Unfolded Protein Responses With or Without Unfolded Proteins?

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Conceptual Model for Sensing Stress
3. Ire1p and IRE1α Structure and Function
3.1. Ire1p/IRE1α Structure
3.2. Ire1p Functional Assays
4. Ire1p/IRE1α Activation Models
4.1. Density Dependent Activation of Ire1p
4.2. Ire1p Sensing of Unfolded Proteins

5. The Regulatory Role of Kar2p/BiP
5.1. BiP Association—Stable or Transient?
6. Ire1p Activation Independent of Unfolded Proteins: The Inositol Sensing Problem
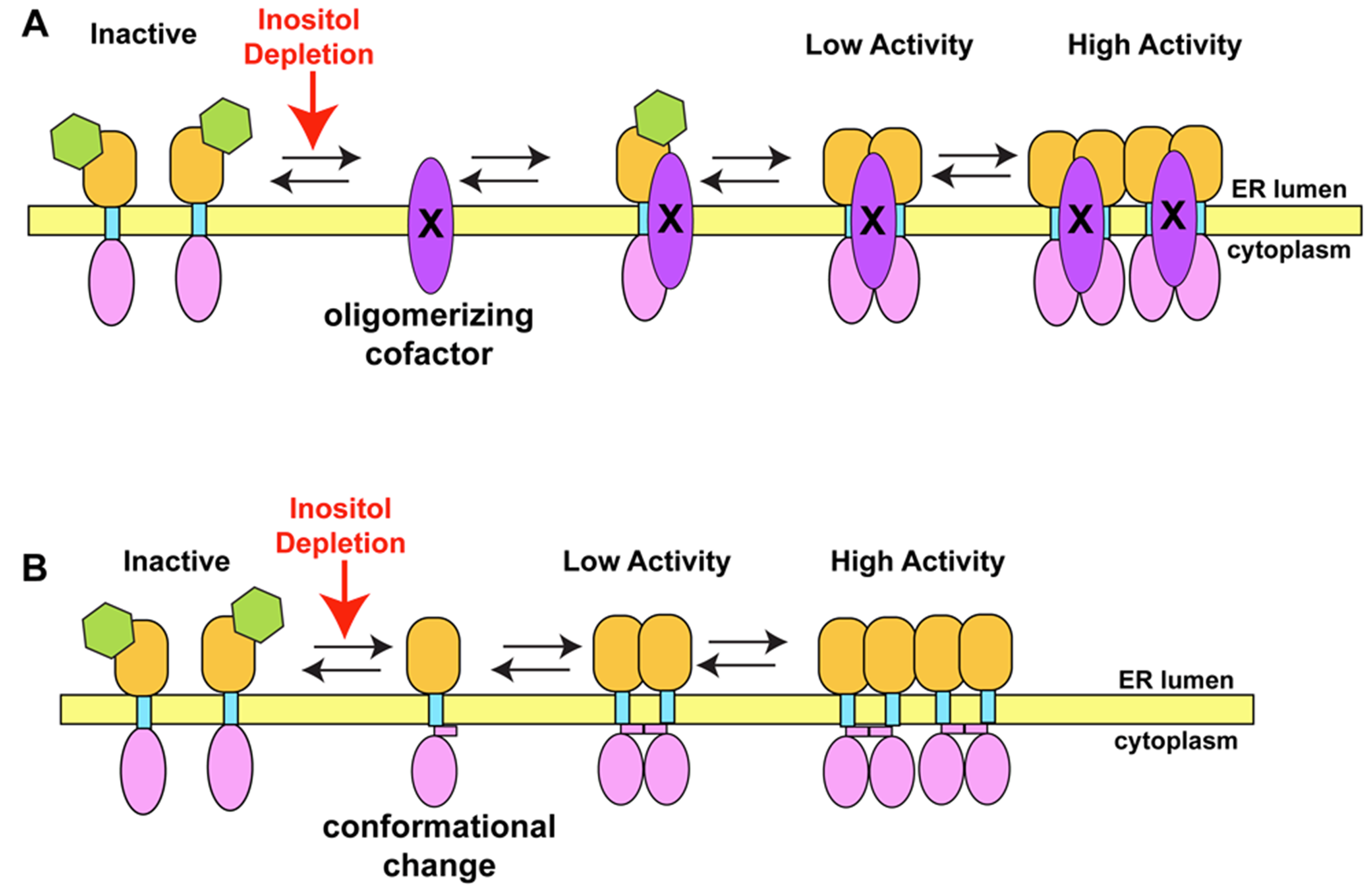
6.1. Ire1p or Protein Co-factor Detects Inositol Changes Model

6.2. How Might Ire1p Be Activated During Inositol Withdrawal or in the Absence of a Luminal Domain?
7. Ire1p Inactivation
8. Conclusion
Acknowledgments
Conflict of Interest
References
- van Anken, E.; Braakman, I.; Craig, E. Versatility of the endoplasmic reticulum protein folding factory. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 191–228. [Google Scholar] [CrossRef]
- Kowarik, M.; Kung, S.; Martoglio, B.; Helenius, A. Protein folding during cotranslational translocation in the endoplasmic reticulum. Mol. Cell. 2002, 10, 769–778. [Google Scholar] [CrossRef]
- Brodsky, J.L.; Skach, W.R. Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Curr. Opin. Cell. Biol. 2011, 23, 464–475. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Feige, M.J.; Groscurth, S.; Marcinowski, M.; Shimizu, Y.; Kessler, H.; Hendershot, L.M.; Buchner, J. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol. Cell. 2009, 34, 569–579. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell. Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Bernales, S.; Papa, F.R.; Walter, P. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell. Dev. Biol. 2006, 22, 487–508. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell. Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Sadighi Akha, A.A.; Raden, D.; Kaufman, R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006, 4, e374. [Google Scholar] [CrossRef]
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.J.; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell. Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded- protein response. Nat. Cell. Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell. 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell. Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell. Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Chawla, A.; Chakrabarti, S.; Ghosh, G.; Niwa, M. Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. J. Cell. Biol. 2011, 193, 41–50. [Google Scholar] [CrossRef]
- Rubio, C.; Pincus, D.; Korennykh, A.; Schuck, S.; El-Samad, H.; Walter, P. Homeostatic adaptation to endoplasmic reticulum stress depends on Ire1 kinase activity. J. Cell. Biol. 2011, 193, 171–184. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Hegde, R.S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell. Biol. 2010, 189, 783–794. [Google Scholar] [CrossRef]
- Sierra, F. Is (your cellular response to) stress killing you? J. Gerontol. 2006, 61, 557–561. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Arsene, F.; Tomoyasu, T.; Bukau, B. The heat shock response of Escherichia coli. Int. J. Food Microbiol. 2000, 55, 3–9. [Google Scholar] [CrossRef]
- Anckar, J.; Sistonen, L. Regulation of HSF1 function in the heat stress response: Implications in aging and disease. Annu. Rev. Biochem. 2011, 80, 1089–1115. [Google Scholar] [CrossRef]
- Nikawa, J.; Yamashita, S. IRE1 encodes a putative protein kinase containing a membrane-spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol. Microbiol. 1992, 6, 1441–1446. [Google Scholar] [CrossRef]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–756. [Google Scholar] [CrossRef]
- Li, W.W.; Alexandre, S.; Cao, X.; Lee, A.S. Transactivation of the grp78 promoter by Ca2+ depletion. A comparative analysis with A23187 and the endoplasmic reticulum Ca(2+)-ATPase inhibitor thapsigargin. J. Biol. Chem. 1993, 268, 12003–12009. [Google Scholar]
- Bork, P.; Sander, C. A hybrid protein kinase-RNase in an interferon-induced pathway? FEBS Lett. 1993, 334, 149–152. [Google Scholar]
- Sidrauski, C.; Cox, J.S.; Walter, P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 1996, 87, 405–413. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Oikawa, D.; Kimata, Y.; Takeuchi, M.; Kohno, K. An essential dimer-forming subregion of the endoplasmic reticulum stress sensor Ire1. Biochem. J. 2005, 391, 135–142. [Google Scholar] [CrossRef]
- Kimata, Y.; Oikawa, D.; Shimizu, Y.; Ishiwata-Kimata, Y.; Kohno, K. A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. J. Cell. Biol. 2004, 167, 445–456. [Google Scholar] [CrossRef]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef]
- Kimata, Y.; Ishiwata-Kimata, Y.; Ito, T.; Hirata, A.; Suzuki, T.; Oikawa, D.; Takeuchi, M.; Kohno, K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J. Cell. Biol. 2007, 179, 75–86. [Google Scholar] [CrossRef]
- Credle, J.J.; Finer-Moore, J.S.; Papa, F.R.; Stroud, R.M.; Walter, P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 18773–18784. [Google Scholar]
- Cox, J.S.; Walter, P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 1996, 87, 391–404. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell. Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande, W.L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; Tsuru, A.; Kohno, K. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell. Biol. 2001, 3, 158–164. [Google Scholar] [CrossRef]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar]
- Lin, J.H.; Li, H.; Zhang, Y.; Ron, D.; Walter, P. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS One 2009, 4, e4170. [Google Scholar] [CrossRef]
- Tsvetanova, N.G.; Riordan, D.P.; Brown, P.O. The Yeast Rab GTPase Ypt1 Modulates Unfolded Protein Response Dynamics by Regulating the Stability of HAC1 RNA. PLoS Gen. 2012, 8, e1002862. [Google Scholar] [CrossRef]
- Chapman, R.E.; Walter, P. Translational attenuation mediated by an mRNA intron. Curr. Biol. 1997, 7, 850–859. [Google Scholar] [CrossRef]
- Kawahara, T.; Yanagi, H.; Yura, T.; Mori, K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol. Biol. Cell. 1997, 8, 1845–1862. [Google Scholar]
- Pincus, D.; Chevalier, M.W.; Aragon, T.; van Anken, E.; Vidal, S.E.; El-Samad, H.; Walter, P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010, 8, e1000415. [Google Scholar] [CrossRef]
- Ruegsegger, U.; Leber, J.H.; Walter, P. Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell 2001, 107, 103–114. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Dis. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Shamu, C.E.; Walter, P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996, 15, 3028–3039. [Google Scholar]
- Lai, C.W.; Aronson, D.E.; Snapp, E.L. BiP availability distinguishes states of homeostasis and stress in the endoplasmic reticulum of living cells. Mol. Biol. Cell. 2010, 21, 1909–19021. [Google Scholar] [CrossRef]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Stroud, R.M.; Zhang, C.; Shokat, K.M.; Walter, P. Cofactor-mediated conformational control in the bifunctional kinase/RNase Ire1. BMC Biol. 2011, 9, 48. [Google Scholar] [CrossRef]
- Lee, K.P.; Dey, M.; Neculai, D.; Cao, C.; Dever, T.E.; Sicheri, F. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 2008, 132, 89–100. [Google Scholar] [CrossRef]
- Liu, C.Y.; Wong, H.N.; Schauerte, J.A.; Kaufman, R.J. The protein kinase/endoribonuclease IRE1alpha that signals the unfolded protein response has a luminal N-terminal ligand-independent dimerization domain. J. Biol. Chem. 2002, 277, 18346–18356. [Google Scholar]
- Aragon, T.; van Anken, E.; Pincus, D.; Serafimova, I.M.; Korennykh, A.V.; Rubio, C.A.; Walter, P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 2009, 457, 736–740. [Google Scholar] [CrossRef]
- Promlek, T.; Ishiwata-Kimata, Y.; Shido, M.; Sakuramoto, M.; Kohno, K.; Kimata, Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol. Biol. Cell. 2011, 22, 3520–3532. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar]
- Ali, M.M.; Bagratuni, T.; Davenport, E.L.; Nowak, P.R.; Silva-Santisteban, M.C.; Hardcastle, A.; McAndrews, C.; Rowlands, M.G.; Morgan, G.J.; Aherne, W.; et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011, 30, 894–905. [Google Scholar] [CrossRef]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The unfolded protein response signals through high-order assembly of Ire1. Nature 2009, 457, 687–693. [Google Scholar] [CrossRef]
- Wiseman, R.L.; Zhang, Y.; Lee, K.P.; Harding, H.P.; Haynes, C.M.; Price, J.; Sicheri, F.; Ron, D. Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Mol. Cell. 2010, 38, 291–304. [Google Scholar] [CrossRef]
- Thibault, G.; Ismail, N.; Ng, D.T. The unfolded protein response supports cellular robustness as a broad-spectrum compensatory pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20597–20602. [Google Scholar] [CrossRef]
- Onn, A.; Ron, D. Modeling the endoplasmic reticulum unfolded protein response. Nat. Struct. Mol. Biol. 2010, 17, 924–925. [Google Scholar] [CrossRef]
- Oikawa, D.; Kimata, Y.; Kohno, K. Self-association and BiP dissociation are not sufficient for activation of the ER stress sensor Ire1. J. Cell. Sci. 2007, 120, 1681–1688. [Google Scholar] [CrossRef]
- Normington, K.; Kohno, K.; Kozutsumi, Y.; Gething, M.J.; Sambrook, J.S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell 1989, 57, 1223–1236. [Google Scholar] [CrossRef]
- Liu, C.Y.; Xu, Z.; Kaufman, R.J. Structure and intermolecular interactions of the luminal dimerization domain of human IRE1alpha. J. Biol. Chem. 2003, 278, 17680–17687. [Google Scholar] [CrossRef]
- Gething, M.J. Role and regulation of the ER chaperone BiP. Semin. Cell. Dev. Biol. 1999, 10, 465–472. [Google Scholar] [CrossRef]
- Dudek, J.; Benedix, J.; Cappel, S.; Greiner, M.; Jalal, C.; Muller, L.; Zimmermann, R. Functions and pathologies of BiP and its interaction partners. Cell. Mol. Life Sci. 2009, 66, 1556–1569. [Google Scholar] [CrossRef]
- Hendershot, L.M. The ER function BiP is a master regulator of ER function. Mt. Sinai. J. Med. 2004, 71, 289–297. [Google Scholar]
- Otero, J.H.; Lizak, B.; Hendershot, L.M. Life and death of a BiP substrate. Semin. Cell. Dev. Biol. 2010, 21, 472–478. [Google Scholar] [CrossRef]
- Kohno, K.; Normington, K.; Sambrook, J.; Gething, M.J.; Mori, K. The promoter region of the yeast KAR2 (BiP) gene contains a regulatory domain that responds to the presence of unfolded proteins in the endoplasmic reticulum. Mol. Cell. Biol. 1993, 13, 877–890. [Google Scholar]
- Schuldiner, M.; Collins, S.R.; Thompson, N.J.; Denic, V.; Bhamidipati, A.; Punna, T.; Ihmels, J.; Andrews, B.; Boone, C.; Greenblatt, J.F.; et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 2005, 123, 507–519. [Google Scholar] [CrossRef]
- Okamura, K.; Kimata, Y.; Higashio, H.; Tsuru, A.; Kohno, K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem. Biophys. Res. Commun. 2000, 279, 445–450. [Google Scholar] [CrossRef]
- Mintz, M.; Vanderver, A.; Brown, K.J.; Lin, J.; Wang, Z.; Kaneski, C.; Schiffmann, R.; Nagaraju, K.; Hoffman, E.P.; Hathout, Y. Time series proteome profiling to study endoplasmic reticulum stress response. J. Proteom. Res. 2008, 7, 2435–2444. [Google Scholar] [CrossRef]
- Wiest, D.L.; Burkhardt, J.K.; Hester, S.; Hortsch, M.; Meyer, D.I.; Argon, Y. Membrane biogenesis during B cell differentiation: Most endoplasmic reticulum proteins are expressed coordinately. J. Cell. Biol. 1990, 110, 1501–1511. [Google Scholar] [CrossRef]
- Hsu, C.L.; Prasad, R.; Blackman, C.; Ng, D.T. ER stress regulation of the Kar2p/BiP chaperone alleviates proteotoxicity via dual degradation pathways. Mol. Biol. Cell. 2012, 23, 630–641. [Google Scholar] [CrossRef]
- Kimata, Y.; Kimata, Y.I.; Shimizu, Y.; Abe, H.; Farcasanu, I.C.; Takeuchi, M.; Rose, M.D.; Kohno, K. Genetic evidence for a role of BiP/Kar2 that regulates Ire1 in response to accumulation of unfolded proteins. Mol. Biol. Cell. 2003, 14, 2559–2569. [Google Scholar] [CrossRef]
- Liu, C.Y.; Schroder, M.; Kaufman, R.J. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 2000, 275, 24881–24885. [Google Scholar]
- Morris, J.A.; Dorner, A.J.; Edwards, C.A.; Hendershot, L.M.; Kaufman, R.J. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J. Biol. Chem. 1997, 272, 4327–4334. [Google Scholar]
- Guth, S.; Volzing, C.; Muller, A.; Jung, M.; Zimmermann, R. Protein transport into canine pancreatic microsomes: a quantitative approach. Eur. J. Biochem. 2004, 271, 3200–3207. [Google Scholar] [CrossRef]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar]
- Ghaemmaghami, S.; Huh, W.K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O'Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef]
- Lai, C.W.; Otero, J.H.; Hendershot, L.M.; Snapp, E. ERdj4 protein is a soluble endoplasmic reticulum (ER) DnaJ family protein that interacts with ER-associated degradation machinery. J. Biol. Chem. 2012, 287, 7969–7978. [Google Scholar]
- Oikawa, D.; Kimata, Y.; Kohno, K.; Iwawaki, T. Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell. Res. 2009, 315, 2496–2504. [Google Scholar] [CrossRef]
- Marcinowski, M.; Holler, M.; Feige, M.J.; Baerend, D.; Lamb, D.C.; Buchner, J. Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat. Struct. Mol. Biol. 2011, 18, 150–158. [Google Scholar] [CrossRef]
- Vanhove, M.; Usherwood, Y.K.; Hendershot, L.M. Unassembled Ig heavy chains do not cycle from BiP in vivo but require light chains to trigger their release. Immunity 2001, 15, 105–114. [Google Scholar] [CrossRef]
- Lajoie, P.; Moir, R.D.; Willis, I.M.; Snapp, E.L. Kar2p Availability Defines Distinct Forms of Endoplasmic Reticulum Stress in Living Cells. Mol. Biol. Cell. 2012. [Google Scholar] [CrossRef]
- Kang, S.W.; Rane, N.S.; Kim, S.J.; Garrison, J.L.; Taunton, J.; Hegde, R.S. Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell 2006, 127, 999–1013. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Kuznetsov, G.; Chen, L.B.; Nigam, S.K. Multiple molecular chaperones complex with misfolded large oligomeric glycoproteins in the endoplasmic reticulum. J. Biol. Chem. 1997, 272, 3057–3063. [Google Scholar] [CrossRef]
- Chambers, J.E.; Petrova, K.; Tomba, G.; Vendruscolo, M.; Ron, D. ADP ribosylation adapts an ER chaperone response to short-term fluctuations in unfolded protein load. J. Cell. Biol. 2012, 198, 371–385. [Google Scholar] [CrossRef]
- Freiden, P.J.; Gaut, J.R.; Hendershot, L.M. Interconversion of three differentially modified and assembled forms of BiP. EMBO J. 1992, 11, 63–70. [Google Scholar]
- Leno, G.H.; Ledford, B.E. ADP-ribosylation of the 78-kDa glucose-regulated protein during nutritional stress. FEBS J. 1989, 186, 205–211. [Google Scholar] [CrossRef]
- Hendershot, L.M.; Ting, J.; Lee, A.S. Identity of the immunoglobulin heavy-chain-binding protein with the 78,000-dalton glucose-regulated protein and the role of posttranslational modifications in its binding function. Mol. Cell. Biol. 1988, 8, 4250–4256. [Google Scholar]
- Laitusis, A.L.; Brostrom, M.A.; Brostrom, C.O. The dynamic role of GRP78/BiP in the coordination of mRNA translation with protein processing. J. Biol. Chem. 1999, 274, 486–493. [Google Scholar]
- Carlsson, L.; Lazarides, E. ADP-ribosylation of the Mr 83,000 stress-inducible and glucose-regulated protein in avian and mammalian cells: modulation by heat shock and glucose starvation. Proc. Natl. Acad. Sci. USA 1983, 80, 4664–4668. [Google Scholar] [CrossRef]
- Griffiths, G.; Warren, G.; Quinn, P.; Mathieu-Costello, O.; Hoppeler, H. Density of newly synthesized plasma membrane proteins in intracellular membranes. I. Stereological studies. J. Cell. Biol. 1984, 98, 2133–2141. [Google Scholar] [CrossRef]
- Todd-Corlett, A.; Jones, E.; Seghers, C.; Gething, M.J. Lobe IB of the ATPase domain of Kar2p/BiP interacts with Ire1p to negatively regulate the unfolded protein response in Saccharomyces cerevisiae. J. Mol. Biol. 2007, 67, 770–787. [Google Scholar]
- Cox, J.S.; Chapman, R.E.; Walter, P. The unfolded protein response coordinates the production of endoplasmic reticulum membrane. Mol. Biol. Cell. 1997, 8, 1805–1814. [Google Scholar]
- Nikawa, J.; Akiyoshi, M.; Hirata, S.; Fukuda, T. Saccharomyces cerevisiae IRE2/HAC1 is involved in IRE1-mediated KAR2 expression. Nucleic. Acids Res. 1996, 24, 4222–4226. [Google Scholar] [CrossRef]
- Mori, K.; Ogawa, N.; Kawahara, T.; Yanagi, H.; Yura, T. mRNA splicing-mediated C-terminal replacement of transcription factor Hac1p is required for efficient activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2000, 97, 4660–4665. [Google Scholar]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Brickner, J.H.; Walter, P. Gene recruitment of the activated INO1 locus to the nuclear membrane. PLoS Biol. 2004, 2, e342. [Google Scholar] [CrossRef] [Green Version]
- Contreras, F.X.; Ernst, A.M.; Haberkant, P.; Bjorkholm, P.; Lindahl, E.; Gonen, B.; Tischer, C.; Elofsson, A.; von Heijne, G.; Thiele, C.; et al. Molecular recognition of a single sphingolipid species by a protein's transmembrane domain. Nature 2012, 481, 525–529. [Google Scholar] [CrossRef]
- Hetz, C.; Glimcher, L.H. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol. Cell. 2009, 35, 551–561. [Google Scholar] [CrossRef]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1alpha. Physiol Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef]
- Lisbona, F.; Rojas-Rivera, D.; Thielen, P.; Zamorano, S.; Todd, D.; Martinon, F.; Glavic, A.; Kress, C.; Lin, J.H.; Walter, P.; et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol. Cell. 2009, 33, 679–691. [Google Scholar] [CrossRef]
- Marcu, M.G.; Doyle, M.; Bertolotti, A.; Ron, D.; Hendershot, L.; Neckers, L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol. Cell. Biol. 2002, 22, 8506–8513. [Google Scholar] [CrossRef]
- Lamb, H.K.; Mee, C.; Xu, W.; Liu, L.; Blond, S.; Cooper, A.; Charles, I.G.; Hawkins, A.R. The affinity of a major Ca2+ binding site on GRP78 is differentially enhanced by ADP and ATP. J. Biol. Chem. 2006, 281, 8796–8805. [Google Scholar]
- Guo, J.; Polymenis, M. Dcr2 targets Ire1 and downregulates the unfolded protein response in Saccharomyces cerevisiae. EMBO Rep. 2006, 7, 1124–1127. [Google Scholar] [CrossRef]
- Welihinda, A.A.; Tirasophon, W.; Green, S.R.; Kaufman, R.J. Protein serine/threonine phosphatase Ptc2p negatively regulates the unfolded-protein response by dephosphorylating Ire1p kinase. Mol. Cell. Biol. 1998, 18, 1967–1977. [Google Scholar]
- Frost, A.; Elgort, M.G.; Brandman, O.; Ives, C.; Collins, S.R.; Miller-Vedam, L.; Weibezahn, J.; Hein, M.Y.; Poser, I.; Mann, M.; et al. Functional repurposing revealed by comparing S. pombe and S. cerevisiae genetic interactions. Cell 2012, 149, 1339–1352. [Google Scholar] [CrossRef]
- Liu, Y.; Chang, A. Heat shock response relieves ER stress. EMBO J. 2008, 27, 1049–1059. [Google Scholar] [CrossRef]
- DuRose, J.B.; Tam, A.B.; Niwa, M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol. Biol. Cell. 2006, 17, 3095–3107. [Google Scholar] [CrossRef]
- Maiuolo, J.; Bulotta, S.; Verderio, C.; Benfante, R.; Borgese, N. Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc. Natl. Acad. Sci. USA. 2011, 108, 7832–7837. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Snapp, E.L. Unfolded Protein Responses With or Without Unfolded Proteins? Cells 2012, 1, 926-950. https://doi.org/10.3390/cells1040926
Snapp EL. Unfolded Protein Responses With or Without Unfolded Proteins? Cells. 2012; 1(4):926-950. https://doi.org/10.3390/cells1040926
Chicago/Turabian StyleSnapp, Erik L. 2012. "Unfolded Protein Responses With or Without Unfolded Proteins?" Cells 1, no. 4: 926-950. https://doi.org/10.3390/cells1040926
APA StyleSnapp, E. L. (2012). Unfolded Protein Responses With or Without Unfolded Proteins? Cells, 1(4), 926-950. https://doi.org/10.3390/cells1040926