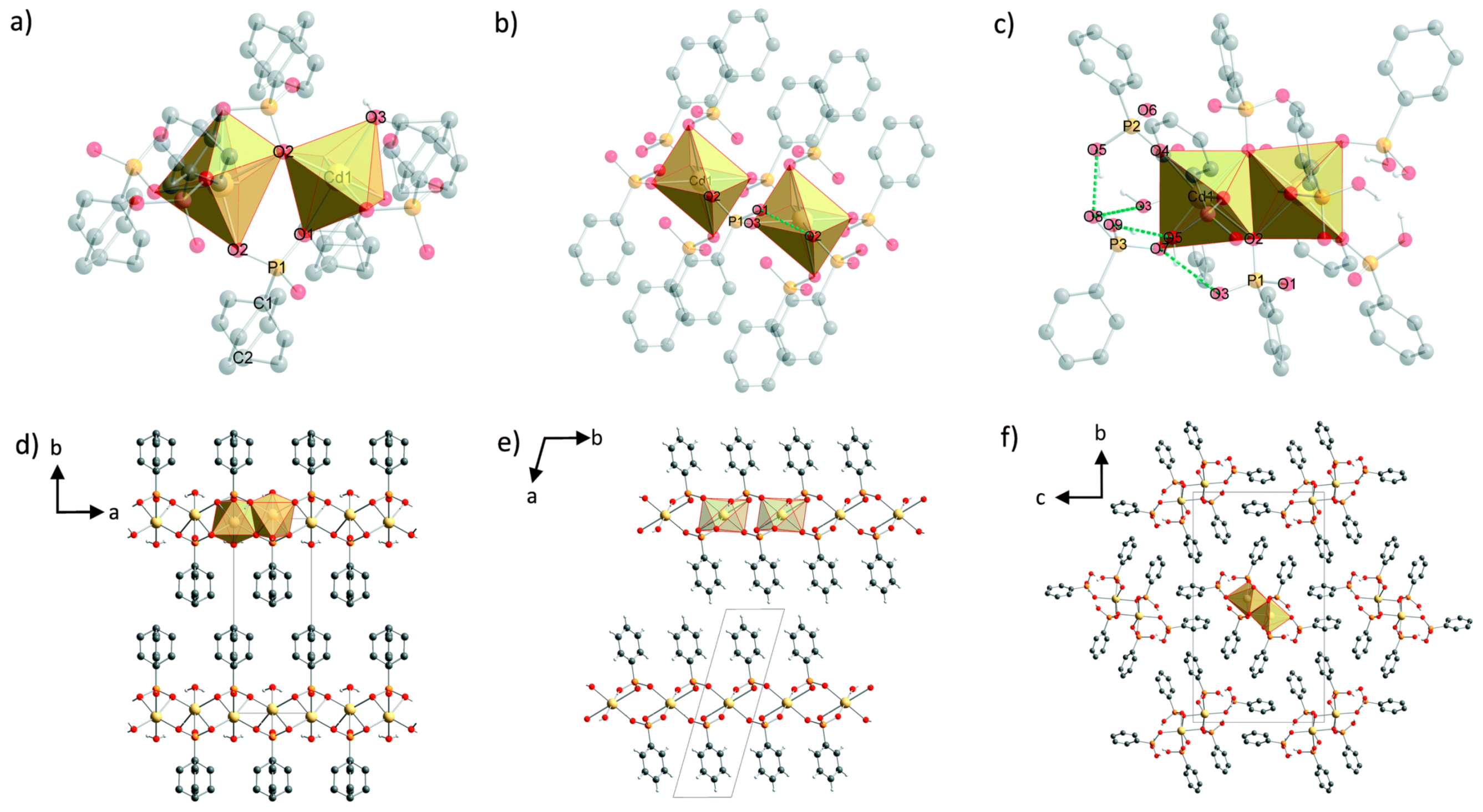
- Wilke, M.; Batzdorf, L.; Fischer, F.; Rademann, K.; Emmerling, F. Cadmium phenylphosphonates: Preparation, characterisation and in situ investigation. RSC Adv. 2016, 6, 36011–36019. [Google Scholar] [CrossRef]
- Wilke, M.; Buzanich, A.G.; Reinholz, U.; Rademann, K.; Emmerling, F. The structure and In Situ synthesis investigation of isomorphic mononuclear molecular metal phenylphosphonates. Dalton Trans. 2016, 45, 9460–9467. [Google Scholar] [CrossRef]
- Wilke, M.; Kabelitz, A.; Zimathies, A.; Rademann, K.; Emmerling, F. Crystal structure and in situ investigation of a mechanochemical synthesized 3D zinc N-(phosphonomethyl)glycinate. J. Mater. Sci. 2017, 52, 12013–12020. [Google Scholar] [CrossRef]
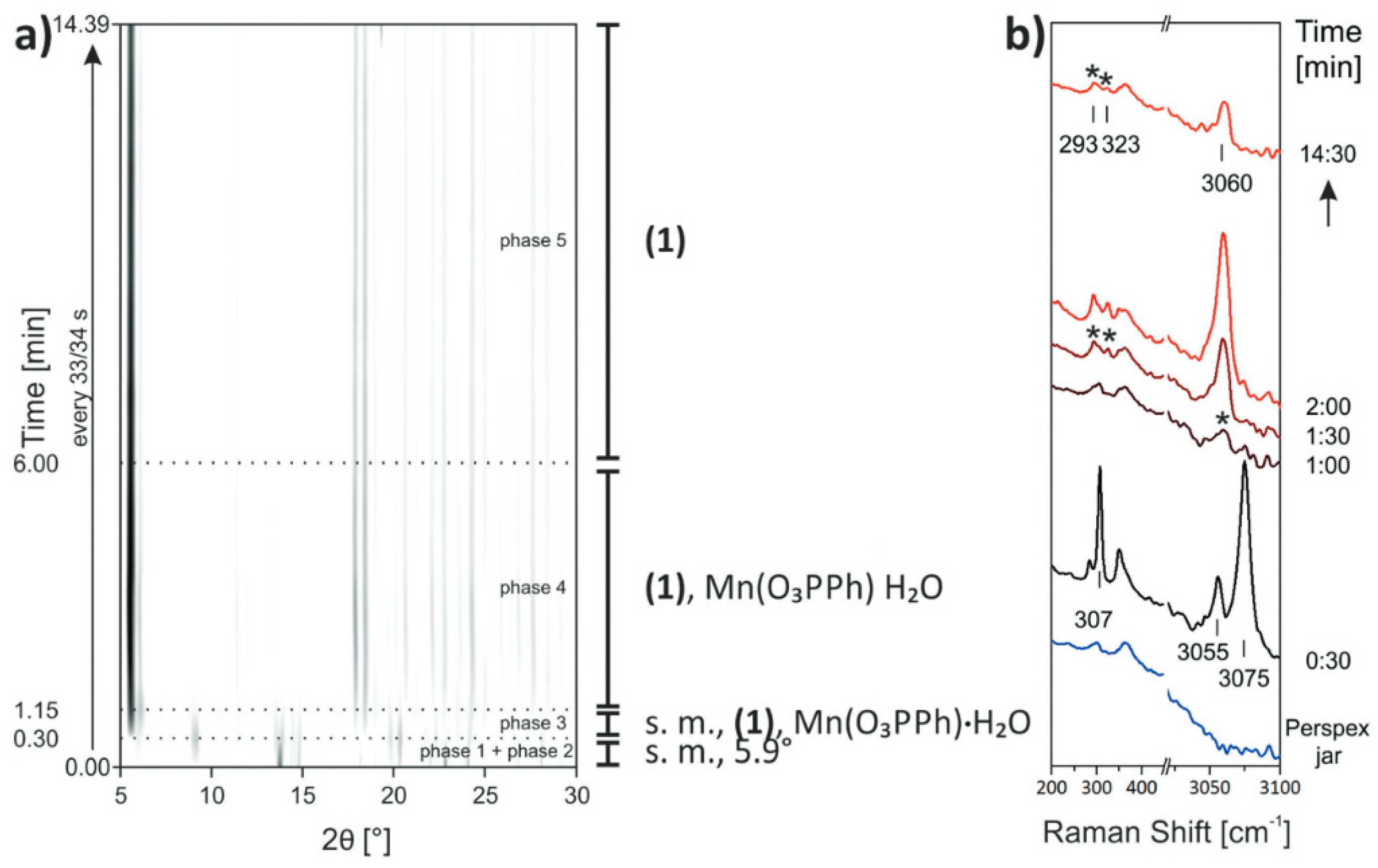
- Akhmetova, I.; Schutjajew, K.; Wilke, M.; Buzanich, A.; Rademann, K.; Roth, C.; Emmerling, F. Synthesis, characterization and in situ monitoring of the mechanochemical reaction process of two manganese(II)-phosphonates with N-containing ligands. J. Mater. Sci. 2018, 53, 13390–13399. [Google Scholar] [CrossRef]
- Maeda, K.; Kiyozumi, Y.; Mizukami, F. Characterization and Gas Adsorption Properties of Aluminum Methylphosphonates with Organically Lined Unidimensional Channels. J. Phys. Chem. B 2002, 101, 4402–4412. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Chui, S.S.Y.; Lo, S.M.F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A chemically functionalizable nanoporous material [Cu3(TMA)2(H2O)3](n). Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef]
- Marsolier, G.; Serre, C.; Thouvenot, C.; Noguès, M.; Louër, D.; Millange, F.; Férey, G. Very Large Breathing Effect in the First Nanoporous Chromium(III)-Based Solids: MIL-53 or CrIII(OH)·{O2C−C6H4−CO2}·{HO2C−C6H4−CO2H}x·H2Oy. J. Am. Chem. Soc. 2002, 124, 13519–13526. [Google Scholar] [CrossRef]
- Long, J.R.; Yaghi, O.M. The pervasive chemistry of metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1213. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-C.; Long, J.R.; Yaghi, O.M. Introduction to Metal–Organic Frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef] [PubMed]
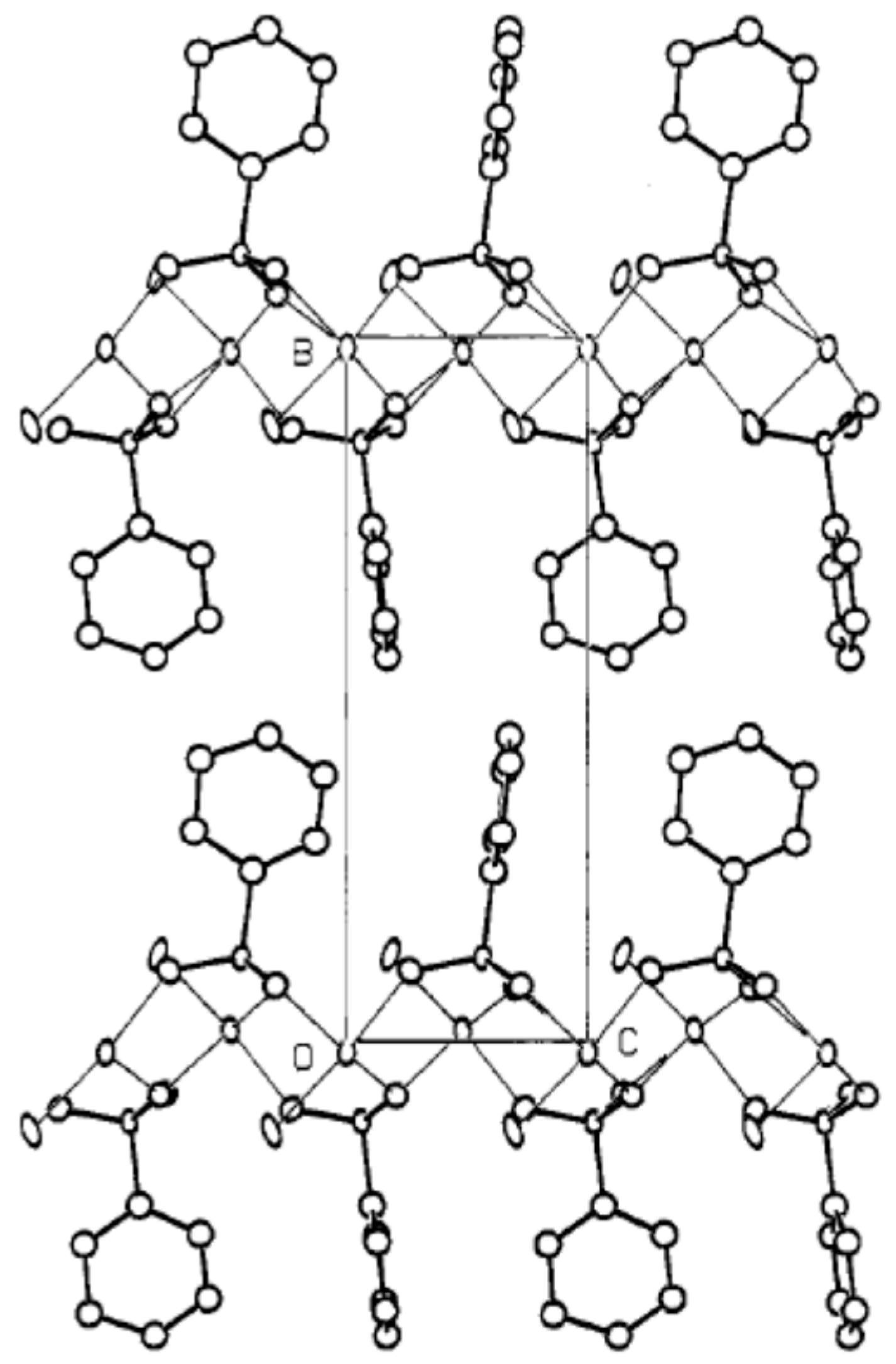
- Rueff, J.M.; Hix, G.B.; Jaffrès, P.A. Rigid Phosphonic Acids as Building Blocks for Crystalline Hybrid Materials. In Tailored Organic-Inorganic Materials; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2015; pp. 341–393. ISBN 9781118792223. [Google Scholar]
- Serre, C.; Groves, J.A.; Lightfoot, P.; Slawin, A.M.Z.; Wright, P.A.; Stock, N.; Bein, T.; Haouas, M.; Taulelle, F.; Férey, G. Synthesis, structure and properties of related microporous N,N′-piperazinebismethylenephosphonates of aluminum and titanium. Chem. Mater. 2006, 18, 1451–1457. [Google Scholar] [CrossRef]
- Benoit, V.; Pillai, R.S.; Orsi, A.; Normand, P.; Jobic, H.; Nouar, F.; Billemont, P.; Bloch, E.; Bourrelly, S.; Devic, T.; et al. MIL-91(Ti), a small pore metal-organic framework which fulfils several criteria: An upscaled green synthesis, excellent water stability, high CO2 selectivity and fast CO2 transport. J. Mater. Chem. A 2016, 4, 1383–1389. [Google Scholar] [CrossRef]
- Llewellyn, P.L.; Garcia-Rates, M.; Gaberová, L.; Miller, S.R.; Devic, T.; Lavalley, J.C.; Bourrelly, S.; Bloch, E.; Filinchuk, Y.; Wright, P.A.; et al. Structural origin of unusual CO2 adsorption behavior of a small-pore aluminum bisphosphonate MOF. J. Phys. Chem. C 2015, 119, 4208–4216. [Google Scholar] [CrossRef]
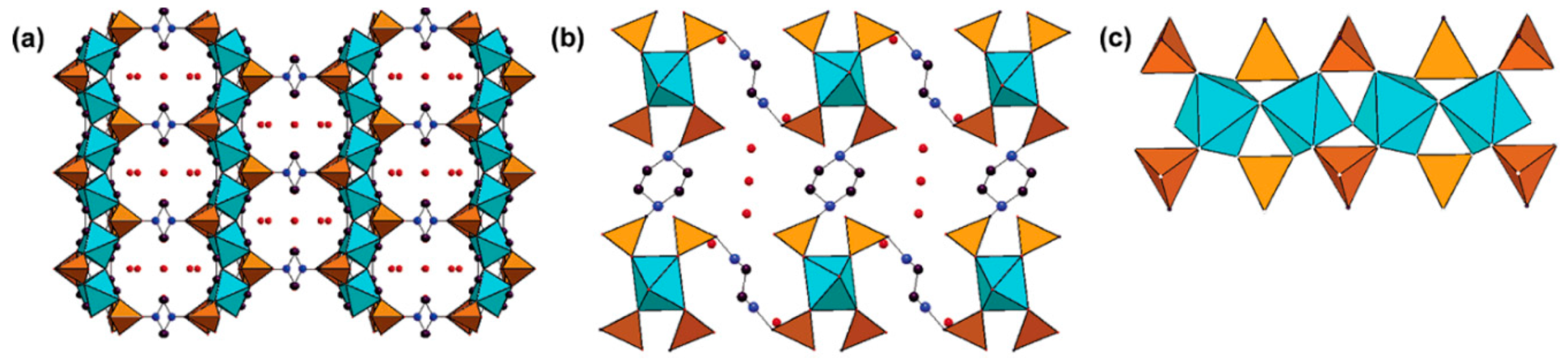
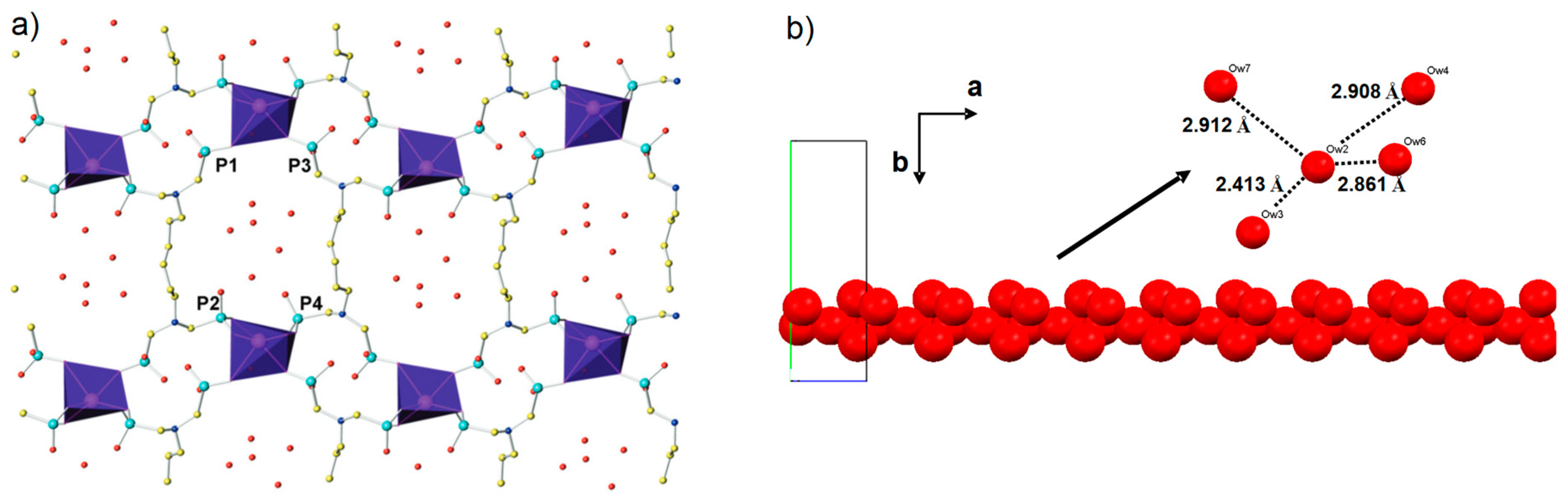
- Groves, J.A.; Miller, S.R.; Warrender, S.J.; Mellot-Draznieks, C.; Lightfoot, P.; Wright, P.A. The first route to large pore metal phosphonates. Chem. Commun. 2006, 1, 3305–3307. [Google Scholar] [CrossRef] [PubMed]
- Wharmby, M.T.; Pearce, G.M.; Mowat, J.P.S.; Griffin, J.M.; Ashbrook, S.E.; Wright, P.A.; Schilling, L.-H.; Lieb, A.; Stock, N.; Chavan, S.; et al. Synthesis and crystal chemistry of the STA-12 family of metal N,N′-piperazinebis(methylenephosphonate)s and applications of STA-12(Ni) in the separation of gases. Microporous Mesoporous Mater. 2011, 157, 3–17. [Google Scholar] [CrossRef]
- Wharmby, M.T.; Mowat, J.P.S.; Thompson, S.P.; Wright, P.A. Extending the pore size of crystalline metal phosphonates toward the mesoporous regime by isoreticular synthesis. J. Am. Chem. Soc. 2011, 133, 1266–1269. [Google Scholar] [CrossRef]
- Taddei, M.; Costantino, F.; Vivani, R. Robust Metal-Organic Frameworks Based on Tritopic Phosphonoaromatic Ligands. Eur. J. Inorg. Chem. 2016, 2016, 4300–4309. [Google Scholar] [CrossRef]
- Firmino, A.D.G.; Figueira, F.; Tomé, J.P.C.; Paz, F.A.A.; Rocha, J. Metal–Organic Frameworks assembled from tetraphosphonic ligands and lanthanides. Coord. Chem. Rev. 2018, 355, 133–149. [Google Scholar] [CrossRef]
- Zaręba, J.K. Tetraphenylmethane and tetraphenylsilane as building units of coordination polymers and supramolecular networks—A focus on tetraphosphonates. Inorg. Chem. Commun. 2017, 86, 172–186. [Google Scholar] [CrossRef]
- Zheng, T.; Yang, Z.; Gui, D.; Liu, Z.; Wang, X.; Dai, X.; Liu, S.; Zhang, L.; Gao, Y.; Chen, L.; et al. Overcoming the crystallization and designability issues in the ultrastable zirconium phosphonate framework system. Nat. Commun. 2017, 8, 15369. [Google Scholar] [CrossRef]
- Erkal, T.S.; Bulut, A.; Zorlu, Y.; Beckmann, J.; Yücesan, G.; Erbahar, D.; Yazaydin, A.O.; Çetinkaya, A. A cobalt arylphosphonate MOF—Superior stability, sorption and magnetism. Chem. Commun. 2019, 55, 3053–3056. [Google Scholar] [CrossRef]
- Wang, B.; Huang, Z.; Yang, T.; Rhauderwiek, T.; Xu, H.; Stock, N.; Inge, A.K.; Zou, X. A Porous Cobalt Tetraphosphonate Metal-Organic Framework: Accurate Structure and Guest Molecule Location Determined by Continuous-Rotation Electron Diffraction. Chem. Eur. J. 2018, 24, 17429–17433. [Google Scholar] [CrossRef] [PubMed]
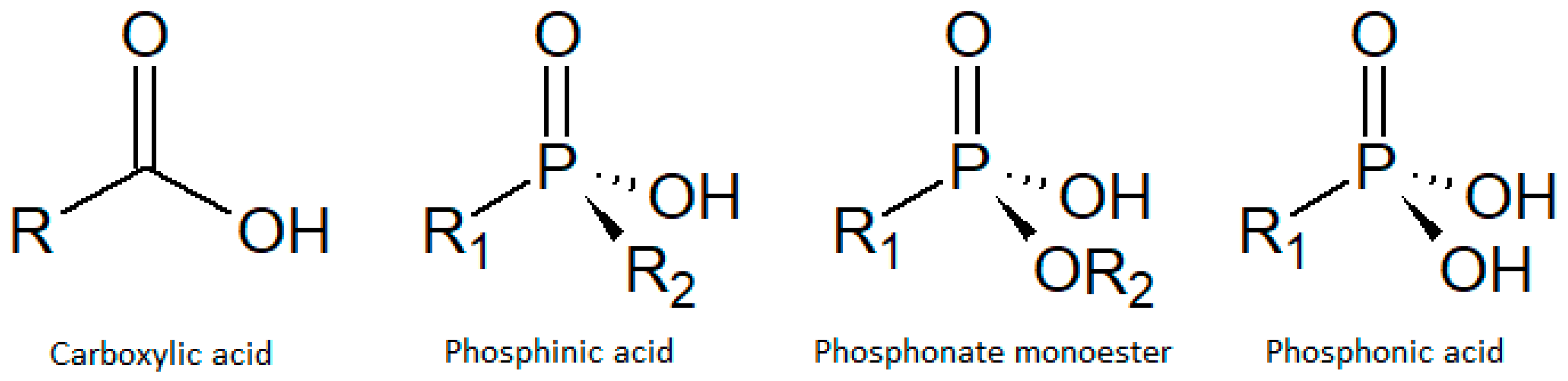
- Carson, I.; Healy, M.R.; Doidge, E.D.; Love, J.B.; Morrison, C.A.; Tasker, P.A. Metal-binding motifs of alkyl and aryl phosphinates; versatile mono and polynucleating ligands. Coord. Chem. Rev. 2017, 335, 150–171. [Google Scholar] [CrossRef] [Green Version]
- Costantino, F.; Ienco, A.; Taddei, M. Hybrid Multifunctional Materials Based on Phosphonates, Phosphinates and Auxiliary Ligands. In Tailored Organic-Inorganic Materials; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 193–244. ISBN 9781118792223. [Google Scholar]
- Shimizu, G.K.H.; Martens, I.; Woo, T.K.; Vaidhyanathan, R.; Daff, T.D.; Yeganegi, S.; Aghaji, M.Z.; Iremonger, S.S.; Liang, J. Phosphonate Monoesters as Carboxylate-like Linkers for Metal Organic Frameworks. J. Am. Chem. Soc. 2011, 133, 20048–20051. [Google Scholar] [CrossRef]
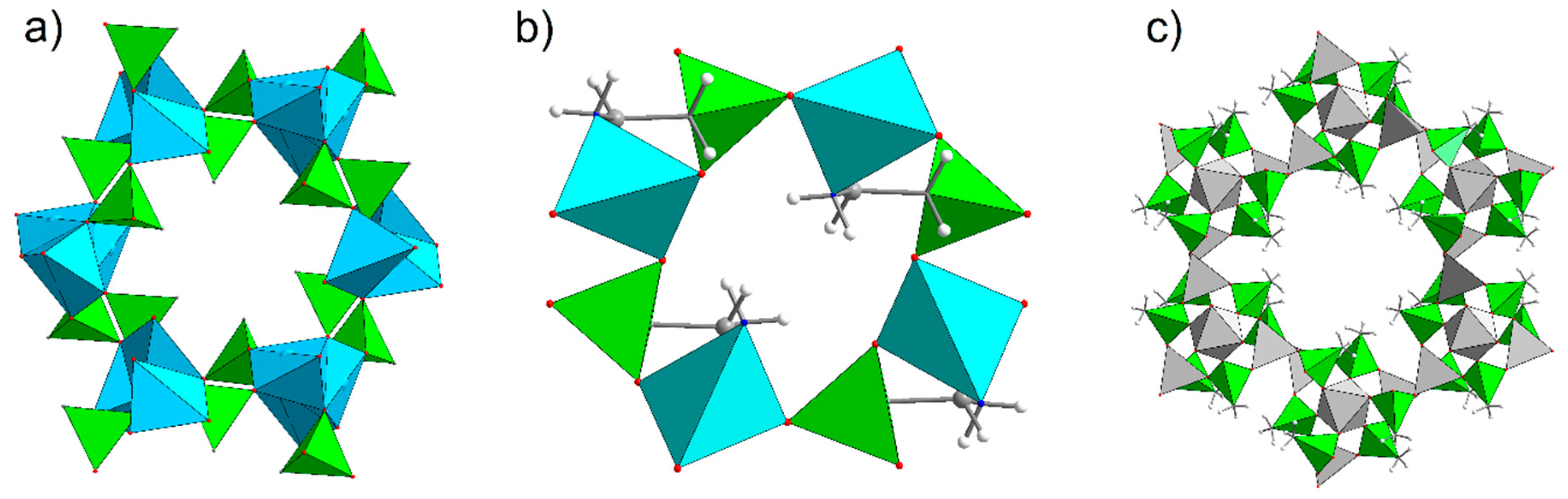
- Hynek, J.; Brázda, P.; Rohlíček, J.; Londesborough, M.G.S.; Demel, J. Phosphinic Acid Based Linkers: Building Blocks in Metal–Organic Framework Chemistry. Angew. Chem. Int. Ed. 2018, 57, 5016–5019. [Google Scholar] [CrossRef]
- Taylor, J.M.; Vaidhyanathan, R.; Iremonger, S.S.; Shimizu, G.K.H. Enhancing water stability of metal-organic frameworks via phosphonate monoester linkers. J. Am. Chem. Soc. 2012, 134, 14338–14340. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, B.S.; Lin, J.B.; Shimizu, G.K.H. Design of a Humidity-stable metal-organic framework using a phosphonate monoester ligand. Inorg. Chem. 2015, 54, 1185–1187. [Google Scholar] [CrossRef]
- Taddei, M.; Costantino, F.; Vivani, R. Synthesis and crystal structure from X-ray powder diffraction data of Two zirconium diphosphonates containing piperazine groups. Inorg. Chem. 2010, 49, 9664–9670. [Google Scholar] [CrossRef]
- Gonen, T.; Hattne, J.; Rodriguez, J.A.; Martynowycz, M.W.; Stoltz, B.M.; Fulton, T.J.; Jones, C.G.; Nelson, H.M. The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Cent. Sci. 2018, 4, 1587–1592. [Google Scholar] [CrossRef]
- Gruene, T.; Wennmacher, J.T.C.; Zaubitzer, C.; Holstein, J.J.; Heidler, J.; Fecteau-Lefebvre, A.; De Carlo, S.; Müller, E.; Goldie, K.N.; Regeni, I.; et al. Rapid Structure Determination of Microcrystalline Molecular Compounds Using Electron Diffraction. Angew. Chem. Int. Ed. 2018, 57, 16313–16317. [Google Scholar] [CrossRef] [Green Version]
- Feyand, M.; Mugnaioli, E.; Vermoortele, F.; Bueken, B.; Dieterich, J.M.; Reimer, T.; Kolb, U.; De Vos, D.; Stock, N. Automated diffraction tomography for the structure elucidation of twinned, sub-micrometer crystals of a highly porous, catalytically active bismuth metal-organic framework. Angew. Chem. Int. Ed. 2012, 51, 10373–10376. [Google Scholar] [CrossRef]
- Kolb, U.; Mugnaioli, E.; Gorelik, T.E. Automated electron diffraction tomography—A new tool for nano crystal structure analysis. Cryst. Res. Technol. 2011, 46, 542–554. [Google Scholar] [CrossRef]
- Mintova, S.; Petit, S.; Zaarour, M.; Boullay, P.; Perez, O.; Brázda, P.; Eigner, V.; Palatinus, L.; Klementová, M. Hydrogen positions in single nanocrystals revealed by electron diffraction. Science 2017, 355, 166–169. [Google Scholar] [CrossRef]
- Wan, W.; Sun, J.; Su, J.; Hovmöller, S.; Zou, X. Three-dimensional rotation electron diffraction: Software RED for automated data collection and data processing. J. Appl. Crystallogr. 2013, 46, 1863–1873. [Google Scholar] [CrossRef]
- Walton, R.I.; Millange, F. In Situ Studies of the Crystallization of Metal-Organic Frameworks. In The Chemistry of Metal-Organic Frameworks: Synthesis, Characterization, and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; pp. 729–764. [Google Scholar]
- Heidenreich, N.; Rütt, U.; Suren, R.; Stock, N.; Inge, A.K.; Dippel, A.-C.; Köppen, M.; Beier, S. A multi-purpose reaction cell for the investigation of reactions under solvothermal conditions. Rev. Sci. Instrum. 2017, 88, 104102. [Google Scholar] [CrossRef] [PubMed]
- Van Vleet, M.J.; Weng, T.; Li, X.; Schmidt, J.R. In Situ, Time-Resolved, and Mechanistic Studies of Metal-Organic Framework Nucleation and Growth. Chem. Rev. 2018, 118, 3681–3721. [Google Scholar] [CrossRef] [PubMed]
- Feyand, M.; Näther, C.; Rothkirch, A.; Stock, N. Systematic and in situ energy dispersive X-ray diffraction investigations on the formation of lanthanide phosphonatobutanesulfonates: Ln(O3P-C4H8-SO3)(H2O) (Ln = La-Gd). Inorg. Chem. 2010, 49, 11158–11163. [Google Scholar] [CrossRef]
- Feyand, M.; Hübner, A.; Rothkirch, A.; Wragg, D.S.; Stock, N. Copper phosphonatoethanesulfonates: Temperature dependent in situ energy dispersive x-ray diffraction study and influence of the pH on the crystal structures. Inorg. Chem. 2012, 51, 12540–12547. [Google Scholar] [CrossRef] [PubMed]
- Batzdorf, L.; Fischer, F.; Wilke, M.; Wenzel, K.J.; Emmerling, F. Direct in situ investigation of milling reactions using combined X-ray diffraction and Raman spectroscopy. Angew. Chem. Int. Ed. 2015, 54, 1799–1802. [Google Scholar] [CrossRef]
- Rothenberg, G. Catalysis: Concepts and Green Applications; Wiley-VCH: Weinheim, Germany, 2017; ISBN 3527808884. [Google Scholar]
- Schlögl, R. Heterogeneous catalysis. Angew. Chem. Int. Ed. 2015, 54, 3465–3520. [Google Scholar] [CrossRef]
- Thomas, J.M.; Thomas, W.J. Principles and Practice of Heterogeneous Catalysis, 2nd ed.; Wiley: Hoboken, NJ, USA, 2015; ISBN 9783527314584. [Google Scholar]
- White, R.J.; Luque, R.; Budarin, V.L.; Clark, J.H.; MacQuarrie, D.J. Supported metal nanoparticles on porous materials. Methods and applications. Chem. Soc. Rev. 2009, 38, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D. (Ed.) Nanoparticles and Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; ISBN 9783527315727. [Google Scholar]
- Hu, Z.; Zhao, D. Metal-organic frameworks with Lewis acidity: Synthesis, characterization, and catalytic applications. CrystEngComm 2017, 19, 4066–4081. [Google Scholar] [CrossRef]
- Ranocchiari, M.; van Bokhoven, J.A. Catalysis by metal-organic frameworks: Fundamentals and opportunities. Phys. Chem. Chem. Phys. 2011, 13, 6388–6396. [Google Scholar] [CrossRef]
- Clearfielda, A.; Thakurb, D.S. Zirconium and titanium phosphates as catalysts: A review. Appl. Catal. 1986, 26, 1–26. [Google Scholar] [CrossRef]
- Wang, Z.; Heising, J.M.; Clearfield, A. Sulfonated microporous organic-inorganic hybrids as strong bronsted acids. J. Am. Chem. Soc. 2003, 125, 10375–10383. [Google Scholar] [CrossRef]
- Donnadio, A.; Nocchetti, M.; Costantino, F.; Taddei, M.; Casciola, M.; Da Silva Lisboa, F.; Vivani, R. A layered mixed zirconium phosphate/phosphonate with exposed carboxylic and phosphonic groups: X-ray powder structure and proton conductivity properties. Inorg. Chem. 2014, 53, 13220–13226. [Google Scholar] [CrossRef] [PubMed]
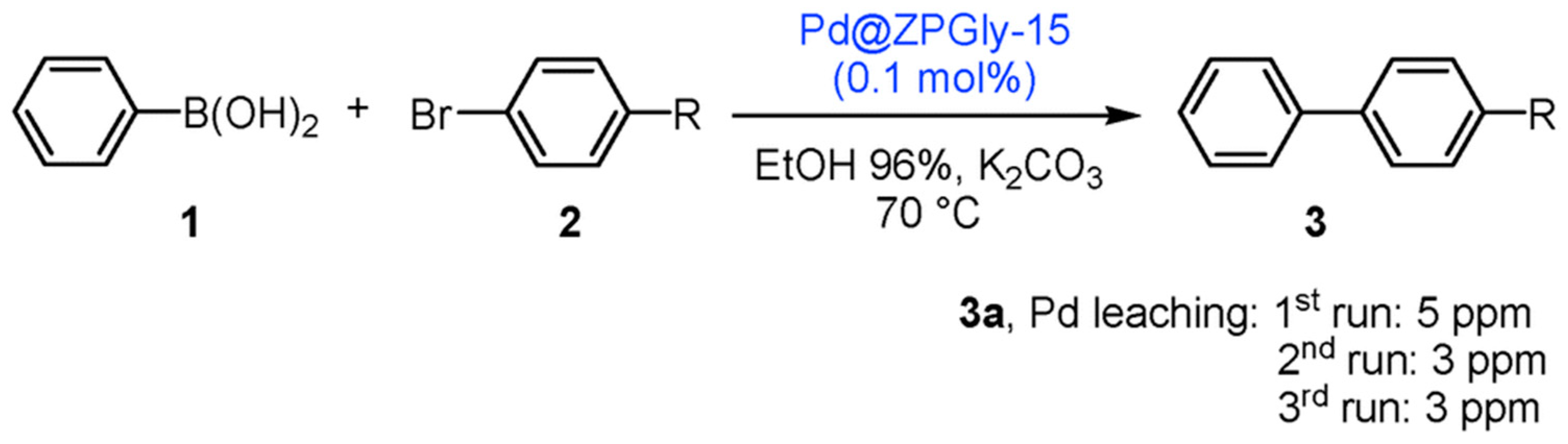
- Costantino, F.; Vivani, R.; Bastianini, M.; Ortolani, L.; Piermatti, O.; Nocchetti, M.; Vaccaro, L. Accessing stable zirconium carboxy-aminophosphonate nanosheets as support for highly active Pd nanoparticles. Chem. Commun. 2015, 51, 15990–15993. [Google Scholar] [CrossRef] [PubMed]
- Bastianini, M.; Costantino, F.; Caporali, M.; Liguori, F.; Nocchetti, M.; Lavacchi, A. Robust Zirconium Phosphate–Phosphonate Nanosheets Containing Palladium Nanoparticles as Efficient Catalyst for Alkynes and Nitroarenes Hydrogenation Reactions. ACS Appl. Nano Mater. 2018, 1, 1750–1757. [Google Scholar] [CrossRef]
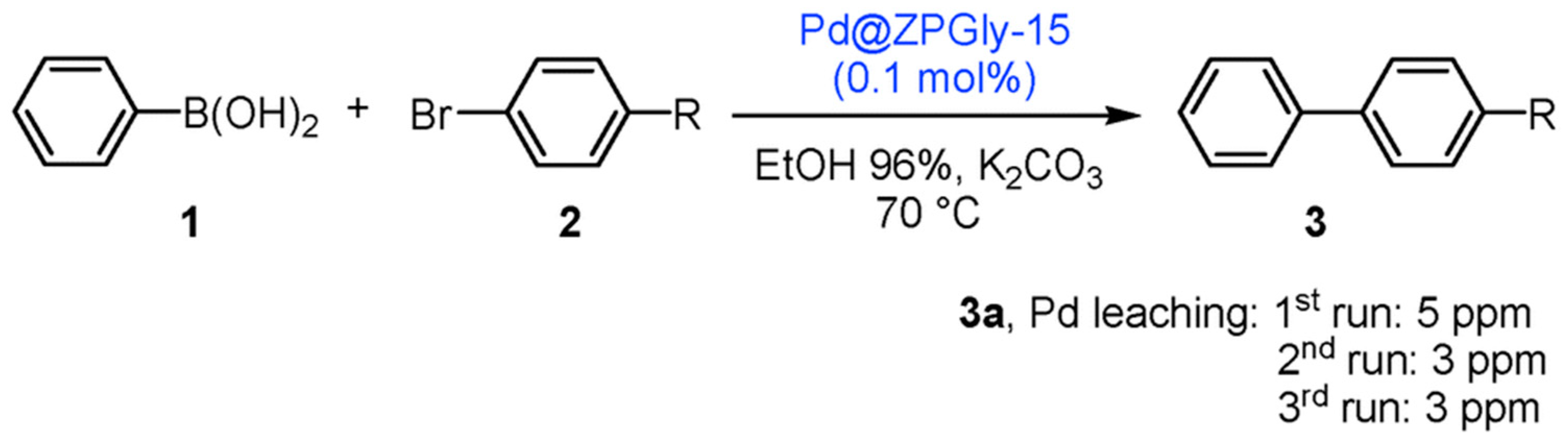
- Nocchetti, M.; Giannoni, T.; Vaccaro, L.; Piermatti, O.; Kozell, V.; Vivani, R. Immobilized Palladium Nanoparticles on Zirconium Carboxy-Aminophosphonates Nanosheets as an Efficient Recoverable Heterogeneous Catalyst for Suzuki–Miyaura and Heck Coupling. Catalysts 2017, 7, 186. [Google Scholar] [CrossRef]
- Ferlin, F.; Cappelletti, M.; Vivani, R.; Pica, M.; Piermatti, O.; Vaccaro, L. Au@zirconium-phosphonate nanoparticles as an effective catalytic system for the chemoselective and switchable reduction of nitroarenes. Green Chem. 2019, 21, 614–626. [Google Scholar] [CrossRef]
- Pica, M.; Donnadio, A.; Capitani, D.; Vivani, R.; Troni, E.; Casciola, M. Advances in the chemistry of nanosized zirconium phosphates: A new mild and quick route to the synthesis of nanocrystals. Inorg. Chem. 2011, 50, 11623–11630. [Google Scholar] [CrossRef]
- Mitchell, L.; Gonzalez-Santiago, B.; Mowat, J.P.S.; Gunn, M.E.; Williamson, P.; Acerbi, N.; Clarke, M.L.; Wright, P.A. Remarkable Lewis acid catalytic performance of the scandium trimesate metal organic framework MIL-100(Sc) for C–C and C=N bond-forming reactions. Catal. Sci. Technol. 2013, 3, 606–617. [Google Scholar] [CrossRef]
- Beier, M.J.; Kleist, W.; Wharmby, M.T.; Kissner, R.; Kimmerle, B.; Wright, P.A.; Grunwaldt, J.D.; Baiker, A. Aerobic epoxidation of olefins catalyzed by the cobalt-based metal-organic framework STA-12(Co). Chem. Eur. J. 2012, 18, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Oschatz, M.; Antonietti, M. A search for selectivity to enable CO2 capture with porous adsorbents. Energy Environ. Sci. 2018, 11, 57–70. [Google Scholar] [CrossRef]
- Li, B.; Chen, B. Fine-Tuning Porous Metal-Organic Frameworks for Gas Separations at Will. Chem 2016, 1, 669–671. [Google Scholar] [CrossRef]
- Hermer, N.; Wharmby, M.T.; Stock, N. Re-Determination of the Crystal Structure of MIL-91(Al). Z. Anorg. Allg. Chem. 2017, 643, 137–140. [Google Scholar] [CrossRef]
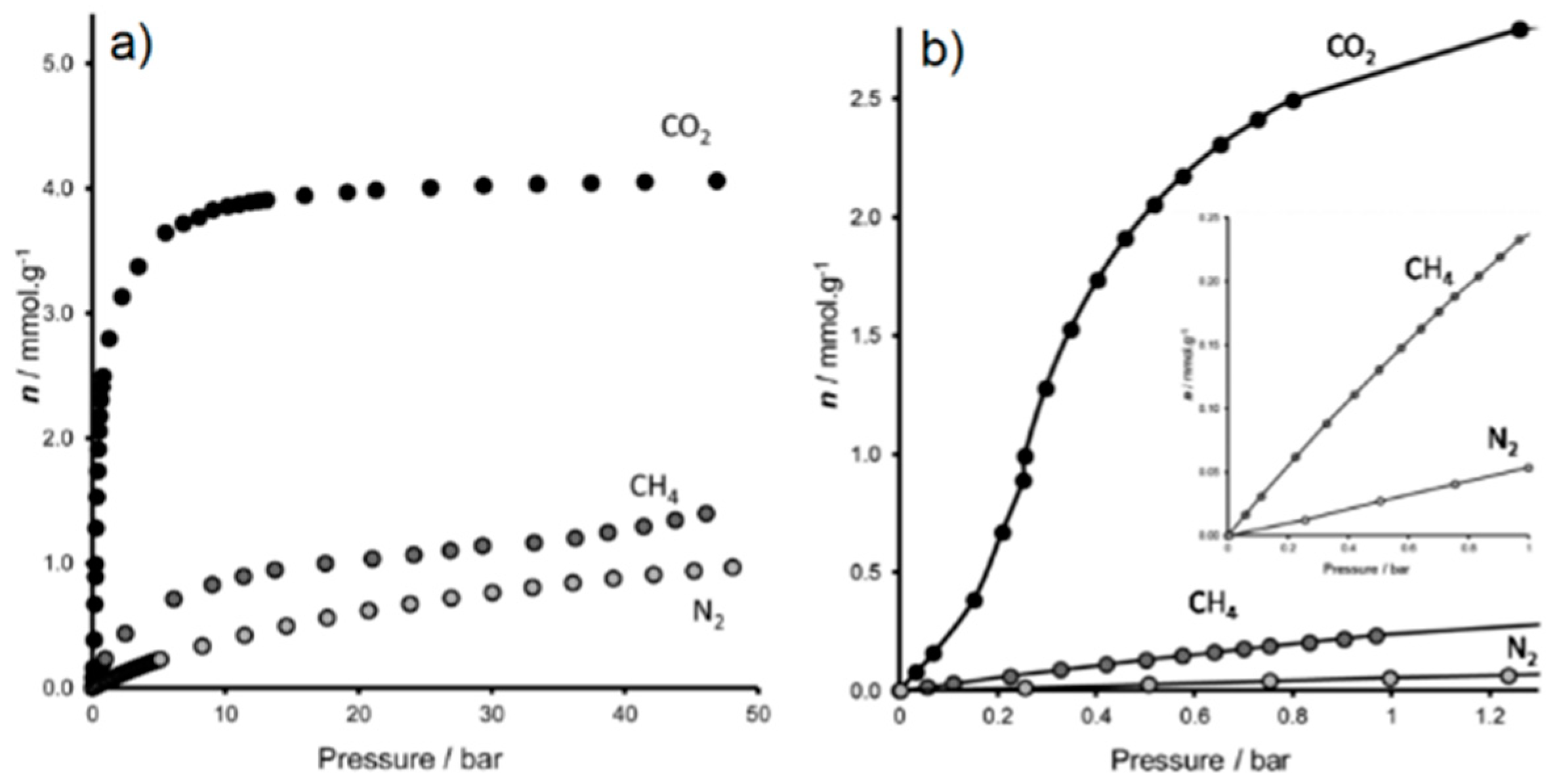
- Miller, S.R.; Pearce, G.M.; Wright, P.A.; Bonino, F.; Chavan, S.; Bordiga, S.; Margiolaki, I.; Guillou, N.; Férey, G.; Bourrelly, S.; et al. Structural transformations and adsorption of fuel-related gases of a structurally responsive nickel phosphonate metal-organic framework, Ni-STA-12. J. Am. Chem. Soc. 2008, 130, 15967–15981. [Google Scholar] [CrossRef]
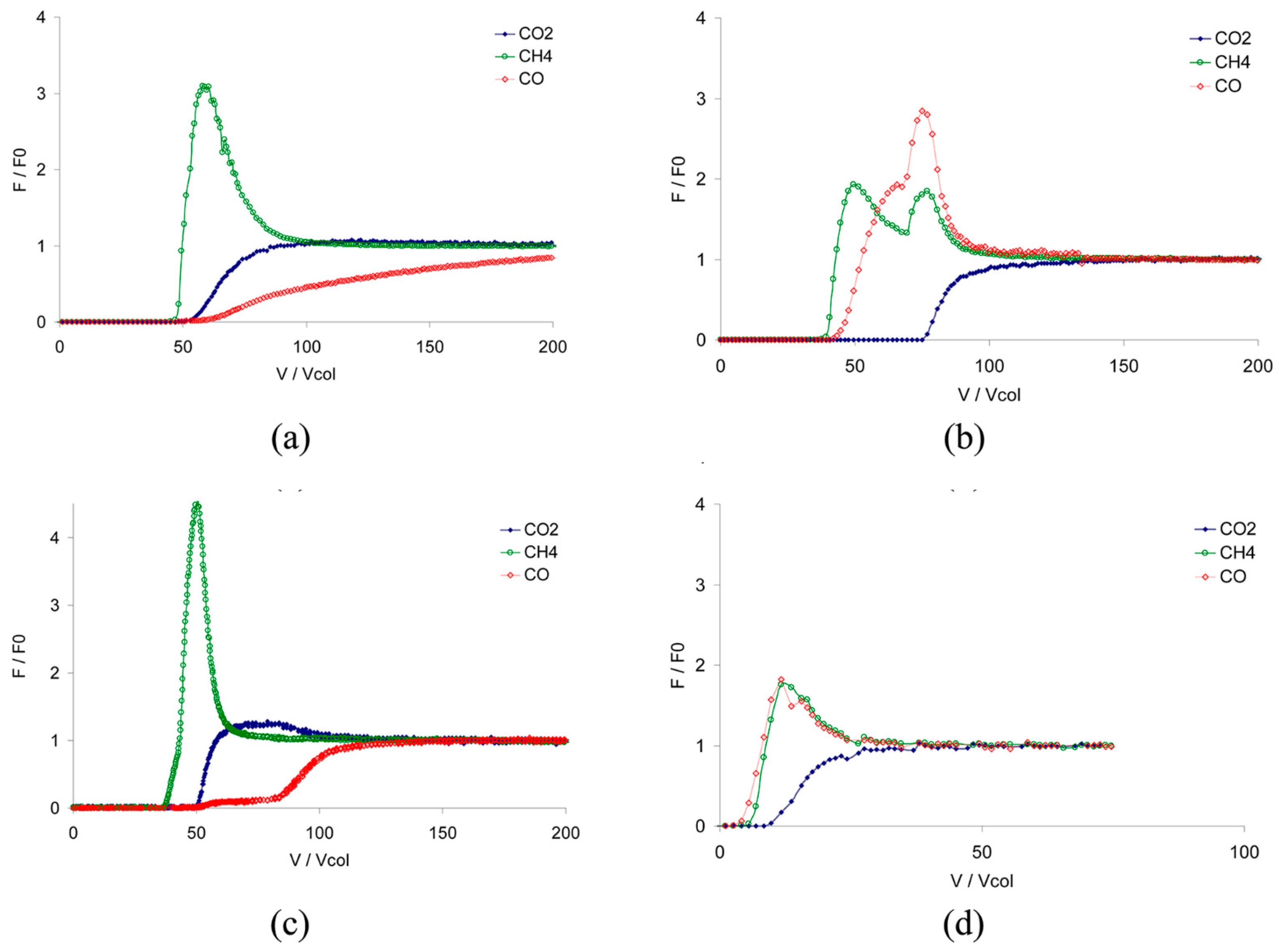
- García, E.J.; Mowat, J.P.S.; Wright, P.A.; Pérez-Pellitero, J.; Jallut, C.; Pirngruber, G.D. Role of structure and chemistry in controlling separations of CO2/CH4 and CO2/CH4/CO mixtures over honeycomb MOFs with coordinatively unsaturated metal sites. J. Phys. Chem. C 2012, 116, 26636–26648. [Google Scholar] [CrossRef]
- World Energy Outlook 2017—Executive Summary; International Energy Agency: Paris, France, 2017.
- Winter, M.; Brodd, R.J. What Are Batteries, Fuel Cells, and Supercapacitors? Chem. Rev. 2004, 104, 4245–4270. [Google Scholar] [CrossRef] [Green Version]
- Bagotsky, V.S.; Skundin, A.M.; Volfkovich, Y.M. Electrochemical Power Sources: Batteries, Fuel Cells, and Supercapacitors; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; ISBN 9781118942857. [Google Scholar]
- Peng, B.; Chen, J. Functional materials with high-efficiency energy storage and conversion for batteries and fuel cells. Coord. Chem. Rev. 2009, 253, 2805–2813. [Google Scholar] [CrossRef]
- Kreuer, K.D. Proton conductivity: Materials and applications. Chem. Mater. 1996, 8, 610–641. [Google Scholar] [CrossRef]
- Alberti, G.; Casciola, M. Solid state protonic conductors, present main applications and future prospects. Solid State Ion. 2001, 145, 3–16. [Google Scholar] [CrossRef]
- Vivani, R.; Casciola, M.; Alberi, G.; Costantino, U.; Peraio, A. Proton conductivity of zirconium carboxy n-alkyl phosphonates with an α-layered structure. Solid State Ion. 1991, 46, 61–68. [Google Scholar] [CrossRef]
- Alberti, G.; Casciola, M.; Palombari, R.; Peraio, A. Protonic conductivity of layered zirconium phosphonates containing –SO3H groups. II. Ac conductivity of zirconium alkyl-sulphophenyl phosphonates in the range 100–200 °C, in the presence or absence of water vapour. Solid State Ion. 1992, 58, 339–344. [Google Scholar] [CrossRef]
- Shimizu, G.K.H.; Taylor, J.M.; Dawson, K.W. Metal Organophosphonate Proton Conductors. In Metal Phosphonate Chemistry; Royal Society of Chemistry: Cambridge, UK, 2011; pp. 493–524. [Google Scholar]
- Colodrero, R.M.P.; Olivera-Pastor, P.; Losilla, E.R.; Aranda, M.A.G.; Leon-Reina, L.; Papadaki, M.; McKinlay, A.C.; Morris, R.E.; Demadis, K.D.; Cabeza, A. Multifunctional lanthanum tetraphosphonates: Flexible, ultramicroporous and proton-conducting hybrid frameworks. Dalton Trans. 2012, 41, 4045–4051. [Google Scholar] [CrossRef]
- Bazaga-García, M.; Colodrero, R.M.; Papadaki, M.; Garczarek, P.; Zoń, J.; Olivera-Pastor, P.; Losilla, E.R.; León-Reina, L.; Aranda, M.A.G.; Choquesillo-Lazarte, D.; et al. Guest Molecule-Responsive Functional Calcium Phosphonate Frameworks for Tuned Proton Conductivity. J. Am. Chem. Soc. 2014, 136, 5731–5739. [Google Scholar] [CrossRef]
- Colodrero, R.M.P.; Papathanasiou, K.E.; Stavgianoudaki, N.; Olivera-Pastor, P.; Losilla, E.R.; Aranda, M.A.G.; León-Reina, L.; Sanz, J.; Sobrados, I.; Choquesillo-Lazarte, D.; et al. Multifunctional luminescent and proton-conducting lanthanide carboxyphosphonate open-framework hybrids exhibiting crystalline-to-amorphous-to-crystalline transformations. Chem. Mater. 2012, 24, 3780–3792. [Google Scholar] [CrossRef]
- Colodrero, R.M.P.; Olivera-Pastor, P.; Losilla, E.R.; Hernández-Alonso, D.; Aranda, M.A.G.; Leon-Reina, L.; Rius, J.; Demadis, K.D.; Moreau, B.; Villemin, D.; et al. High Proton Conductivity in a Flexible, Cross-Linked, Ultramicroporous Magnesium Tetraphosphonate Hybrid Framework. Inorg. Chem. 2012, 51, 7689–7698. [Google Scholar] [CrossRef]
- Bazaga-García, M.; Papadaki, M.; Colodrero, R.M.P.; Olivera-Pastor, P.; Losilla, E.R.; Nieto-Ortega, B.; Aranda, M.Á.G.; Choquesillo-Lazarte, D.; Cabeza, A.; Demadis, K.D. Tuning proton conductivity in alkali metal phosphonocarboxylates by cation size-induced and water-facilitated proton transfer pathways. Chem. Mater. 2015, 27, 424–435. [Google Scholar] [CrossRef]
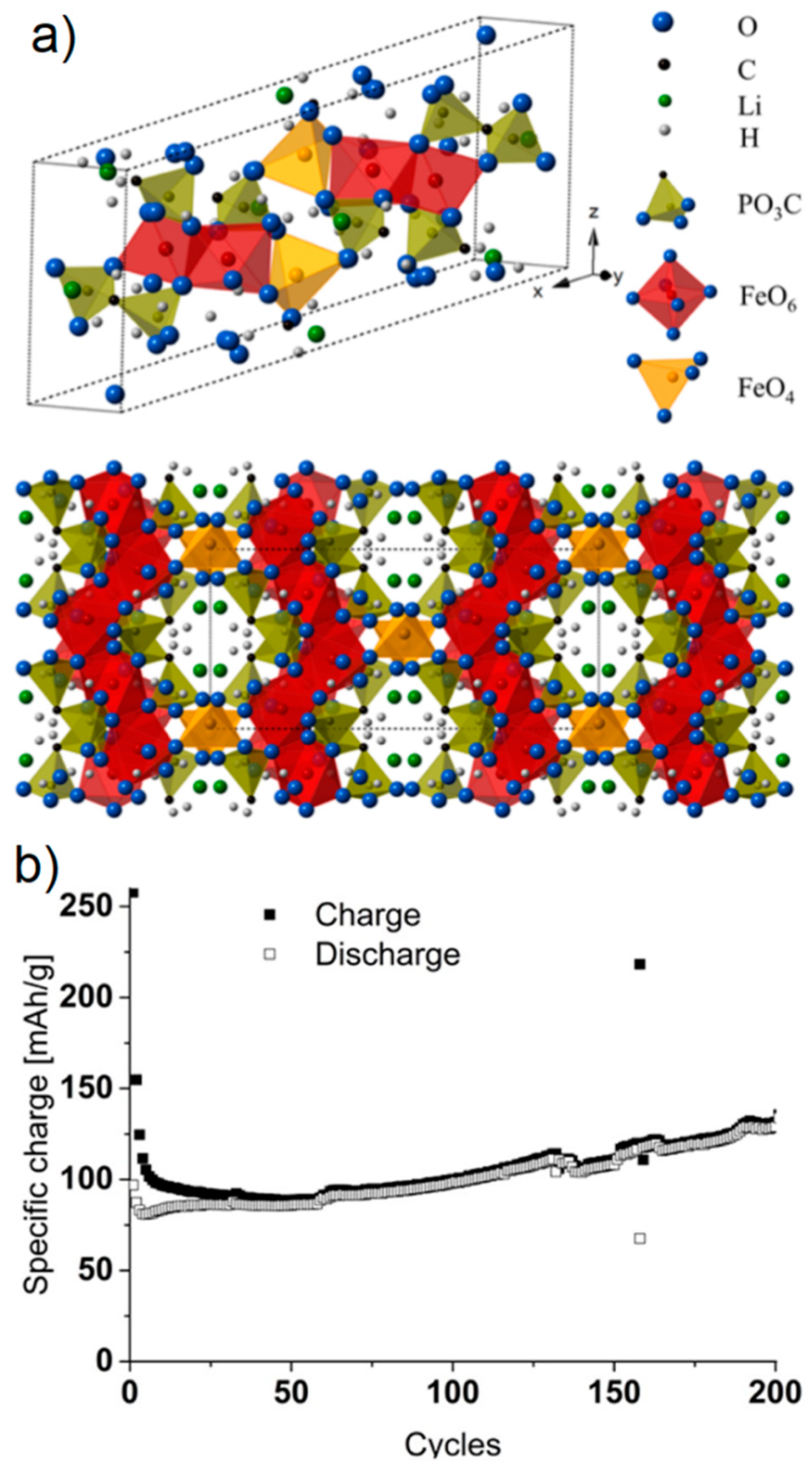
- Schmidt, S.; Sheptyakov, D.; Jumas, J.C.; Medarde, M.; Benedek, P.; Novák, P.; Sallard, S.; Villevieille, C. Lithium Iron Methylenediphosphonate: A Model Material for New Organic-Inorganic Hybrid Positive Electrode Materials for Li Ion Batteries. Chem. Mater. 2015, 27, 7889–7895. [Google Scholar] [CrossRef]
- Schmidt, S.; Sallard, S.; Sheptyakov, D.; Nachtegaal, M.; Novák, P.; Villevieille, C. Fe and Co methylene diphosphonates as conversion materials for Li-ion batteries. J. Power Sources 2017, 342, 879–885. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.; Sallard, S.; Sheptyakov, D.; Novák, P.; Villevieille, C. Ligand influence in Li-ion battery hybrid active materials: Ni methylenediphosphonate: Vs. Ni dimethylamino methylenediphosphonate. Chem. Commun. 2017, 53, 5420–5423. [Google Scholar] [CrossRef]
- Galezowska, J.; Gumienna-Kontecka, E. Phosphonates, their complexes and bio-applications: A spectrum of surprising diversity. Coord. Chem. Rev. 2012, 256, 105–124. [Google Scholar] [CrossRef]
- Pazianas, M.; Abrahamsen, B.; Ferrari, S.; Russell, R.G.G. Eliminating the need for fasting with oral administration of bisphosphonates. Ther. Clin. Risk Manag. 2013, 9, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Papathanasiou, K.E.; Vassaki, M.; Spinthaki, A.; Alatzoglou, F.-E.G.; Tripodianos, E.; Turhanen, P.; Demadis, K.D. Phosphorus chemistry: From small molecules, to polymers, to pharmaceutical and industrial applications. Pure Appl. Chem. 2018, 91, 421–441. [Google Scholar] [CrossRef]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
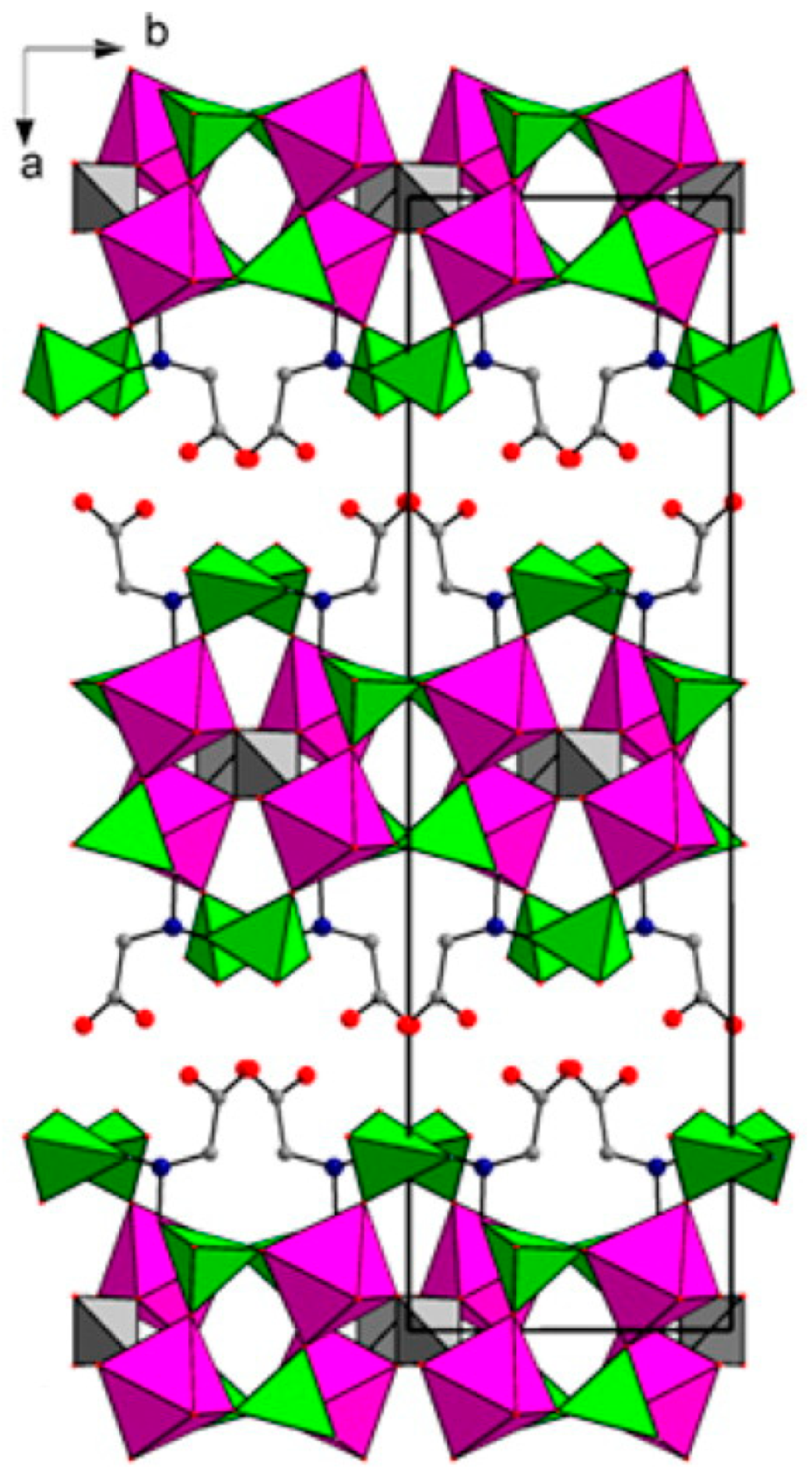{kind=link}
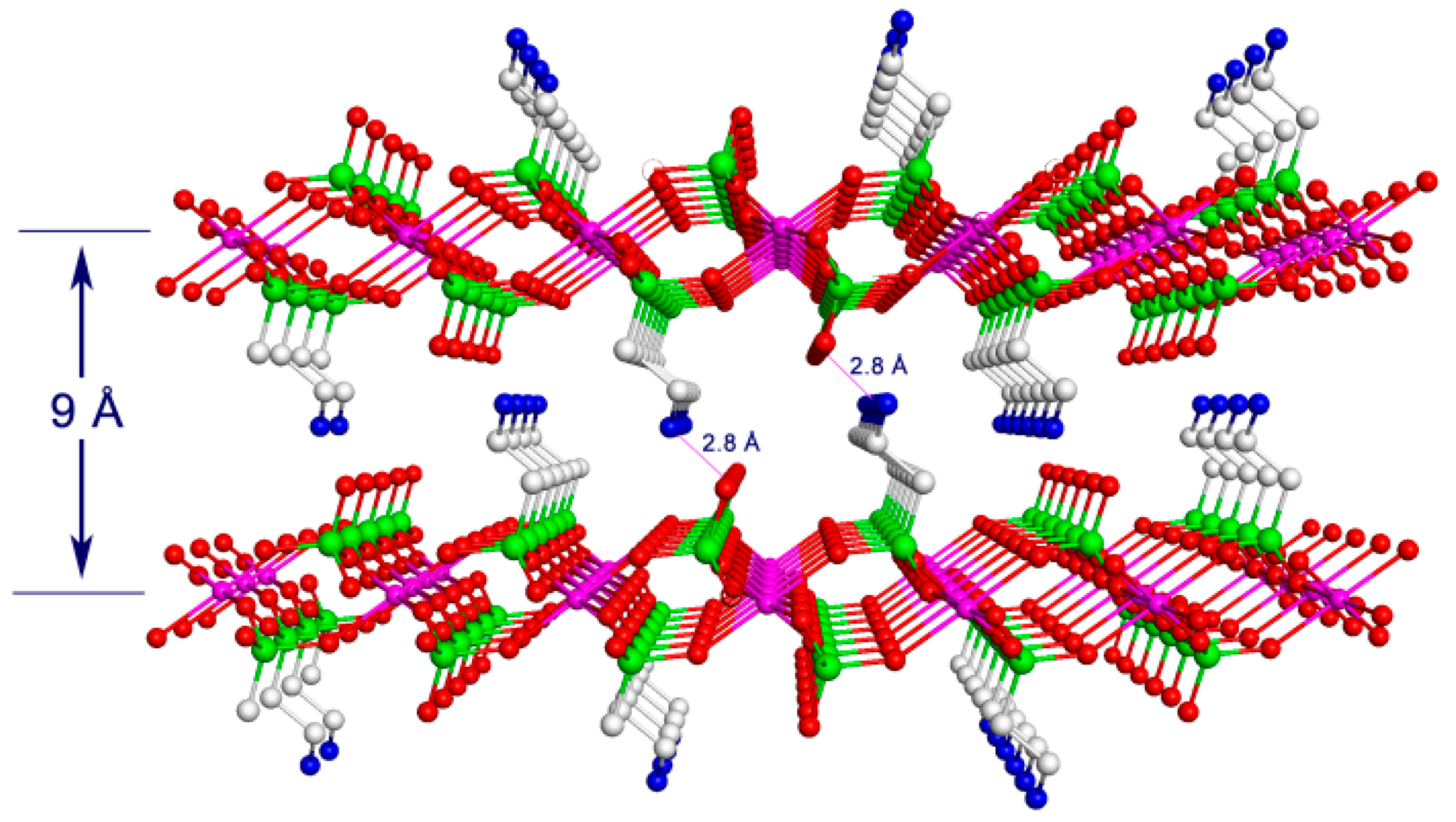{kind=link}
{kind=link}
{kind=link}
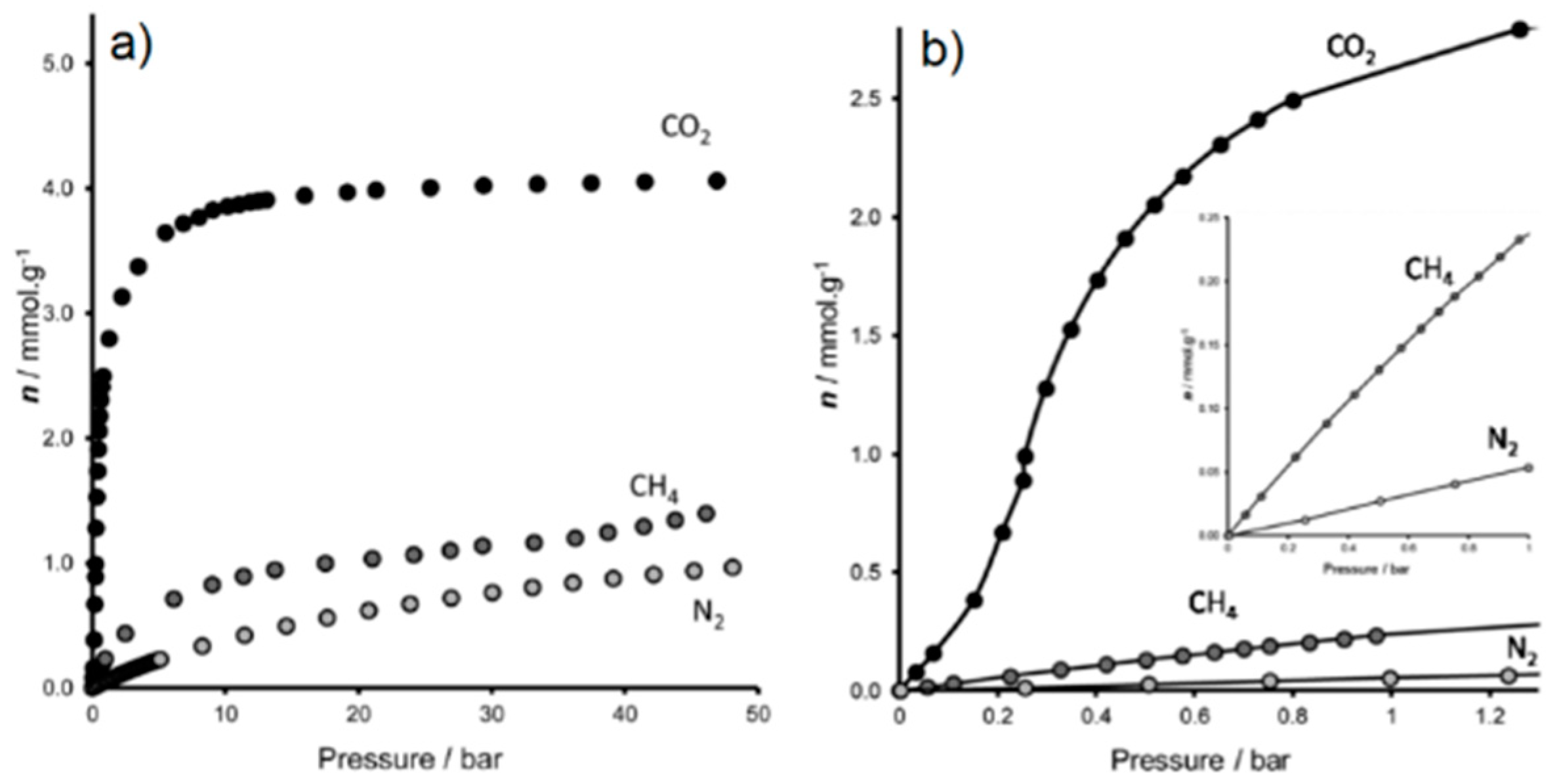{kind=link}
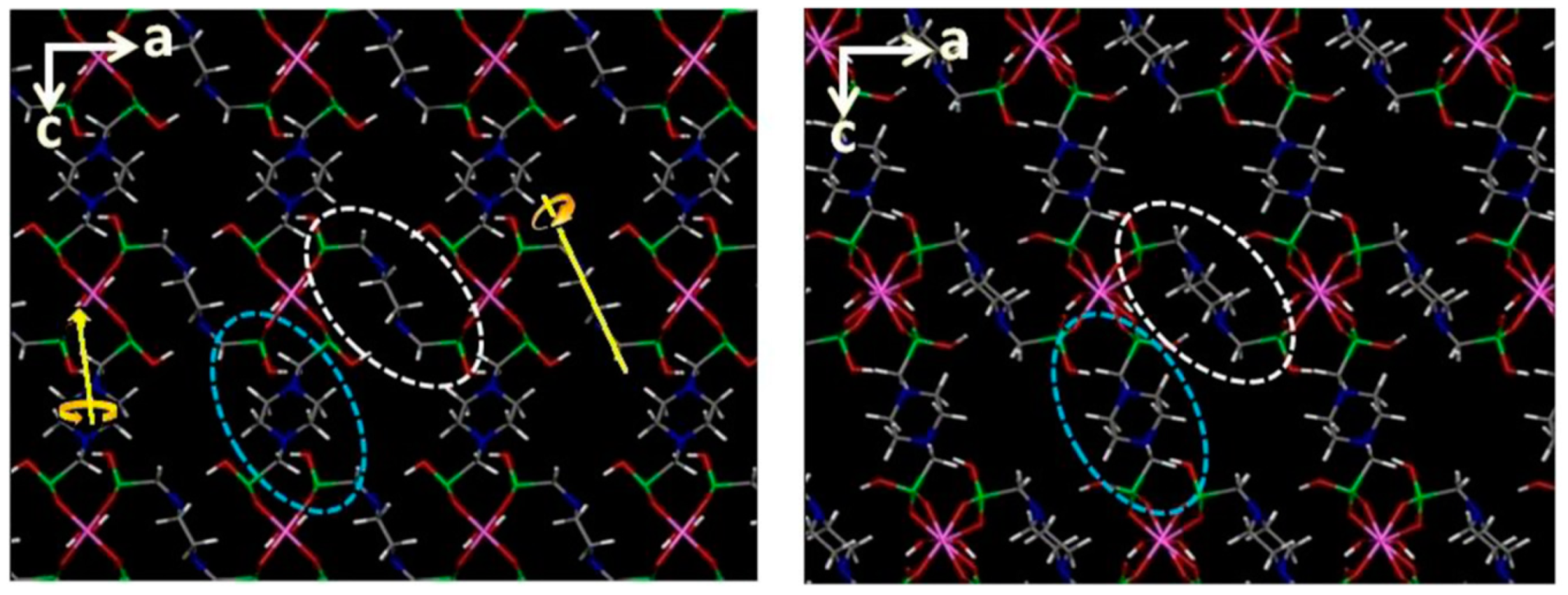{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Aryl Bromide | R | t (h) | Yield (%) |
|---|---|---|---|---|
| 1 | 2a (1st) | Me | 30 | 97 |
| 2 | 2a (2nd) | Me | 30 | 97 |
| 3 | 2a (3rd) | Me | 30 | 97 |
| 4 | 2b | H | 30 | 96 |
| 5 | 2c | CHO | 10 | 98 |
| 6 | 2d | NO2 | 10 | 98 |
| Entry a | Run | Medium | t (h) | C b (%) | 2 : 3 : 4 Ratio b |
|---|---|---|---|---|---|
| 1 | Run 1 | 96% EtOH | 3 | 98 | 96 : 4 : 0 |
| 2 | Run 2 | 96% EtOH | 3 | 96 | 97 : 3 : 0 |
| 3 | Run 3 | 96% EtOH | 3 | 94 | 97 : 3 : 0 |
| 4 | Run 4 | 96% EtOH | 3 | 93 | 96 : 4 : 0 |
| 5 | Run 5 | 96% EtOH | 3 | 87 | 98 : 2 : 0 |
| 6 | Run 1 | EtOHabs | 2 | >99 | 0 : 0 : 100 |
| 7 | Run 2 | EtOHabs | 2 | >99 | 0 : 0 : 100 |
| 8 | Run 3 | EtOHabs | 2 | >99 | 0 : 0 : 100 |
| 9 | Run 4 | EtOHabs | 2 | >99 | 0 : 0 : 100 |
| 10 | Run 5 | EtOHabs | 2 | >99 | 0 : 0 : 100 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shearan, S.J.I.; Stock, N.; Emmerling, F.; Demel, J.; Wright, P.A.; Demadis, K.D.; Vassaki, M.; Costantino, F.; Vivani, R.; Sallard, S.; et al. New Directions in Metal Phosphonate and Phosphinate Chemistry. Crystals 2019, 9, 270. https://doi.org/10.3390/cryst9050270
Shearan SJI, Stock N, Emmerling F, Demel J, Wright PA, Demadis KD, Vassaki M, Costantino F, Vivani R, Sallard S, et al. New Directions in Metal Phosphonate and Phosphinate Chemistry. Crystals. 2019; 9(5):270. https://doi.org/10.3390/cryst9050270
Chicago/Turabian StyleShearan, Stephen J.I., Norbert Stock, Franziska Emmerling, Jan Demel, Paul A. Wright, Konstantinos D. Demadis, Maria Vassaki, Ferdinando Costantino, Riccardo Vivani, Sébastien Sallard, and et al. 2019. "New Directions in Metal Phosphonate and Phosphinate Chemistry" Crystals 9, no. 5: 270. https://doi.org/10.3390/cryst9050270
APA StyleShearan, S. J. I., Stock, N., Emmerling, F., Demel, J., Wright, P. A., Demadis, K. D., Vassaki, M., Costantino, F., Vivani, R., Sallard, S., Ruiz Salcedo, I., Cabeza, A., & Taddei, M. (2019). New Directions in Metal Phosphonate and Phosphinate Chemistry. Crystals, 9(5), 270. https://doi.org/10.3390/cryst9050270