Structural Insights into the Two-Step Spin-Crossover Compound Fe(3,4-dimethyl-pyridine)2[Ag(CN)2]2

 , ,
, , 
Abstract
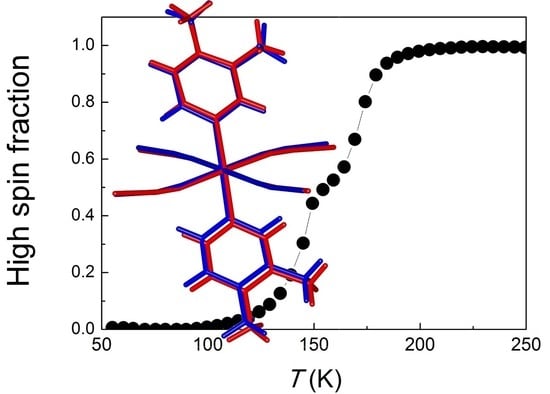
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Linert, W.; Verdaguer, M. Molecular Magnets: Recent Highlights; Springer: Wien, Austria, 2003. [Google Scholar]
- Gutlich, P.; Goodwin, H.A. Topics in Current Chemistry. In Spin-Crossover in Transition Metal Compounds; Springer: Berlin, Germany, 2004. [Google Scholar]
- Muñoz, M.C.; Real, J.A. Thermo-, piezo-, photo- and chemoswitchable spin crossover iron(II) metallocyanate based coordination polymers. Coord. Chem. Rev. 2011, 255, 2068–2093. [Google Scholar] [CrossRef]
- Halcrow, M.A. Spin-Crossover Materials: Properties and Applications; John Wiley & Sons: Chichester, UK, 2013. [Google Scholar]
- Senthil Kumar, K.; Ruben, M. Emerging trends in spin crossover (SCO) based functional materials and devices. Coord. Chem. Rev. 2017, 346, 176–205. [Google Scholar] [CrossRef]
- Hauser, A. Ligand Field Theoretical Considerations. Top. Curr. Chem. 2004, 233, 49–58. [Google Scholar]
- Gütlich, P.; Goodwin, H. Spin Crossover—An Overall Perspective. Top. Curr. Chem. 2004, 233, 1–47. [Google Scholar]
- König, E. Nature and dynamics of the spin-state interconversion in metal complexes. Struct. Bond. 1991, 76, 51–152. [Google Scholar]
- Collet, E.; Guionneau, P. Structural analysis of spin-crossover materials: From molecules to materials. C. R. Chim. 2018, 21, 1133–1151. [Google Scholar] [CrossRef]
- Guionneau, P.; Marchivie, M.; Bravic, G.; Létard, J.-F.; Chasseau, D. Structural Aspects of Spin Crossover. Example of the [FeIILn(NCS)2] Complexes. Top. Curr. Chem. 2004, 234, 97–128. [Google Scholar]
- Garcia, Y.; Niel, V.; Muñoz, M.C.; Real, J.A. Spin crossover in 1D, 2D and 3D polymeric Fe(II) networks. Top. Curr. Chem. 2004, 233, 229–257. [Google Scholar]
- Sciortino, N.F.; Neville, S.M. Two-Dimensional Coordination Polymers with Spin Crossover Functionality. Aust. J. Chem. 2014, 67, 1553–1562. [Google Scholar] [CrossRef]
- Kitazawa, T.; Gomi, Y.; Takahasi, M.; Takeda, M.; Enomoto, M.; Miyazaki, A.; Enoki, T. Spin-crossover behaviour of the coordination polymer FeII(C5H5N)2NiII(CN)4. J. Mater. Chem. 1996, 6, 119–121. [Google Scholar] [CrossRef]
- Ni, Z.-P.; Liu, J.-L.; Hogue, N.; Liu, W.; Li, J.-Y.; Chen, Y.-C.; Tong, M.-L. Recent advances in guest effects on spin-crossover behavior in Hofmann-type metal-organic frameworks. Coord. Chem. Rev. 2017, 335, 28–43. [Google Scholar] [CrossRef]
- Rodríguez-Velamazán, J.A.; Castro, M.; Palacios, E.; Burriel, R.; Kitazawa, T.; Kawasaki, T. A Two-Step Spin Transition with a Disordered Intermediate State in a New Two-Dimensional Coordination Polymer. J. Phys. Chem. B 2007, 111, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Velamazán, J.A.; Carbonera, C.; Castro, M.; Palacios, E.; Kitazawa, T.; Létard, J.F.; Burriel, R. Two-Step Thermal Spin Transition and LIESST Relaxation of the Polymeric Spin-Crossover Compounds Fe(X-py)2[Ag(CN)2]2 (X=H, 3-methyl, 4-methyl, 3,4-dimethyl, 3-Cl). Chem. Eur. J. 2010, 16, 8785–8796. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.C.; Gaspar, A.B.; Galet, A.; Real, J.A. Spin-Crossover Behavior in Cyanide-Bridged Iron(II)-Silver(I) Bimetallic 2D Hofmann-like Metal-Organic Frameworks. Inorg. Chem. 2007, 46, 8182–8192. [Google Scholar] [CrossRef] [PubMed]
- Agustí, C.; Muñoz, M.C.; Gaspar, A.B.; Real, J.A. Spin-Crossover Behavior in Cyanide-bridged Iron(II)-Gold(I) Bimetallic 2D Hofmann-like Metal-Organic Frameworks. Inorg. Chem. 2008, 47, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Hiiuk, V.M.; Shova, S.; Rotaru, A.; Ksenofontov, V.; Fritsky, I.O.; Gural’skiy, I.A. Room temperature hysteretic spin crossover in a new cyanoheterometallic framework. Chem. Commun. 2019, 55, 3359–3362. [Google Scholar] [CrossRef] [PubMed]
- Kosone, T.; Kawasaki, T.; Tomori, I.; Okabayashi, J.; Kitazawa, T. Modification of Cooperativity and Critical Temperatures on a Hofmann-Like Template Structure by Modular Substituent. Inorganics 2017, 5, 55. [Google Scholar] [CrossRef]
- Kosone, T.; Kitazawa, T. Guest-dependent spin transition with long range intermediate state for 2-dimensional Hofmann-like coordination polymer. Inorg. Chim. Acta 2016, 439, 159–163. [Google Scholar] [CrossRef]
- Sciortino, N.F.; Scherl-Gruenwald, K.R.; Chastanet, G.; Halder, G.J.; Chapman, K.W.; Letard, J.-F.; Kepert, C.J. Hysteretic Three-Step Spin Crossover in a Thermo- and Photochromic 3D Pillared Hofmann-type Metal-Organic Framework. Angew. Chem. Int. Ed. 2012, 51, 10154–10158. [Google Scholar] [CrossRef]
- Kucheriv, O.I.; Shylin, S.I.; Ksenofontov, V.; Dechert, S.; Haukka, M.; Fritsky, I.O.; Gural’skiy, I.A. Spin Crossover in Fe(II)–M(II) Cyanoheterobimetallic Frameworks (M = Ni, Pd, Pt) with 2-Substituted Pyrazines. Inorg. Chem. 2016, 55, 4906–4914. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS Program for Empirical Absorption Correction for Area Detector Data; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXTL Program for the Solution of Crystal Structure; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Schier, A. Argentophilic Interactions. Angew. Chem. Int. Ed. 2015, 54, 746–784. [Google Scholar] [CrossRef] [PubMed]
- Romstedt, H.; Hauser, A.; Spiering, H.; Gutlich, P. Modelling of two step high spin⇌ low spin transitions using the cluster variation method. J. Phys. Chem. Solids 1998, 59, 1353–1362. [Google Scholar] [CrossRef]




{kind=link}
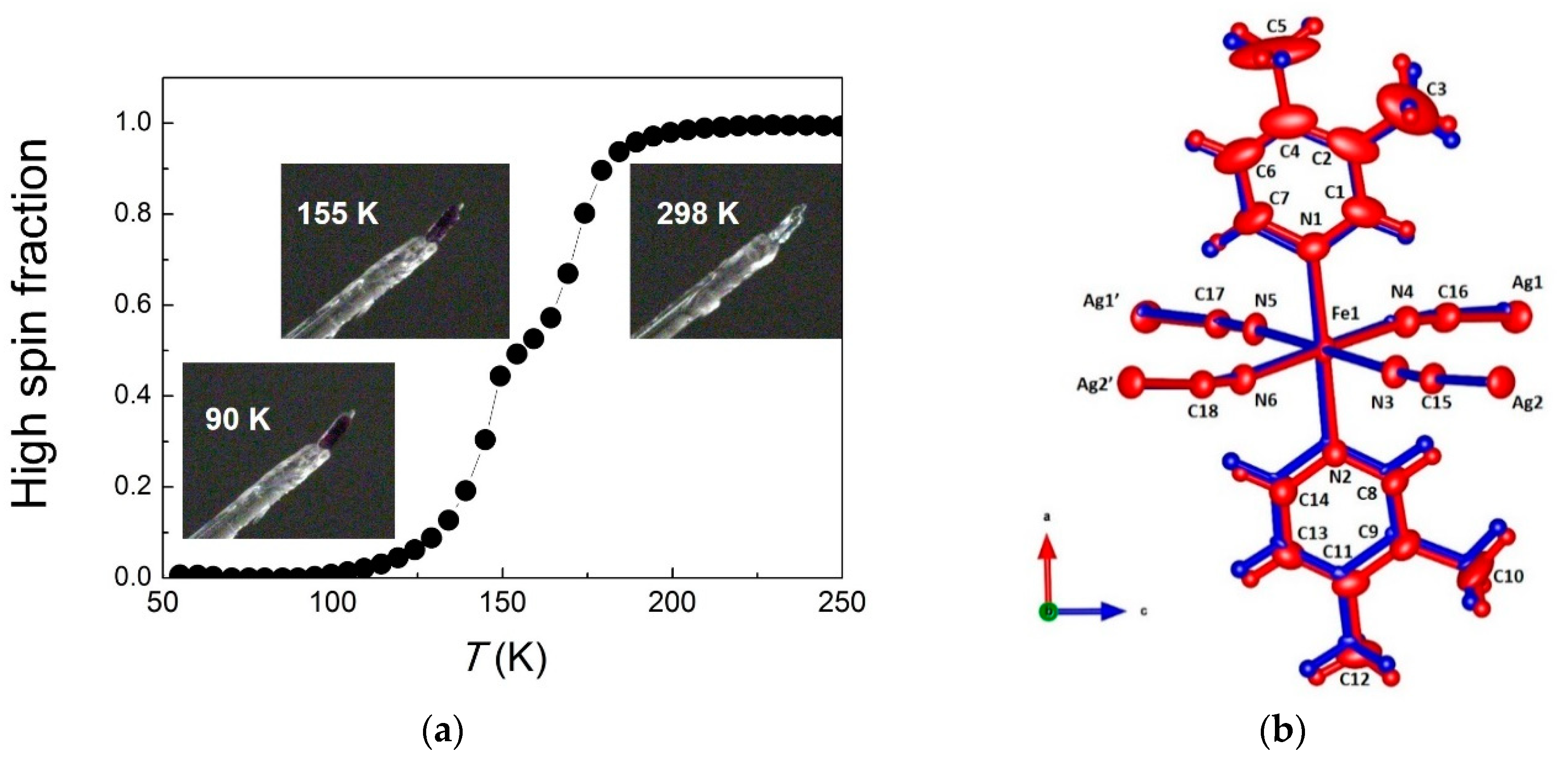{kind=link}
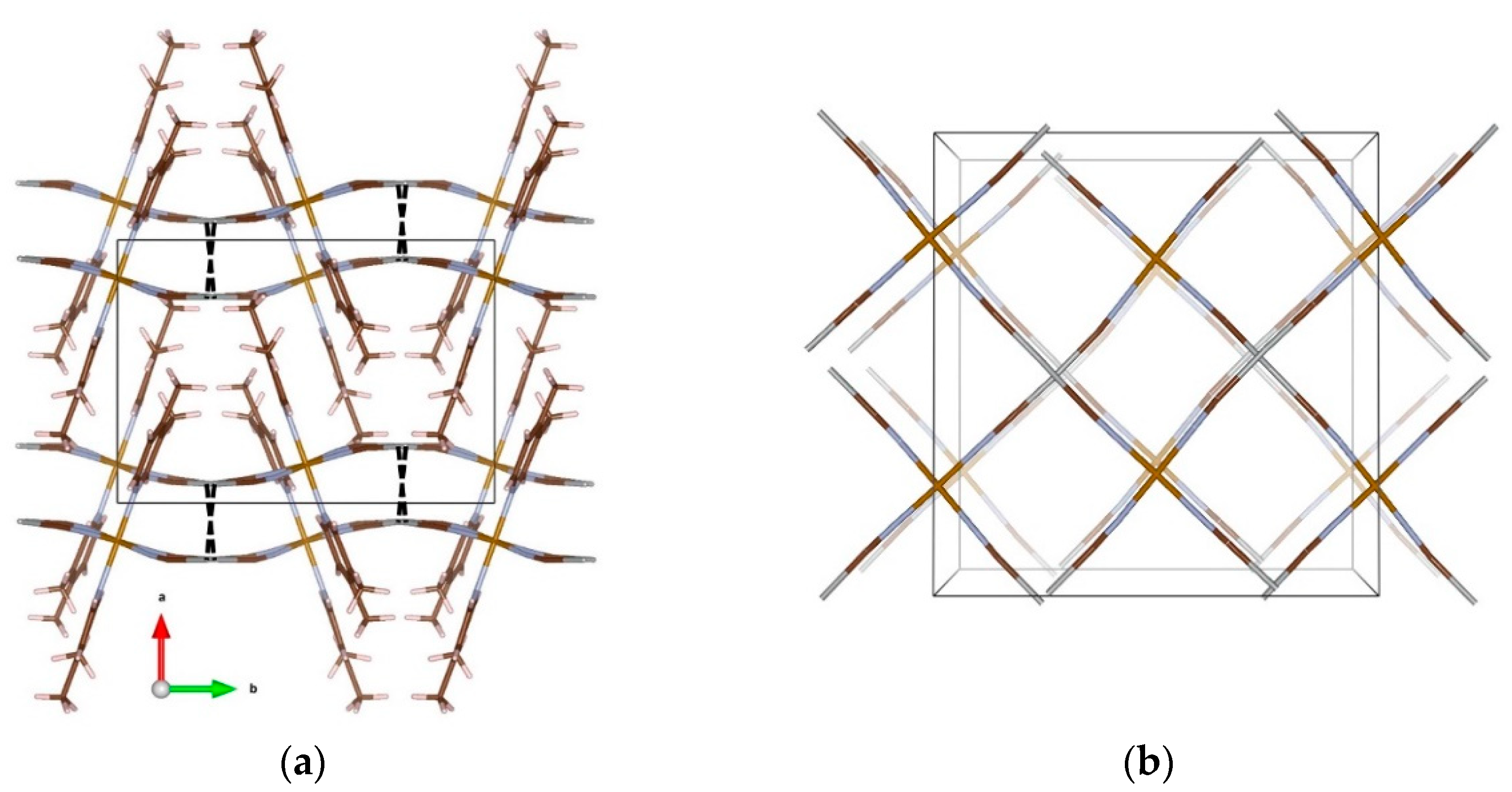{kind=link}
{kind=link}
| Formula | C18 H18 Ag2 Fe N6 | ||
|---|---|---|---|
| Formula weight | 589.97 | ||
| T (K) | 298 K | 155 K | 90 K |
| Crystal system | monoclinic | monoclinic | monoclinic |
| Space Group | P 21/c | P 21/c | P 21/c |
| Z | 4 | 4 | 4 |
| a (Å) | 10.0093(8) | 9.898(8) | 9.9550(13) |
| b (Å) | 15.0039(11) | 14.609(12) | 14.2900(19) |
| c (Å) | 14.8435(11) | 14.573(12) | 14.3979(19) |
| β (°) | 91.4390(10) | 91.356(10) | 91.090(2) |
| V (Å3) | 2228.5(3) | 2107(3) | 2047.8(5) |
| Fe–N(1) (Å) | 2.211(4) | 2.09(2) | 1.997(5) |
| Fe–N(2) (Å) | 2.208(4) | 2.08(2) | 2.001(4) |
| Fe–N(3) (Å) | 2.155(4) | 2.01(2) | 1.939(4) |
| Fe–N(4) (Å) | 2.162(4) | 2.08(3) | 1.943(4) |
| Fe–N(5) (Å) | 2.162(4) | 2.03(3) | 1.942(4) |
| Fe–N(6) (Å) | 2.180(4) | 2.04(3) | 1.940(3) |
| No. of measured, independent and observed [I > 2σ (I)] reflections (Rint) | 10391, 3404, 2672 (0.023) | 9951, 3900, 2788 (0.084) | 13965, 4884, 4043 (0.030) |
| R[F2 > 2σ(F2)], wR(F2), S | 0.030, 0.067, 1.04 | 0.109, 0.313, 1.85 | 0.040, 0.141, 1.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Velamazán, J.A.; Kitase, K.; Palacios, E.; Castro, M.; Fernández-Blanco, Á.; Burriel, R.; Kitazawa, T. Structural Insights into the Two-Step Spin-Crossover Compound Fe(3,4-dimethyl-pyridine)2[Ag(CN)2]2. Crystals 2019, 9, 316. https://doi.org/10.3390/cryst9060316
Rodríguez-Velamazán JA, Kitase K, Palacios E, Castro M, Fernández-Blanco Á, Burriel R, Kitazawa T. Structural Insights into the Two-Step Spin-Crossover Compound Fe(3,4-dimethyl-pyridine)2[Ag(CN)2]2. Crystals. 2019; 9(6):316. https://doi.org/10.3390/cryst9060316
Chicago/Turabian StyleRodríguez-Velamazán, José Alberto, Kosuke Kitase, Elías Palacios, Miguel Castro, Ángel Fernández-Blanco, Ramón Burriel, and Takafumi Kitazawa. 2019. "Structural Insights into the Two-Step Spin-Crossover Compound Fe(3,4-dimethyl-pyridine)2[Ag(CN)2]2" Crystals 9, no. 6: 316. https://doi.org/10.3390/cryst9060316
APA StyleRodríguez-Velamazán, J. A., Kitase, K., Palacios, E., Castro, M., Fernández-Blanco, Á., Burriel, R., & Kitazawa, T. (2019). Structural Insights into the Two-Step Spin-Crossover Compound Fe(3,4-dimethyl-pyridine)2[Ag(CN)2]2. Crystals, 9(6), 316. https://doi.org/10.3390/cryst9060316