β2-Adrenergic Signaling Modulates Mitochondrial Function and Morphology in Skeletal Muscle in Response to Aerobic Exercise

 ,
, 
 , and
, and
Abstract
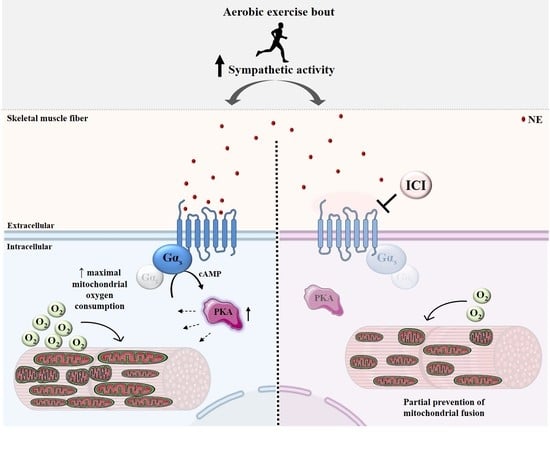
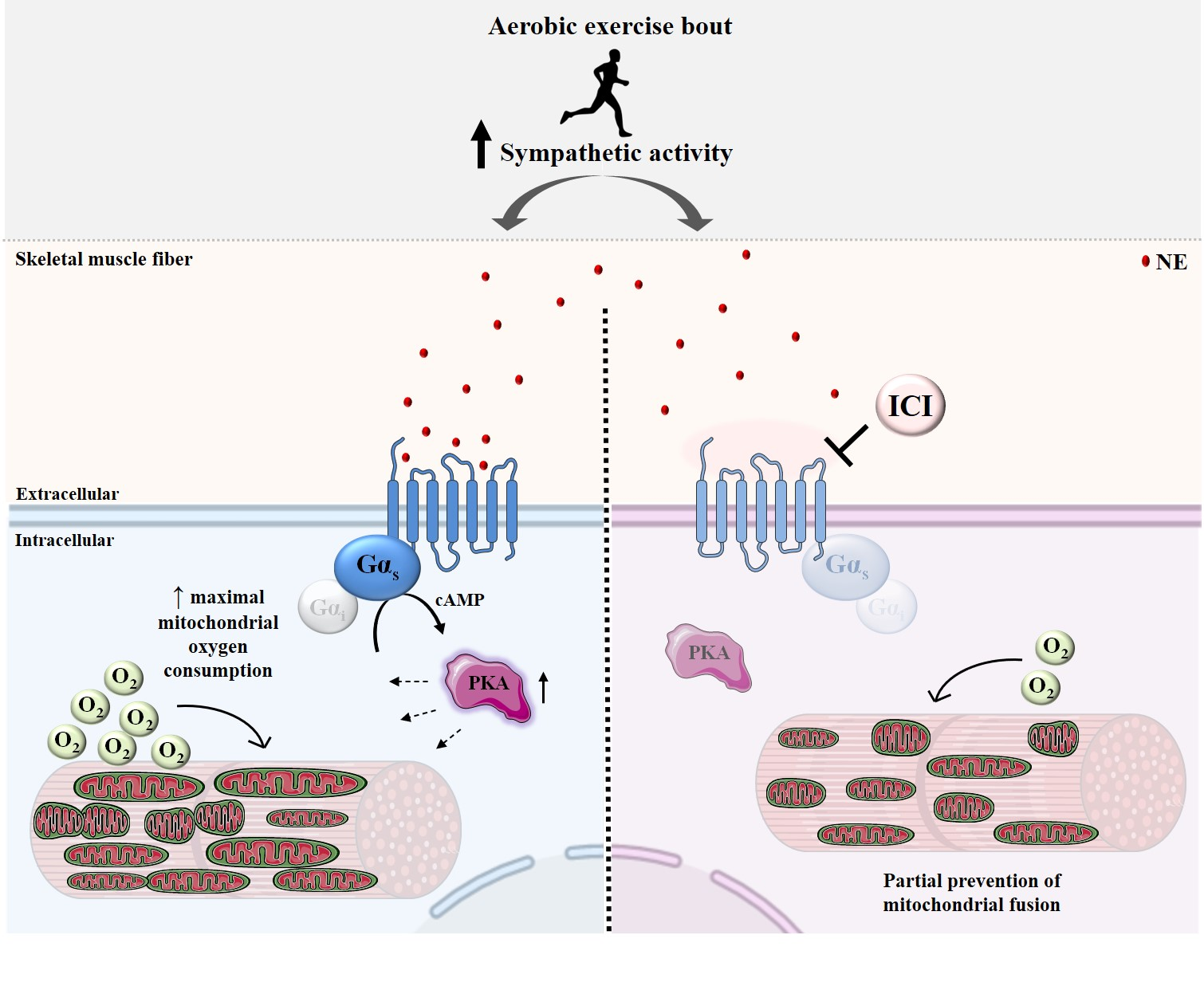
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
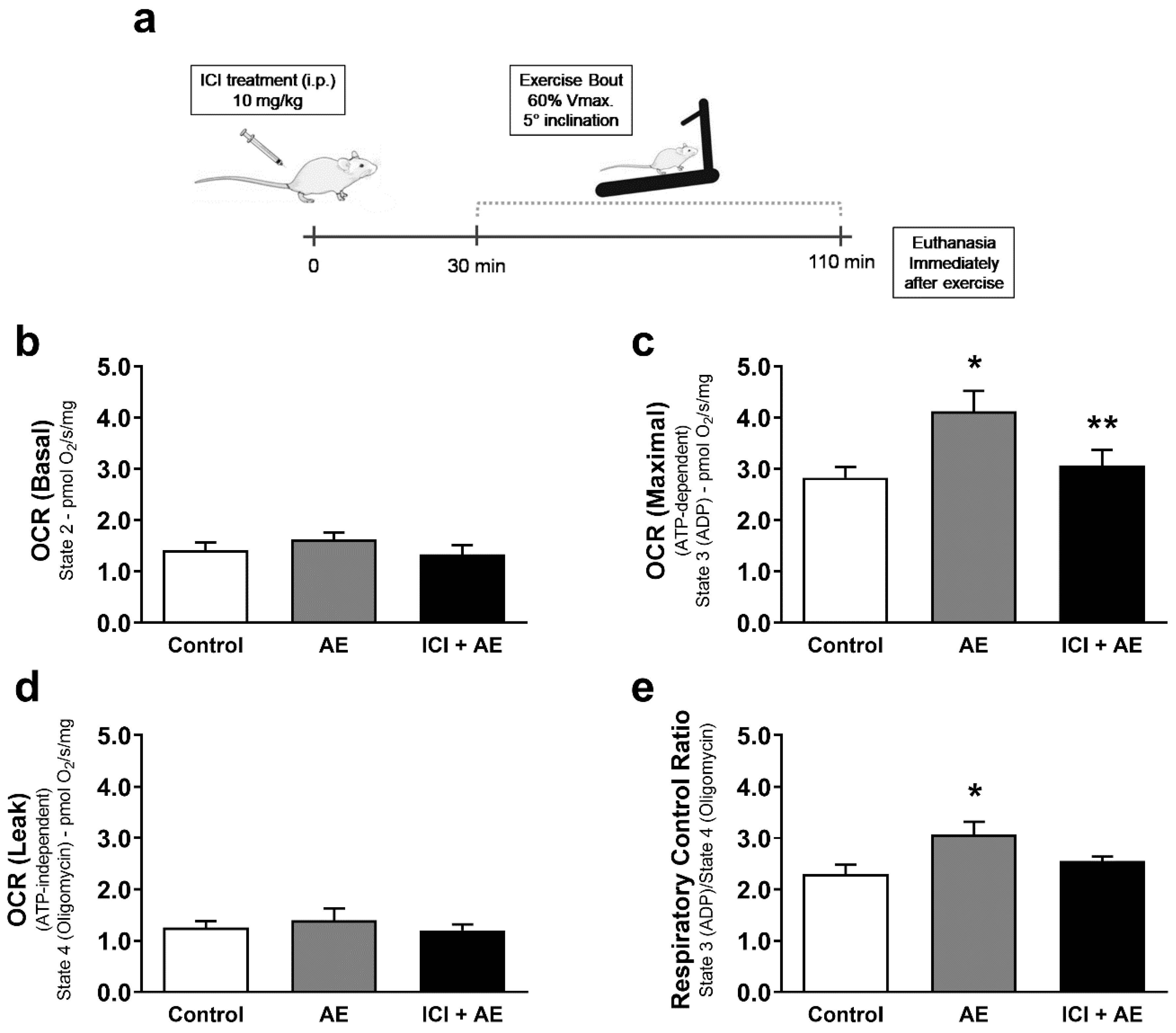
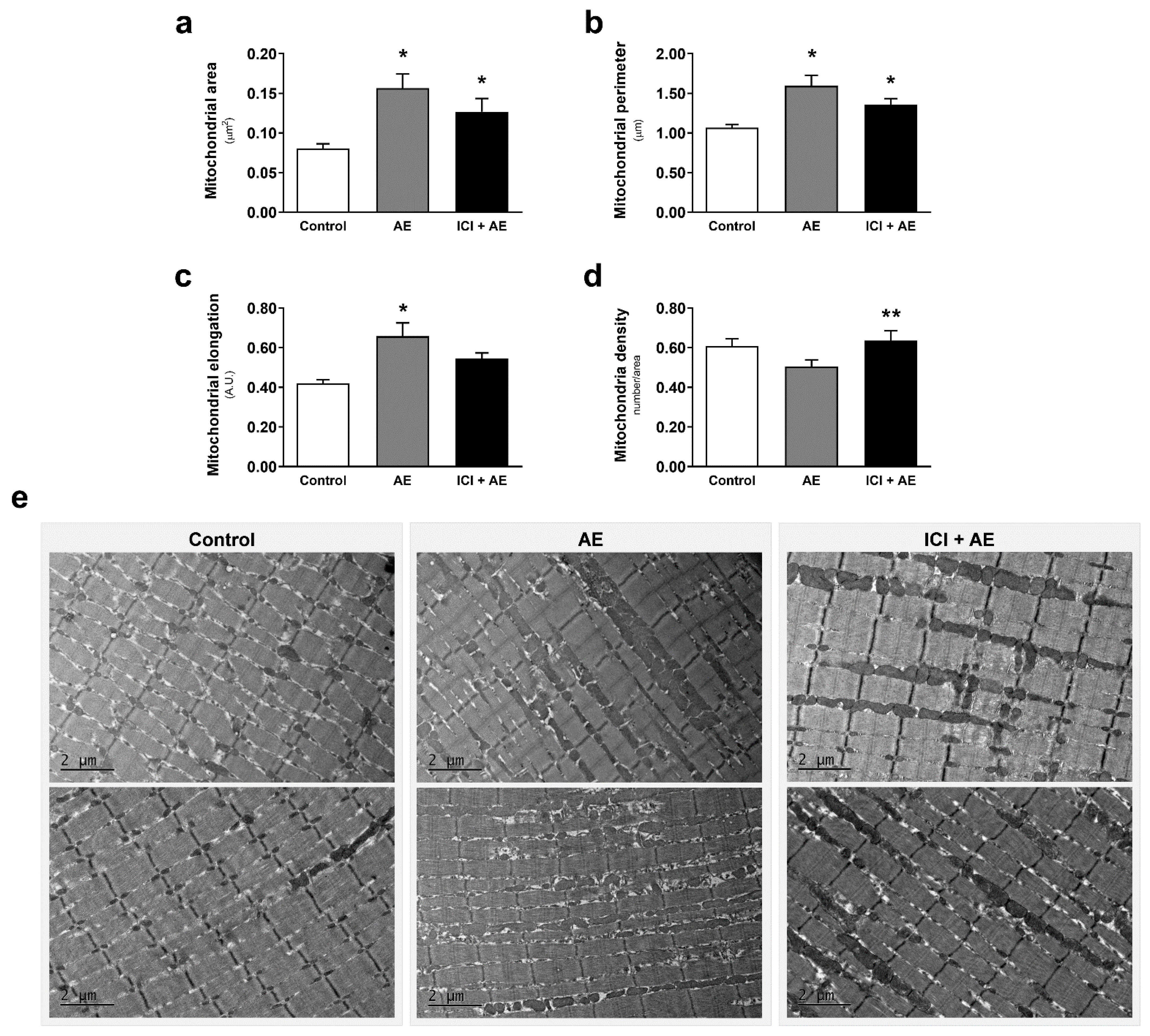
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloszy, J.O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar] [CrossRef]
- Hood, D. A Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle. Appl. Physiol. Nutr. Metab. 2009, 34, 465–472. [Google Scholar] [CrossRef]
- Cartoni, R.; Léger, B.; Hock, M.B.; Praz, M.; Crettenand, A.; Pich, S.; Ziltener, J.-L.; Luthi, F.; Dériaz, O.; Zorzano, A.; et al. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J. Physiol. 2005, 567, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Jiang, N.; Liu, H.; Liu, X.; Liu, D.; Zhao, F.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim. Biophys. Acta 2010, 1800, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.; Fortin, D.; Zoll, J.; N’Guessan, B.; Mettauer, B.; Lampert, E.; Veksler, V.; Ventura-Clapier, R. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. FASEB J. 2005, 19, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.C.; Baehr, L.M.; Gomes, K.M.S.; Bechara, L.R.G.; Voltarelli, V.A.; Bozi, L.H.M.; Ribeiro, M.A.C.; Ferreira, N.D.; Moreira, J.B.N.; Brum, P.C.; et al. Exercise prevents impaired autophagy and proteostasis in a model of neurogenic myopathy. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Campos, J.C.; Queliconi, B.B.; Bozi, L.H.M.; Bechara, L.R.G.; Dourado, P.M.M.; Andres, A.M.; Jannig, P.R.; Gomes, K.M.S.; Zambelli, V.O.; Rocha-Resende, C.; et al. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy 2017. [Google Scholar] [CrossRef] [Green Version]
- Miura, S.; Kai, Y.; Kamei, Y.; Ezaki, O. Isoform-Specific Increases in Murine Skeletal Muscle Peroxisome Proliferator-Activated Receptor- Coactivator-1 (PGC-1) mRNA in Response to 2-Adrenergic Receptor Activation and Exercise. Endocrinology 2008, 149, 4527–4533. [Google Scholar] [CrossRef] [Green Version]
- Wills, L.P.; Trager, R.E.; Beeson, G.C.; Lindsey, C.C.; Peterson, Y.K.; Beeson, C.C.; Schnellmann, R.G. The β2-adrenoceptor agonist formoterol stimulates mitochondrial biogenesis. J. Pharmacol. Exp. Ther. 2012. [Google Scholar] [CrossRef] [Green Version]
- Campos, J.C.; Baehr, L.M.; Ferreira, N.D.; Bozi, L.H.M.; Andres, A.M.; Ribeiro, M.A.C.; Gottlieb, R.A.; Bodine, S.C.; Ferreira, J.C.B. β2-adrenoceptor activation improves skeletal muscle autophagy in neurogenic myopathy. FASEB J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Lustrino, D.; Silveira, W.A.; Wild, F.; Straka, T.; Issop, Y.; O’Connor, E.; Cox, D.; Reischl, M.; Marquardt, T.; et al. Sympathetic innervation controls homeostasis of neuromuscular junctions in health and disease. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef] [Green Version]
- Elfellah, M.; Dalling, R.; Kantola, I.; Reid, J. Beta-adrenoceptors and human skeletal muscle characterisation of receptor subtype and effect of age. Br. J. Clin. Pharmacol. 1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, S.J.; Dyck, D.J.; Bonen, A.; Spriet, L.L. Effects of epinephrine on lipid metabolism in resting skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.H.; Barnes, W.S. The positive inotropic effect of epinephrine on skeletal muscle: A brief review. Muscle Nerve 1989. [Google Scholar] [CrossRef] [PubMed]
- Coronado, M.; Fajardo, G.; Nguyen, K.; Zhao, M.; Kooiker, K.; Jung, G.; Hu, D.Q.; Reddy, S.; Sandoval, E.; Stotland, A.; et al. Physiological mitochondrial fragmentation is a normal cardiac adaptation to increased energy demand. Circ. Res. 2018. [Google Scholar] [CrossRef]
- Ferreira, J.C.B.; Rolim, N.P.L.; Bartholomeu, J.B.; Gobatto, C.A.; Kokubun, E.; Brum, P.C. Maximal lactate steady state in running mice: Effect of exercise training. Clin. Exp. Pharmacol. Physiol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Urhausen, A.; Weiler, B.; Coen, B.; Kindermann, W. Plasma catecholamines during endurance exercise of different intensities as related to the individual anaerobic threshold. Eur. J. Appl. Physiol. Occup. Physiol. 1994. [Google Scholar] [CrossRef]
- Zouhal, H.; Jacob, C.; Delamarche, P.; Gratas-Delamarche, A. Catecholamines and the effects of exercise, training and gender. Sport. Med. 2008, 38, 401–423. [Google Scholar] [CrossRef]
- Menconi, M.; Gonnella, P.; Petkova, V.; Lecker, S.; Hasselgren, P.O. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J. Cell. Biochem. 2008. [Google Scholar] [CrossRef] [Green Version]
- Ryall, J.G.; Sillence, M.N.; Lynch, G.S. Systemic administration of beta2-adrenoceptor agonists, formoterol and salmeterol, elicit skeletal muscle hypertrophy in rats at micromolar doses. Br. J. Pharmacol. 2006, 147, 587–595. [Google Scholar] [CrossRef] [Green Version]
- Richards, D.A.; Bao, W.; Rambo, M.V.; Burgert, M.; Jucker, B.M.; Lenhard, S.C. Examining the relationship between exercise tolerance and isoproterenol-based cardiac reserve in murine models of heart failure. J. Appl. Physiol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Engle, S.K.; Jordan, W.H.; Pritt, M.L.; Chiang, A.Y.; Davis, M.A.; Zimmermann, J.L.; Rudmann, D.G.; Heinz-Taheny, K.M.; Irizarry, A.R.; Yamamoto, Y.; et al. Qualification of Cardiac Troponin I Concentration in Mouse Serum Using Isoproterenol and Implementation in Pharmacology Studies to Accelerate Drug Development. Toxicol. Pathol. 2009. [Google Scholar] [CrossRef]
- Brooks, W.W.; Conrad, C.H. Isoproterenol-induced myocardial injury and diastolic dysfunction in mice: Structural and functional correlates. Comp. Med. 2009, 59, 339–343. [Google Scholar]
- Burniston, J.G.; Tan, L.B.; Goldspink, D.F. Relative myotoxic and haemodynamic effects of the β-agonists fenoterol and clenbuterol measured in conscious unrestrained rats. Exp. Physiol. 2006. [Google Scholar] [CrossRef] [Green Version]
- Shivachar, A.C.; Eikenburg, D.C. Differential effects of epinephrine and norepinephrine on cAMP response and G(i3)α protein expression in cultured sympathetic neurons. J. Pharmacol. Exp. Ther. 1999, 291, 258–264. [Google Scholar] [PubMed]
- Xiang, Y.; Kobilka, B. The PDZ-binding motif of the β2-adrenoceptor is essential for physiologic signaling and trafficking in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisel, A.S.; Michel, M.C.; Insel, P.A.; Ennis, C.; Ziegler, M.G.; Phillips, C. Pertussis toxin treatment of whole blood: A novel approach to assess g protein function in congestive heart failure. Circulation 1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009. [Google Scholar] [CrossRef] [PubMed]
- Dimauro, I.; Pearson, T.; Caporossi, D.; Jackson, M.J. A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res. Notes 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnovsky, M.J. The Localization of Cholinesterase Activity in Rat Cardiac Muscle by Electron Microscopy. J. Cell Biol. 1964. [Google Scholar] [CrossRef] [PubMed]
- Madsen, K.; Ertbjerg, P.; Djurhuus, M.S.; Pedersen, P.K. Calcium content and respiratory control index of skeletal muscle mitochondria during exercise and recovery. Am. J. Physiol. Endocrinol. Metab. 1996. [Google Scholar] [CrossRef] [PubMed]
- Tonkonogi, M.; Harris, B.; Sahlin, K. Mitochondrial oxidative function in human saponin-skinned muscle fibres: Effects of prolonged exercise. J. Physiol. 1998. [Google Scholar] [CrossRef] [PubMed]
- Tonkonogi, M.; Walsh, B.; Tiivel, T.; Saks, V.; Sahlin, K. Mitochondrial function in human skeletal muscle is not impaired by high intensity exercise. Pflugers Arch. Eur. J. Physiol. 1999. [Google Scholar] [CrossRef] [PubMed]
- Fernström, M.; Tonkonogi, M.; Sahlin, K. Effects of acute and chronic endurance exercise on mitochondrial uncoupling in human skeletal muscle. J. Physiol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Zhang, G.; Bo, H.; Qu, J.; Ma, G.; Cao, D.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Upregulation of uncoupling protein-3 in skeletal muscle during exercise: A potential antioxidant function. Free Radic. Biol. Med. 2009. [Google Scholar] [CrossRef]
- Ryall, J.G.; Gregorevic, P.; Plant, D.R.; Sillence, M.N.; Lynch, G.S. β2-agonist fenoterol has greater effects on contractile function of rat skeletal muscles than clenbuterol. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002. [Google Scholar] [CrossRef] [Green Version]
- Navegantes, L.C.C.; Resano, N.M.Z.; Baviera, A.M.; Migliorini, R.H.; Kettelhut, I.C. Effect of sympathetic denervation on the rate of protein synthesis in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2004. [Google Scholar] [CrossRef]
- Kim, Y.S.; Sainz, R.D.; Summers, R.J.; Molenaar, P. Cimaterol reduces beta-adrenergic receptor density in rat skeletal muscles. J. Anim. Sci. 1992. [Google Scholar] [CrossRef]
- Jensen, J.; Brennesvik, E.O.; Bergersen, L.H.; Oseland, H.; Jebens, E.; Brørs, O. Quantitative determination of cell surface β-adrenoceptors in different rat skeletal muscles. Pflugers Arch. Eur. J. Physiol. 2002. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, G.S.; Ryall, J.G. Role of β-adrenoceptor signaling in skeletal muscle: Implications for muscle wasting and disease. Physiol. Rev. 2008, 88, 729–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, S.P.; Borrani, F. β-Adrenergic modulation of skeletal muscle contraction: Key role of excitation-contraction coupling. J. Physiol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voltarelli, V.A.; Bechara, L.R.G.; Bacurau, A.V.N.; Mattos, K.C.; Dourado, P.M.M.; Bueno, C.R.; Casarini, D.E.; Negrao, C.E.; Brum, P.C. Lack of β2-adrenoceptors aggravates heart failure-induced skeletal muscle myopathy in mice. J. Cell. Mol. Med. 2014, 18. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.F.; Nakamura, K. Central neural pathways for thermoregulation. Front. Biosci. 2011. [Google Scholar] [CrossRef] [Green Version]
- van Marken Lichtenbelt, W.D.; Schrauwen, P. Implications of nonshivering thermogenesis for energy balance regulation in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R285–R296. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yang, X. Inter-organ regulation of adipose tissue browning. Cell. Mol. Life Sci. 2017, 74, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Sprague, J.E.; Yang, X.; Sommers, J.; Gilman, T.L.; Mills, E.M. Roles of norepinephrine, free fatty acids, thyroid status, and skeletal muscle uncoupling protein 3 expression in sympathomimetic-induced thermogenesis. J. Pharmacol. Exp. Ther. 2007. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta-Bioenerg. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zhang, L.; Qi, Y.; Xu, H. Mitochondrial cAMP signaling. Cell. Mol. Life Sci. 2016, 73, 4577–4590. [Google Scholar] [CrossRef] [Green Version]
- Eisner, V.; Lenaers, G.; Hajnóczky, G. Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J. Cell Biol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Moore, T.M.; Zhou, Z.; Cohn, W.; Norheim, F.; Lin, A.J.; Kalajian, N.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; et al. The impact of exercise on mitochondrial dynamics and the role of Drp1 in exercise performance and training adaptations in skeletal muscle. Mol. Metab. 2019. [Google Scholar] [CrossRef]
- Picard, M.; Gentil, B.J.; McManus, M.J.; White, K.; St Louis, K.; Gartside, S.E.; Wallace, D.C.; Turnbull, D.M. Acute exercise remodels mitochondrial membrane interactions in mouse skeletal muscle. J. Appl. Physiol. 2013, 115, 1562–1571. [Google Scholar] [CrossRef]
- Huertas, J.R.; Ruiz-Ojeda, F.J.; Plaza-Díaz, J.; Nordsborg, N.B.; Martín-Albo, J.; Rueda-Robles, A.; Casuso, R.A. Human muscular mitochondrial fusion in athletes during exercise. FASEB J. 2019. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem. Sci. 2001, 26, 23–29. [Google Scholar] [CrossRef]
- Drake, J.C.; Wilson, R.J.; Yan, Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J. 2016, 30, 13–22. [Google Scholar] [CrossRef]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.H.; Dourado, P.M.M.; et al. A selective inhibitor of mitofusin 1-βIIPKC association improves heart failure outcome in rats. Nat. Commun. 2019. [Google Scholar] [CrossRef]
- Bozi, L.H.M.; Campos, J.C.; Zambelli, V.O.; Ferreira, N.D.; Ferreira, J.C.B. Mitochondrially-targeted treatment strategies. Mol. Aspects Med. 2020, 71, 100836. [Google Scholar] [CrossRef]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001. [Google Scholar] [CrossRef]
- Sebastián, D.; Hernández-Alvarez, M.I.; Segalés, J.; Sorianello, E.; Muñoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef] [Green Version]
- Lira, V.A.; Benton, C.R.; Yan, Z.; Bonen, A. PGC-1α regulation by exercise training and its influences on muscle function and insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, 145–161. [Google Scholar] [CrossRef] [Green Version]
- Agudelo, L.Z.; Ferreira, D.M.S.; Dadvar, S.; Cervenka, I.; Ketscher, L.; Izadi, M.; Zhengye, L.; Furrer, R.; Handschin, C.; Venckunas, T.; et al. Skeletal muscle PGC-1α1 reroutes kynurenine metabolism to increase energy efficiency and fatigue-resistance. Nat. Commun. 2019. [Google Scholar] [CrossRef]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011. [Google Scholar] [CrossRef] [Green Version]
- García-Bermúdez, J.; Sánchez-Aragó, M.; Soldevilla, B.; del Arco, A.; Nuevo-Tapioles, C.; Cuezva, J.M. PKA Phosphorylates the ATPase Inhibitory Factor 1 and Inactivates Its Capacity to Bind and Inhibit the Mitochondrial H+-ATP Synthase. Cell Rep. 2015. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Valsecchi, F.; Ramos-Espiritu, L.S.; Buck, J.; Levin, L.R.; Manfredi, G. cAMP and mitochondria. Physiology 2013, 28, 199–209. [Google Scholar] [CrossRef]
- Feliciello, A.; Gottesman, M.E.; Avvedimento, E.V. cAMP-PKA signaling to the mitochondria: Protein scaffolds, mRNA and phosphatases. Cell. Signal. 2005, 17, 279–287. [Google Scholar] [CrossRef]
- Lark, D.S.; Reese, L.R.; Ryan, T.E.; Torres, M.J.; Smith, C.D.; Lin, C.T.; Neufer, P.D. Protein kinase A governs oxidative phosphorylation kinetics and oxidant emitting potential at complex I. Front. Physiol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Dagda, R.K.; Gusdon, A.M.; Pien, I.; Strack, S.; Green, S.; Li, C.; Van Houten, B.; Cherra, S.J.; Chu, C.T. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ. 2011, 18, 1914–1923. [Google Scholar] [CrossRef] [Green Version]
- De Rasmo, D.; Gattoni, G.; Papa, F.; Santeramo, A.; Pacelli, C.; Cocco, T.; Micelli, L.; Sardaro, N.; Larizza, M.; Scivetti, M.; et al. The β-adrenoceptor agonist isoproterenol promotes the activity of respiratory chain complex I and lowers cellular reactive oxygen species in fibroblasts and heart myoblasts. Eur. J. Pharmacol. 2011, 652, 15–22. [Google Scholar] [CrossRef]
- Lefkimmiatis, K.; Leronni, D.; Hofer, A.M. The inner and outer compartments of mitochondria are sites of distinct cAMP/PKA signaling dynamics. J. Cell Biol. 2013, 202, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Negrao, C.E.; Middlekauff, H.R. Adaptations in autonomic function during exercise training in heart failure. Heart Fail. Rev. 2008, 13, 51–60. [Google Scholar] [CrossRef]
- Negrão, C.E.; Moreira, E.D.; Brum, P.C.; Denadai, M.L.; Krieger, E.M. Vagal and sympathetic control of heart rate during exercise by sedentary and exercise-trained rats. Braz. J. Med. Biol. Res. 1992, 25, 1045–1052. [Google Scholar]
- Fernandez-Gonzalo, R.; Lundberg, T.R.; Tesch, P.A. Acute molecular responses in untrained and trained muscle subjected to aerobic and resistance exercise training versus resistance training alone. Acta Physiol. 2013. [Google Scholar] [CrossRef]
- Chapman, M.A.; Arif, M.; Emanuelsson, E.B.; Reitzner, S.M.; Lindholm, M.E.; Mardinoglu, A.; Sundberg, C.J. Skeletal Muscle Transcriptomic Comparison between Long-Term Trained and Untrained Men and Women. Cell Rep. 2020. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azevedo Voltarelli, V.; Coronado, M.; Gonçalves Fernandes, L.; Cruz Campos, J.; Jannig, P.R.; Batista Ferreira, J.C.; Fajardo, G.; Chakur Brum, P.; Bernstein, D. β2-Adrenergic Signaling Modulates Mitochondrial Function and Morphology in Skeletal Muscle in Response to Aerobic Exercise. Cells 2021, 10, 146. https://doi.org/10.3390/cells10010146
Azevedo Voltarelli V, Coronado M, Gonçalves Fernandes L, Cruz Campos J, Jannig PR, Batista Ferreira JC, Fajardo G, Chakur Brum P, Bernstein D. β2-Adrenergic Signaling Modulates Mitochondrial Function and Morphology in Skeletal Muscle in Response to Aerobic Exercise. Cells. 2021; 10(1):146. https://doi.org/10.3390/cells10010146
Chicago/Turabian StyleAzevedo Voltarelli, Vanessa, Michael Coronado, Larissa Gonçalves Fernandes, Juliane Cruz Campos, Paulo Roberto Jannig, Julio Cesar Batista Ferreira, Giovanni Fajardo, Patricia Chakur Brum, and Daniel Bernstein. 2021. "β2-Adrenergic Signaling Modulates Mitochondrial Function and Morphology in Skeletal Muscle in Response to Aerobic Exercise" Cells 10, no. 1: 146. https://doi.org/10.3390/cells10010146
APA StyleAzevedo Voltarelli, V., Coronado, M., Gonçalves Fernandes, L., Cruz Campos, J., Jannig, P. R., Batista Ferreira, J. C., Fajardo, G., Chakur Brum, P., & Bernstein, D. (2021). β2-Adrenergic Signaling Modulates Mitochondrial Function and Morphology in Skeletal Muscle in Response to Aerobic Exercise. Cells, 10(1), 146. https://doi.org/10.3390/cells10010146