Hippo Signaling Pathway in Gliomas


{kind=link}
{kind=link}
Abstract
:1. Introduction
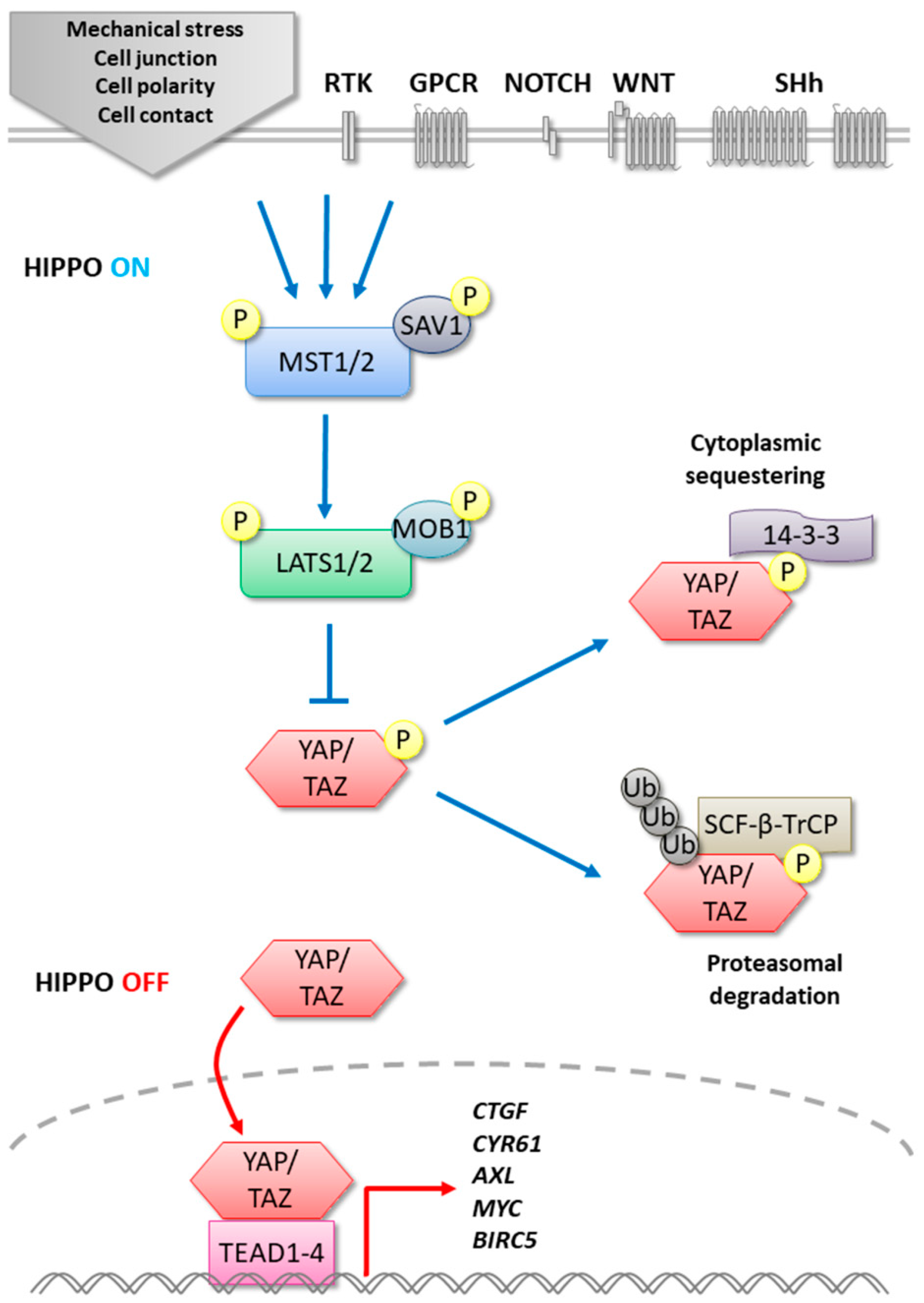
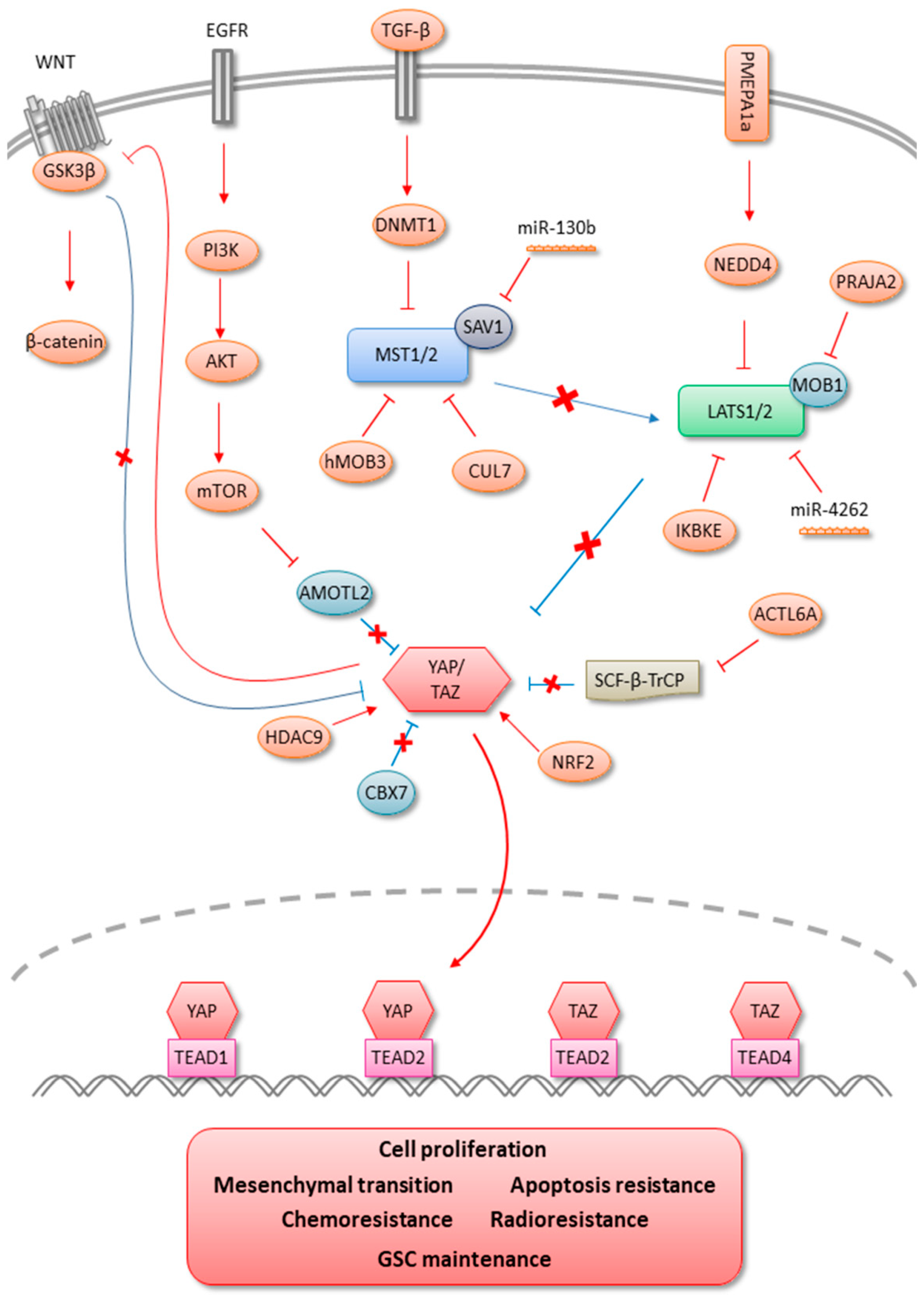
2. MST1/2
3. LATS1/2
4. YAP/TAZ
5. TEADs
6. Hippo Signaling Pathway Signatures
7. Therapeutic Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.J.; Luo, X. Activation mechanisms of the Hippo kinase signaling cascade. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pobbati, A.V.; Hong, W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol. Ther. 2013, 14, 390–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Nakagawa, M.; Olson, E.N.; Nakagawa, O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 18034–18039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [Green Version]
- Lallemand, D.; Curto, M.; Saotome, I.; Giovannini, M.; McClatchey, A.I. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev. 2003, 17, 1090–1100. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 2013, 154, 1342–1355. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.X.; Guan, K.L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.P.; Lee, J.H.; Kim, T.S.; Kim, T.H.; Park, H.D.; Byun, J.S.; Kim, M.C.; Jeong, W.I.; Calvisi, D.F.; Kim, J.M.; et al. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 8248–8253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, T.; Meng, W.; Li, M.; Hong, T.; Zhang, N. Recent Advances of the Hippo/YAP Signaling Pathway in Brain Development and Glioma. Cell. Mol. Neurobiol. 2020, 40, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.; Halder, G.; Finegold, M.J.; Lee, J.S.; et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Mak, K.K.; Topol, L.; Yun, K.; Hu, J.; Garrett, L.; Chen, Y.; Park, O.; Chang, J.; Simpson, R.M.; et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. USA 2010, 107, 1431–1436. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Conrad, C.; Xia, F.; Park, J.S.; Payer, B.; Yin, Y.; Lauwers, G.Y.; Thasler, W.; Lee, J.T.; Avruch, J.; et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009, 16, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. CB 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, T.S.; Yang, T.H.; Koo, B.K.; Oh, S.P.; Lee, K.P.; Oh, H.J.; Lee, S.H.; Kong, Y.Y.; Kim, J.M.; et al. A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J. 2008, 27, 1231–1242. [Google Scholar] [CrossRef] [Green Version]
- Reddy, B.V.; Rauskolb, C.; Irvine, K.D. Influence of fat-hippo and notch signaling on the proliferation and differentiation of Drosophila optic neuroepithelia. Development 2010, 137, 2397–2408. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.Y.; Lin, S.; Ye, J. YAP and TAZ, the conductors that orchestrate eye development, homeostasis, and disease. J. Cell. physiol. 2018, 10.1002/jcp.26870. [Google Scholar] [CrossRef] [Green Version]
- Staley, B.K.; Irvine, K.D. Warts and Yorkie mediate intestinal regeneration by influencing stem cell proliferation. Curr. Biol. CB 2010, 20, 1580–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Pfaff, S.L.; Gage, F.H. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 2008, 22, 3320–3334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, I.; Kim, J.; Okazawa, H.; Zhao, J.; Zhao, B.; Yu, J.; Chinnaiyan, A.; Israel, M.A.; Goldstein, L.S.; Abujarour, R.; et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010, 24, 1106–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.Y.; Yu, J.; Guan, K.L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [Green Version]
- Barry, E.R.; Camargo, F.D. The Hippo superhighway: Signaling crossroads converging on the Hippo/Yap pathway in stem cells and development. Curr. Opin. Cell Biol. 2013, 25, 247–253. [Google Scholar] [CrossRef]
- Barron, D.A.; Kagey, J.D. The role of the Hippo pathway in human disease and tumorigenesis. Clin. Transl. Med. 2014, 3, 25. [Google Scholar] [CrossRef]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Liang, N.; Zhang, C.; Dill, P.; Panasyuk, G.; Pion, D.; Koka, V.; Gallazzini, M.; Olson, E.N.; Lam, H.; Henske, E.P.; et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J. Exp. Med. 2014, 211, 2249–2263. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Mao, B.; Cheng, J.; Gao, Y.; Jiang, K.; Chen, J.; Yuan, Z.; Meng, S. YAP enhances autophagic flux to promote breast cancer cell survival in response to nutrient deprivation. PLoS ONE 2015, 10, e0120790. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.; Dupont, S.; Piccolo, S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nature reviews. Mol. Cell Biol. 2012, 13, 591–600. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Stetson, L.; Virk, S.M.; Barnholtz-Sloan, J.S. Epidemiology of gliomas. Cancer Treat. Res. 2015, 163, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Coons, S.W.; Johnson, P.C.; Scheithauer, B.W.; Yates, A.J.; Pearl, D.K. Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer 1997, 79, 1381–1393. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [Green Version]
- van den Bent, M.J.; Mellinghoff, I.K.; Bindra, R.S. Gray Areas in the Gray Matter: IDH1/2 Mutations in Glioma. American Society of Clinical Oncology educational book. Am. Soc. Clin. Oncol. Annu. Meet. 2020, 40, 1–8. [Google Scholar] [CrossRef]
- Venneti, S.; Huse, J.T. The evolving molecular genetics of low-grade glioma. Adv. Anat. Pathol. 2015, 22, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. J. Pediatr. Oncol. Nurs. 2014, 16, 896–913. [Google Scholar] [CrossRef] [Green Version]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Yuan, X.; Curtin, J.; Xiong, Y.; Liu, G.; Waschsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Yu, J.S. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 2004, 23, 9392–9400. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Zhang, L.; Xue, G.; Hynx, D.; Wang, Y.; Cron, P.D.; Hundsrucker, C.; Hergovich, A.; Frank, S.; Hemmings, B.A.; et al. hMOB3 modulates MST1 apoptotic signaling and supports tumor growth in glioblastoma multiforme. Cancer Res. 2014, 74, 3779–3789. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.; Wang, Y.; Liu, X.; Ma, P.; Shi, Y.; Gao, J.; Shi, Q.; Hu, J.; Yu, R.; Zhou, X. Mst1 regulates glioma cell proliferation via the AKT/mTOR signaling pathway. J. Neuro-Oncol. 2015, 121, 279–288. [Google Scholar] [CrossRef]
- Guo, Z.; Li, G.; Bian, E.; Ma, C.C.; Wan, J.; Zhao, B. TGF-beta-mediated repression of MST1 by DNMT1 promotes glioma malignancy. Biomed. Pharmacother. 2017, 94, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, Y.; Mijiti, M.; Wang, Z.; Wu, P.F.; Jiafu, D. Upregulation of miR-130b enhances stem cell-like phenotype in glioblastoma by inactivating the Hippo signaling pathway. Biochem. Biophys. Res. Commun. 2015, 465, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Sun, C.; Qian, X. MST1 suppresses viability and promotes apoptosis of glioma cells via upregulating SIRT6 expression. J. Integr. Neurosci. 2019, 18, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Z.; Qian, M.; Wang, S.; Qiu, W.; Chen, Z.; Sun, Z.; Xiong, Y.; Wang, C.; Sun, X.; et al. Cullin-7 (CUL7) is overexpressed in glioma cells and promotes tumorigenesis via NF-kappaB activation. J. Exp. Clin. Cancer Res. 2020, 39, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiyama, Y.; Hirota, T.; Morisaki, T.; Hara, T.; Marumoto, T.; Iida, S.; Makino, K.; Yamamoto, H.; Hiraoka, T.; Kitamura, N.; et al. A human homolog of Drosophila warts tumor suppressor, h-warts, localized to mitotic apparatus and specifically phosphorylated during mitosis. FEBS Lett. 1999, 459, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, D.M.; Chen, W.; Xu, T. Human homologue of Drosophila lats, LATS1, negatively regulate growth by inducing G(2)/M arrest or apoptosis. Oncogene 2001, 20, 6516–6523. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Qi, H.; Li, Y.; Pei, J.; Barton, J.; Blackstad, M.; Xu, T.; Tao, W. LATS1 tumor suppressor regulates G2/M transition and apoptosis. Oncogene 2002, 21, 1233–1241. [Google Scholar] [CrossRef] [Green Version]
- Yabuta, N.; Fujii, T.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Nishiguchi, H.; Endo, Y.; Toji, S.; Tanaka, H.; Nishimune, Y.; et al. Structure, expression, and chromosome mapping of LATS2, a mammalian homologue of the Drosophila tumor suppressor gene lats/warts. Genomics 2000, 63, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Hori, T.; Takaori-Kondo, A.; Kamikubo, Y.; Uchiyama, T. Molecular cloning of a novel human protein kinase, kpm, that is homologous to warts/lats, a Drosophila tumor suppressor. Oncogene 2000, 19, 3101–3109. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Pei, J.; Xia, H.; Ke, H.; Wang, H.; Tao, W. Lats2, a putative tumor suppressor, inhibits G1/S transition. Oncogene 2003, 22, 4398–4405. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Li, X.; Hu, J.; Zhou, W.; Jiang, Y.; Li, G.; Lu, D. Promoter hypermethylation-mediated down-regulation of LATS1 and LATS2 in human astrocytoma. Neurosci. Res. 2006, 56, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Liu, D.; Shao, W.; Yang, W.; Wu, H.; Bian, X. Decreased expression of LATS1 is correlated with the progression and prognosis of glioma. J. Exp. Clin. Cancer Res. 2012, 31, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lignitto, L.; Arcella, A.; Sepe, M.; Rinaldi, L.; Delle Donne, R.; Gallo, A.; Stefan, E.; Bachmann, V.A.; Oliva, M.A.; Tiziana Storlazzi, C.; et al. Proteolysis of MOB1 by the ubiquitin ligase praja2 attenuates Hippo signalling and supports glioblastoma growth. Nat. Commun. 2013, 4, 1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, H.; Zhang, H.; Cai, J.; Wu, J.; Yuan, J.; Li, J.; Huang, Z.; Li, M. IKBKE is over-expressed in glioma and contributes to resistance of glioma cells to apoptosis via activating NF-kappaB. J. Pathol. 2011, 223, 436–445. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, J.; Zhang, Z.; Zhu, L.; Dong, S.; Guo, G.; Li, R.; Nan, Y.; Yu, K.; Zhong, Y.; et al. Amlexanox, a selective inhibitor of IKBKE, generates anti-tumoral effects by disrupting the Hippo pathway in human glioblastoma cell lines. Cell Death Dis. 2017, 8, e3022. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wei, Y.; Zhang, L.; Yee, P.P.; Johnson, M.; Zhang, X.; Gulley, M.; Atkinson, J.M.; Trebak, M.; Wang, H.G.; et al. Induction of store-operated calcium entry (SOCE) suppresses glioblastoma growth by inhibiting the Hippo pathway transcriptional coactivators YAP/TAZ. Oncogene 2019, 38, 120–139. [Google Scholar] [CrossRef]
- Liu, C.; Ma, T.; Jiang, T.; Jia, G.; Yang, C.; Peng, Y.; Qian, Y.; Wang, R.; Wang, S. Abnormal increase of miR-4262 promotes cell proliferation and migration by targeting large tumor suppressor 1 in gliomas. Pathol. Res. Pract. 2020, 216, 152778. [Google Scholar] [CrossRef]
- Ji, J.; Ding, K.; Luo, T.; Xu, R.; Zhang, X.; Huang, B.; Chen, A.; Zhang, D.; Miletic, H.; Bjerkvig, R.; et al. PMEPA1 isoform a drives progression of glioblastoma by promoting protein degradation of the Hippo pathway kinase LATS1. Oncogene 2020, 39, 1125–1139. [Google Scholar] [CrossRef] [Green Version]
- Levallet, G.; Creveuil, C.; Bekaert, L.; Peres, E.; Planchard, G.; Lecot-Cotigny, S.; Guillamo, J.S.; Emery, E.; Zalcman, G.; Lechapt-Zalcman, E. Promoter Hypermethylation of Genes Encoding for RASSF/Hippo Pathway Members Reveals Specific Alteration Pattern in Diffuse Gliomas. JMD 2019, 21, 695–704. [Google Scholar] [CrossRef]
- Bhat, K.P.; Salazar, K.L.; Balasubramaniyan, V.; Wani, K.; Heathcock, L.; Hollingsworth, F.; James, J.D.; Gumin, J.; Diefes, K.L.; Kim, S.H.; et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011, 25, 2594–2609. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Li, A.; Lu, H.; Luo, R.; Zhang, M.; Li, Z. TAZ promotes temozolomide resistance by upregulating MCL-1 in human glioma cells. Biochem. Biophys. Res. Commun. 2015, 463, 638–643. [Google Scholar] [CrossRef]
- Yang, R.; Wu, Y.; Zou, J.; Zhou, J.; Wang, M.; Hao, X.; Cui, H. The Hippo transducer TAZ promotes cell proliferation and tumor formation of glioblastoma cells through EGFR pathway. Oncotarget 2016, 7, 36255–36265. [Google Scholar] [CrossRef] [Green Version]
- Yee, P.P.; Wei, Y.; Kim, S.Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, F.; Wei, Y.; Zhang, L.; Guo, D.; Wang, B.; Li, W. Inhibition of TAZ contributes radiation-induced senescence and growth arrest in glioma cells. Oncogene 2019, 38, 2788–2799. [Google Scholar] [CrossRef]
- Yang, R.; Wu, Y.; Wang, M.; Sun, Z.; Zou, J.; Zhang, Y.; Cui, H. HDAC9 promotes glioblastoma growth via TAZ-mediated EGFR pathway activation. Oncotarget 2015, 6, 7644–7656. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, Z.; Patil, V.; Arora, A.; Hegde, A.S.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Cbx7 is epigenetically silenced in glioblastoma and inhibits cell migration by targeting YAP/TAZ-dependent transcription. Sci. Rep. 2016, 6, 27753. [Google Scholar] [CrossRef] [Green Version]
- Escoll, M.; Lastra, D.; Pajares, M.; Robledinos-Anton, N.; Rojo, A.I.; Fernandez-Gines, R.; Mendiola, M.; Martinez-Marin, V.; Esteban, I.; Lopez-Larrubia, P.; et al. Transcription factor NRF2 uses the Hippo pathway effector TAZ to induce tumorigenesis in glioblastomas. Redox Biol. 2020, 30, 101425. [Google Scholar] [CrossRef]
- Liu, M.; Lin, Y.; Zhang, X.C.; Tan, Y.H.; Yao, Y.L.; Tan, J.; Zhang, X.; Cui, Y.H.; Liu, X.; Wang, Y.; et al. Phosphorylated mTOR and YAP serve as prognostic markers and therapeutic targets in gliomas. Lab. Investig. 2017, 97, 1354–1363. [Google Scholar] [CrossRef] [Green Version]
- Orr, B.A.; Bai, H.; Odia, Y.; Jain, D.; Anders, R.A.; Eberhart, C.G. Yes-associated protein 1 is widely expressed in human brain tumors and promotes glioblastoma growth. J. Neuropathol. Exp. Neurol. 2011, 70, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yang, Y.; Guo, G.; Liu, Y.; Zhang, Z.; Dong, S.; Nan, Y.; Zhao, Z.; Zhong, Y.; Huang, Q. IKBKE regulates cell proliferation and epithelial-mesenchymal transition of human malignant glioma via the Hippo pathway. Oncotarget 2017, 8, 49502–49514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Pan, P.; Wang, Z.; Zhang, Y.; Xie, P.; Geng, D.; Jiang, Y.; Yu, R.; Zhou, X. beta-catenin-mediated YAP signaling promotes human glioma growth. J. Exp. Clin. Cancer Res. Cr 2017, 36, 136. [Google Scholar] [CrossRef] [PubMed]
- Artinian, N.; Cloninger, C.; Holmes, B.; Benavides-Serrato, A.; Bashir, T.; Gera, J. Phosphorylation of the Hippo Pathway Component AMOTL2 by the mTORC2 Kinase Promotes YAP Signaling, Resulting in Enhanced Glioblastoma Growth and Invasiveness. J. Biol. Chem. 2015, 290, 19387–19401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Yee, P.P.; Wei, Y.; Liu, Z.; Kawasawa, Y.I.; Li, W. Differential YAP expression in glioma cells induces cell competition and promotes tumorigenesis. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Xu, R.; Zhang, X.; Han, M.; Xu, Y.; Wei, Y.; Ding, K.; Wang, S.; Bin, H.; Chen, A.; et al. Actin like-6A promotes glioma progression through stabilization of transcriptional regulators YAP/TAZ. Cell Death Dis. 2018, 9, 517. [Google Scholar] [CrossRef] [Green Version]
- Guichet, P.O.; Masliantsev, K.; Tachon, G.; Petropoulos, C.; Godet, J.; Larrieu, D.; Milin, S.; Wager, M.; Karayan-Tapon, L. Fatal correlation between YAP1 expression and glioma aggressiveness: Clinical and molecular evidence. J. Pathol. 2018, 246, 205–216. [Google Scholar] [CrossRef]
- Vigneswaran, K.; Boyd, N.H.; Oh, S.Y.; Lallani, S.; Boucher, A.; Neill, S.G.; Olson, J.J.; Read, R.D. YAP/TAZ transcriptional co-activators create therapeutic vulnerability to verteporfin in EGFR mutant glioblastoma. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Castellan, M.; Guarnieri, A.; Fujimura, A.; Zanconato, F.; Battilana, G.; Panciera, T.; Sladitschek, H.L.; Contessotto, P.; Citron, A.; Grilli, A.; et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in glioblastoma. Nat. Cancer 2020. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, Q.; Wang, Y.; Huang, K.; Yang, C.; Li, Y.; Yi, K.; Kang, C. EGFR/c-myc axis regulates TGFbeta/Hippo/Notch pathway via epigenetic silencing miR-524 in gliomas. Cancer Lett. 2017, 406, 12–21. [Google Scholar] [CrossRef]
- Li, W.; Dong, S.; Wei, W.; Wang, G.; Zhang, A.; Pu, P.; Jia, Z. The role of transcriptional coactivator TAZ in gliomas. Oncotarget 2016, 7, 82686–82699. [Google Scholar] [CrossRef] [Green Version]
- Tome-Garcia, J.; Erfani, P.; Nudelman, G.; Tsankov, A.M.; Katsyv, I.; Tejero, R.; Bin, Z.; Walsh, M.; Friedel, R.H.; Zaslavsky, E.; et al. Analysis of chromatin accessibility uncovers TEAD1 as a regulator of migration in human glioblastoma. Nat. Commun. 2018, 9, 4020. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, X.; Maglic, D.; Dill, M.T.; Mojumdar, K.; Ng, P.K.; Jeong, K.J.; Tsang, Y.H.; Moreno, D.; Bhavana, V.H.; et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018, 25, 1304–1317 e1305. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.H.; Sohn, B.H.; Eun, Y.G.; Lee, D.J.; Yim, S.Y.; Kang, S.G.; Lee, J.S. Silence of Hippo Pathway Associates with Pro-Tumoral Immunosuppression: Potential Therapeutic Target of Glioblastomas. Cells 2020, 9, 1761. [Google Scholar] [CrossRef]
- Gravendeel, L.A.; Kouwenhoven, M.C.; Gevaert, O.; de Rooi, J.J.; Stubbs, A.P.; Duijm, J.E.; Daemen, A.; Bleeker, F.E.; Bralten, L.B.; Kloosterhof, N.K.; et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009, 69, 9065–9072. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Hong, W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics 2020, 10, 3622–3635. [Google Scholar] [CrossRef]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [Green Version]
- Dasari, V.R.; Mazack, V.; Feng, W.; Nash, J.; Carey, D.J.; Gogoi, R. Verteporfin exhibits YAP-independent anti-proliferative and cytotoxic effects in endometrial cancer cells. Oncotarget 2017, 8, 28628–28640. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ramakrishnan, S.K.; Triner, D.; Centofanti, B.; Maitra, D.; Gyorffy, B.; Sebolt-Leopold, J.S.; Dame, M.K.; Varani, J.; Brenner, D.E.; et al. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci. Signal. 2015, 8, ra98. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Chan, S.W.; Lee, I.; Song, H.; Hong, W. Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure 2012, 20, 1135–1140. [Google Scholar] [CrossRef] [Green Version]
- Gibault, F.; Coevoet, M.; Sturbaut, M.; Farce, A.; Renault, N.; Allemand, F.; Guichou, J.F.; Drucbert, A.S.; Foulon, C.; Magnez, R.; et al. Toward the Discovery of a Novel Class of YAP(-)TEAD Interaction Inhibitors by Virtual Screening Approach Targeting YAP(-)TEAD Protein(-)Protein Interface. Cancers 2018, 10, 140. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Xie, M.; Scott, A.W.; Jin, J.; Ma, L.; Dong, X.; Skinner, H.D.; Johnson, R.L.; Ding, S.; Ajani, J.A. A Novel YAP1 Inhibitor Targets CSC-Enriched Radiation-Resistant Cells and Exerts Strong Antitumor Activity in Esophageal Adenocarcinoma. Mol. Cancer Ther. 2018, 17, 443–454. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masliantsev, K.; Karayan-Tapon, L.; Guichet, P.-O. Hippo Signaling Pathway in Gliomas. Cells 2021, 10, 184. https://doi.org/10.3390/cells10010184
Masliantsev K, Karayan-Tapon L, Guichet P-O. Hippo Signaling Pathway in Gliomas. Cells. 2021; 10(1):184. https://doi.org/10.3390/cells10010184
Chicago/Turabian StyleMasliantsev, Konstantin, Lucie Karayan-Tapon, and Pierre-Olivier Guichet. 2021. "Hippo Signaling Pathway in Gliomas" Cells 10, no. 1: 184. https://doi.org/10.3390/cells10010184
APA StyleMasliantsev, K., Karayan-Tapon, L., & Guichet, P.-O. (2021). Hippo Signaling Pathway in Gliomas. Cells, 10(1), 184. https://doi.org/10.3390/cells10010184