Simultaneous SAXS-WAXS Experiments on Semi-Crystalline Polymers: Example of PA11 and Its Brill Transition

 ,
, {kind=link}
{kind=link}
{kind=link}
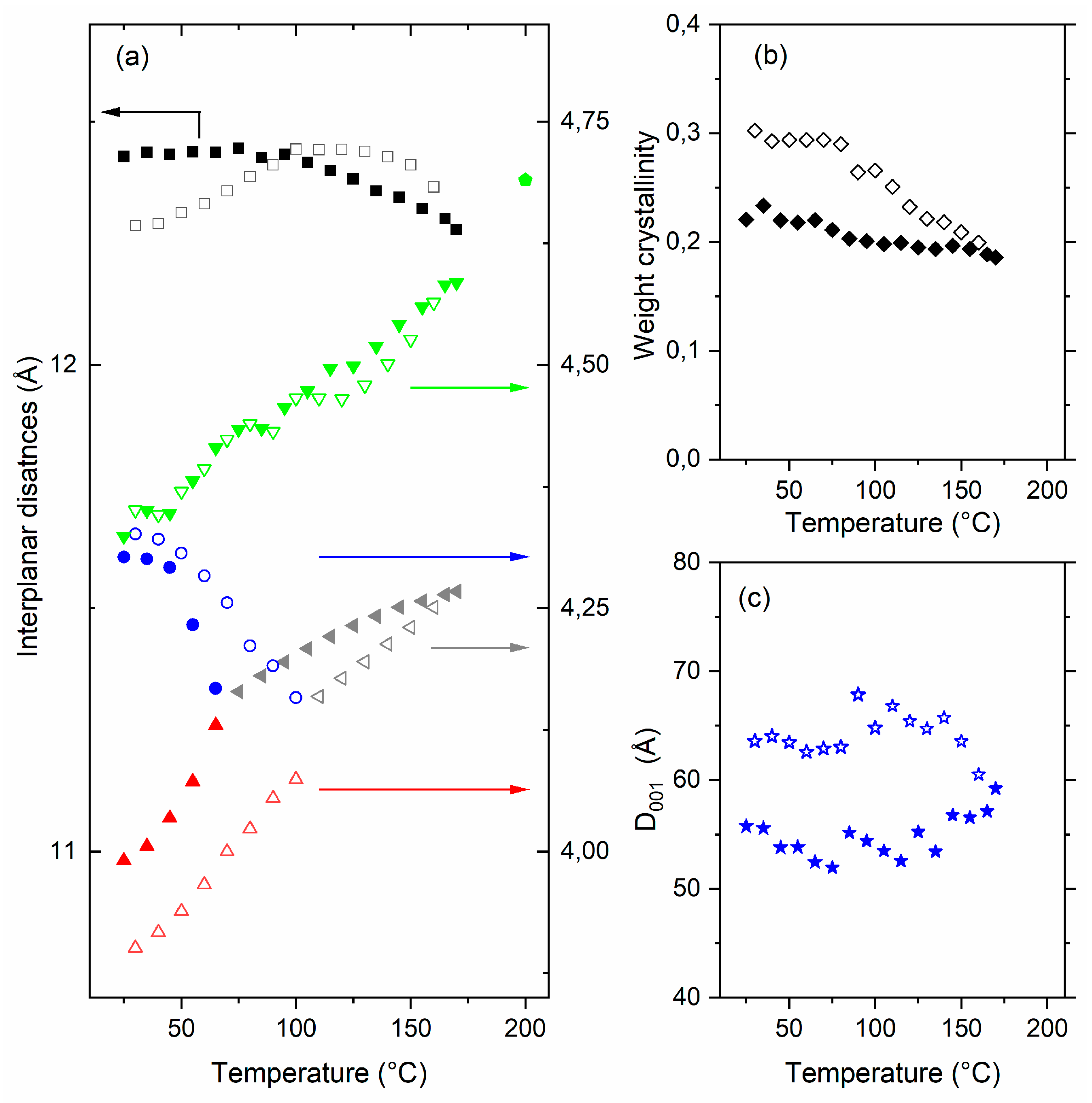{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Share and Cite
Tencé-Girault, S.; Lebreton, S.; Bunau, O.; Dang, P.; Bargain, F. Simultaneous SAXS-WAXS Experiments on Semi-Crystalline Polymers: Example of PA11 and Its Brill Transition. Crystals 2019, 9, 271. https://doi.org/10.3390/cryst9050271
Tencé-Girault S, Lebreton S, Bunau O, Dang P, Bargain F. Simultaneous SAXS-WAXS Experiments on Semi-Crystalline Polymers: Example of PA11 and Its Brill Transition. Crystals. 2019; 9(5):271. https://doi.org/10.3390/cryst9050271
Chicago/Turabian StyleTencé-Girault, Sylvie, Sylvie Lebreton, Oana Bunau, Patrick Dang, and François Bargain. 2019. "Simultaneous SAXS-WAXS Experiments on Semi-Crystalline Polymers: Example of PA11 and Its Brill Transition" Crystals 9, no. 5: 271. https://doi.org/10.3390/cryst9050271
APA StyleTencé-Girault, S., Lebreton, S., Bunau, O., Dang, P., & Bargain, F. (2019). Simultaneous SAXS-WAXS Experiments on Semi-Crystalline Polymers: Example of PA11 and Its Brill Transition. Crystals, 9(5), 271. https://doi.org/10.3390/cryst9050271