GRKs as Modulators of Neurotransmitter Receptors



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction: GRKs in GPCR Signaling and Trafficking
2. Regulation of Non-Visual GPCRs by GRK Phosphorylation
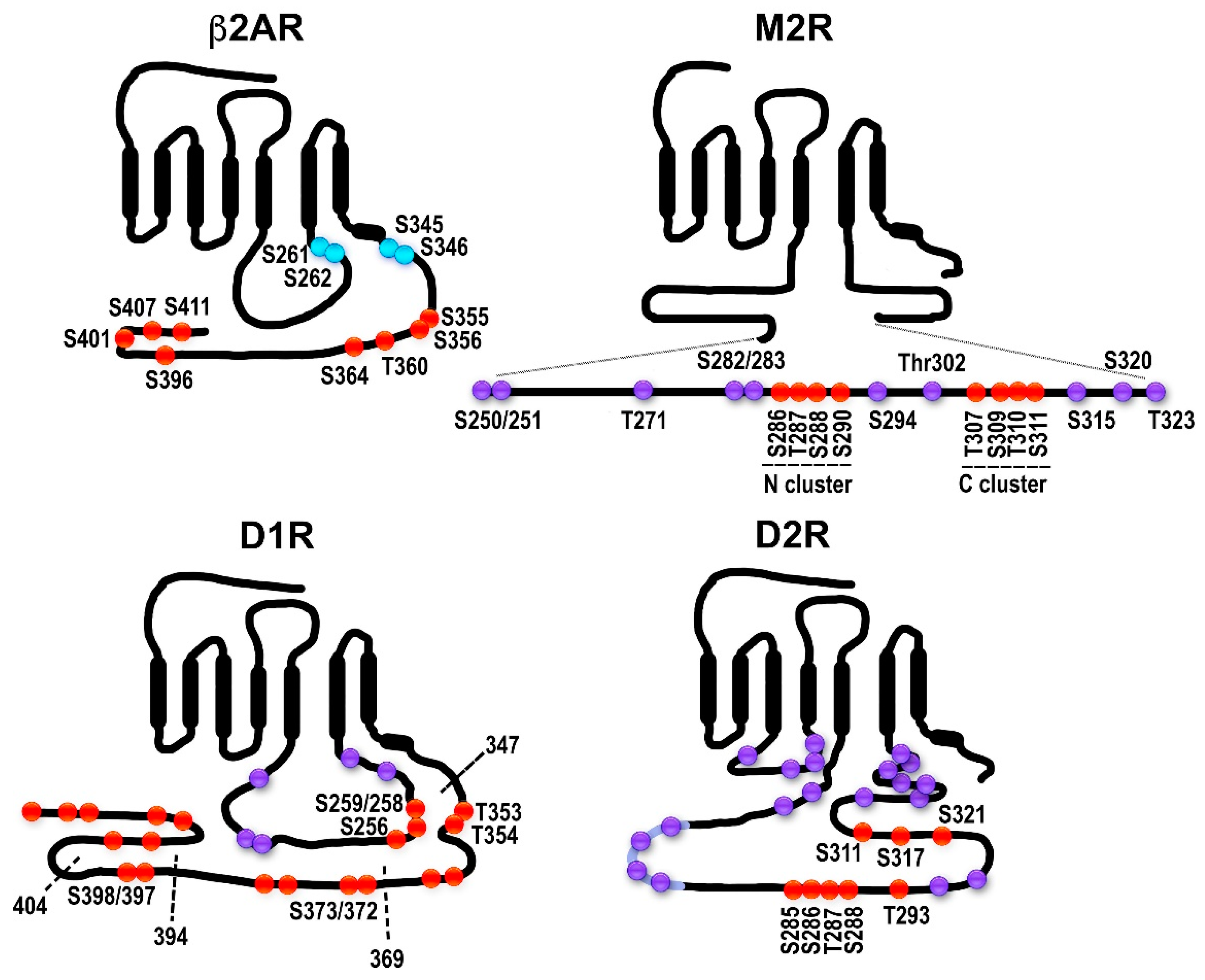
2.1. The Model GPCRs: Adrenergic Receptors
2.2. Dopamine Receptors: Outside of the Classical Paradigm?
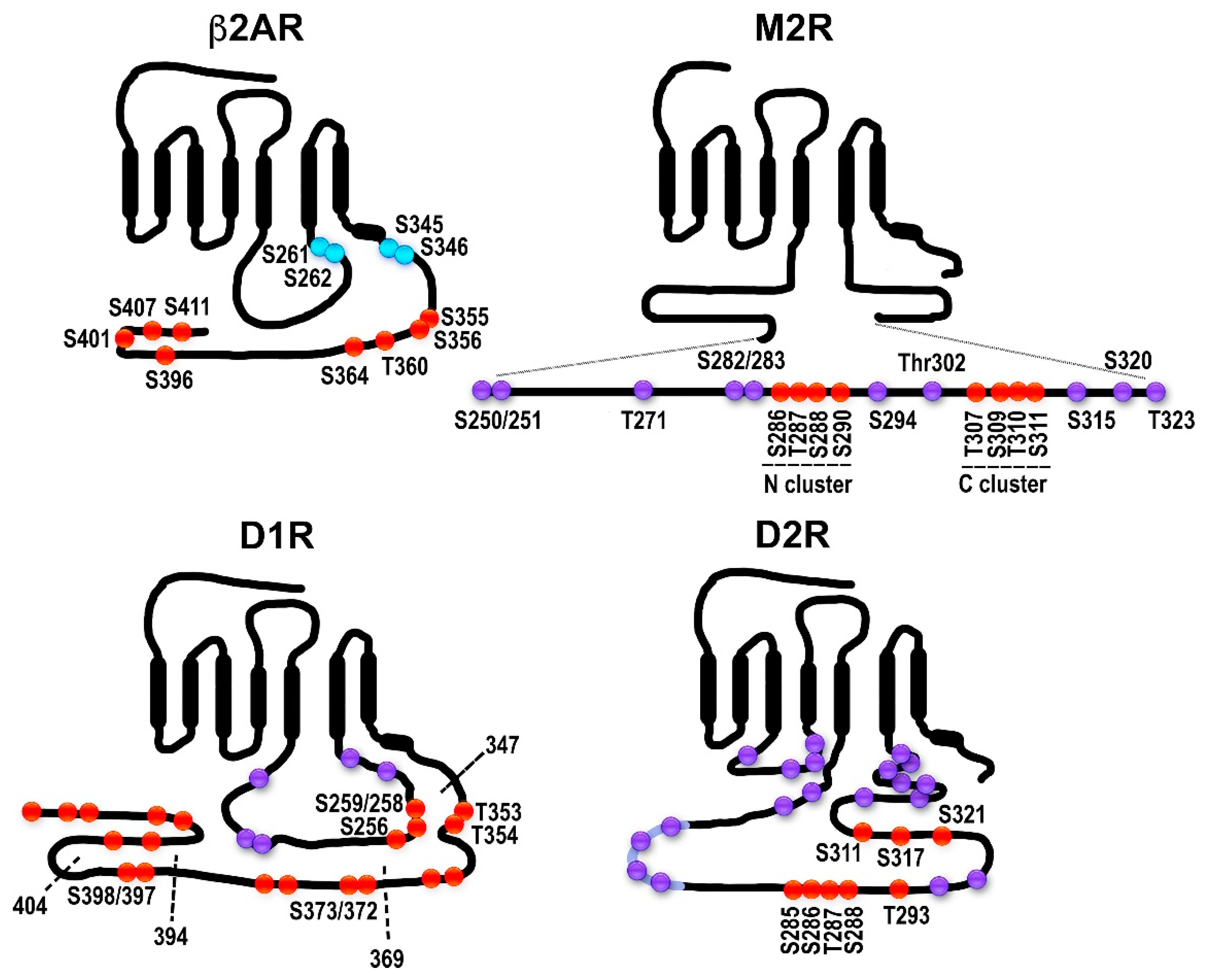
2.3. Muscarinic Acetylcholine Receptors: Phosphorylation Sites Come in Clusters
3. The Order of Phosphorylation: Sequential and Hierarchical?
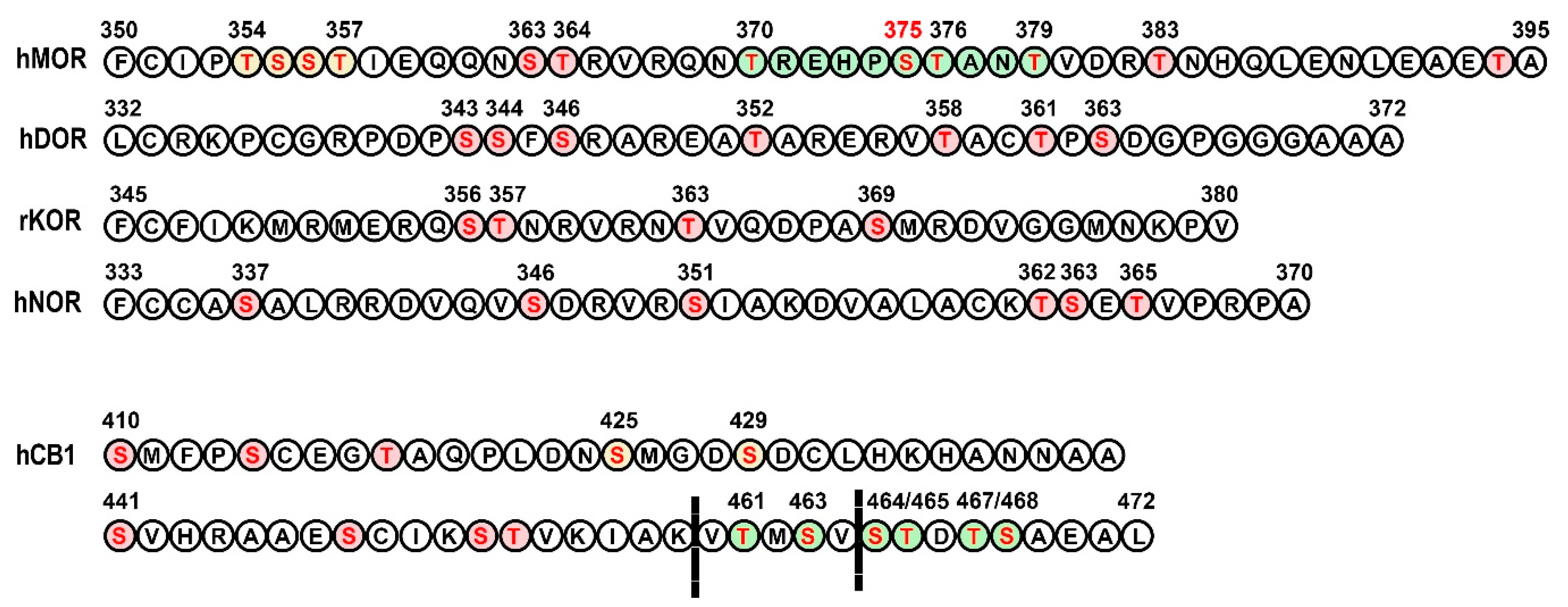
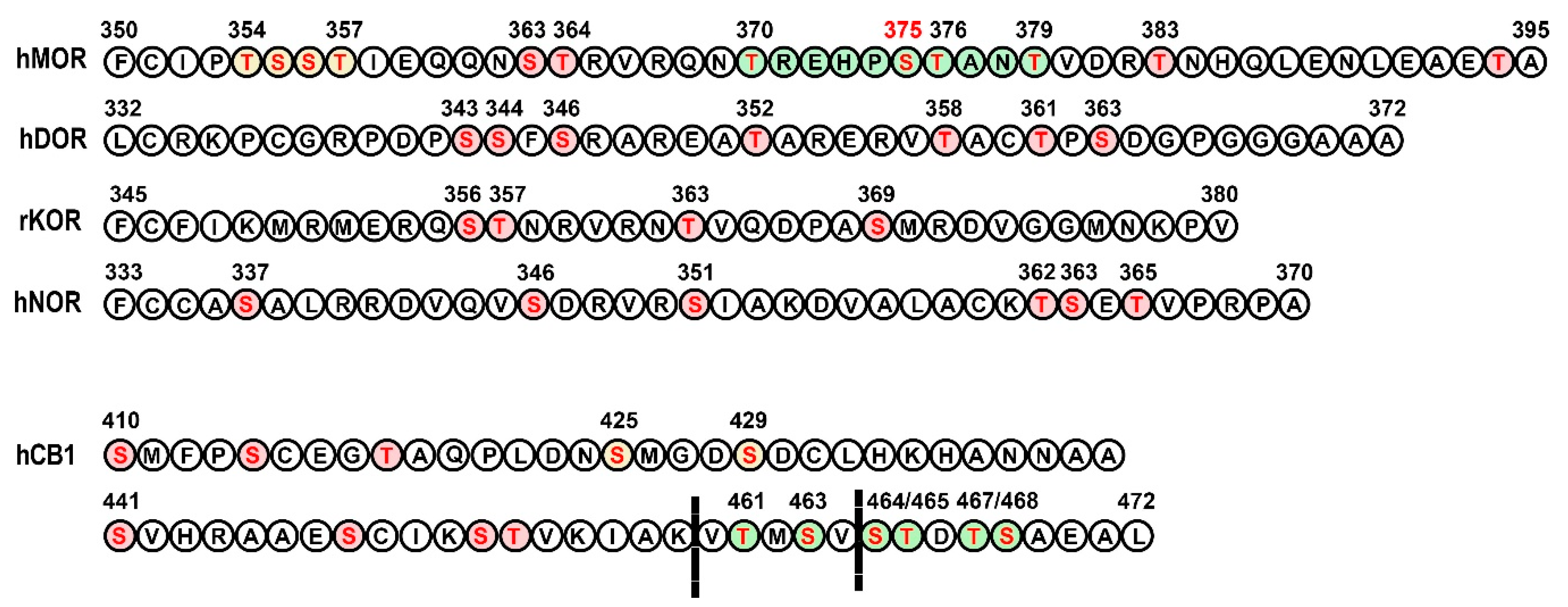
3.1. Opioid Receptors
3.2. Cannabinoid Receptors
3.3. Class A GPCR Oligomers
4. Barcode Hypothesis
5. Agonist Dependence of GRK Action
6. From Neurotransmitter Receptor Regulation to Neural Adaptation: The Role of GRKs
6.1. GRKs in the Regulation of Acute Responsiveness to Neural Stimulation
6.2. GRK-Mediated Rapid Desensitization in Long-Term Neural Adaptations
6.3. Neurotropic Drugs: To Bias or Not to Bias?
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kühn, H.; Dreyer, W.J. Light dependent phosphorylation of rhodopsin by atp. FEBS Lett. 1972, 20, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kühn, H.; Cook, J.H.; Dreyer, W.J. Phosphorylation of rhodopsin in bovine photoreceptor membranes. A dark reaction after illumination. Biochemistry 1973, 12, 2495–2502. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, Y.A.; Abdulaev, N.G.; Feigina, M.Y.; Artamonov, I.D.; Zolotarev, A.S.; Miroshnikov, A.I.; Martynov, V.I.; Kostina, M.B.; Kudelin, A.B.; Bogachuk, A.S. The complete amino acid sequence of visual rhodopsin. Bioorg. Khim. 1982, 8, 1011–1014. [Google Scholar]
- Dixon, R.A.; Kobilka, B.K.; Strader, D.J.; Benovic, J.L.; Dohlman, H.G.; Frielle, T.; Bolanowski, M.A.; Bennett, C.D.; Rands, E.; Diehl, R.E.; et al. Cloning of the gene and cdna for mammalian beta-adrenergic receptor and homology with rhodopsin. Nature 1986, 321, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Tesmer, J.J.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacology 2012, 133, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, H. Light-regulated binding of rhodopsin kinase and other proteins to cattle photoreceptor membranes. Biochemistry 1978, 17, 4389–4395. [Google Scholar] [CrossRef]
- Weller, M.; Virmaux, N.; Mandel, P. Light-stimulated phosphorylation of rhodopsin in the retina: The presence of a protein kinase that is specific for photobleached rhodopsin. Proc. Natl. Acad. Sci. USA 1975, 72, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Wilden, U.; Hall, S.W.; Kühn, H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kda protein of rod outer segments. Proc. Natl. Acad. Sci. USA 1986, 83, 1174–1178. [Google Scholar] [CrossRef] [Green Version]
- Strasser, R.H.; Sibley, D.R.; Lefkowitz, R.J. A novel catecholamine-activated adenosine cyclic 3’,5’-phosphate independent pathway for beta-adrenergic receptor phosphorylation in wild-type and mutant s49 lymphoma cells: Mechanism of homologous desensitization of adenylate cyclase. Biochemistry 1986, 25, 1371–1377. [Google Scholar] [CrossRef]
- Benovic, J.L.; DeBlasi, A.; Stone, W.C.; Caron, M.G.; Lefkowitz, R.J. Beta-adrenergic receptor kinase: Primary structure delineates a multigene family. Science 1989, 246, 235–240. [Google Scholar] [CrossRef]
- Benovic, J.L.; Mayor, F.J.; Somers, R.L.; Caron, M.G.; Lefkowitz, R.J. Light-dependent phosphorylation of rhodopsin by beta-adrenergic receptor kinase. Nature 1986, 321, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, W.; Inglese, J.; Palczewski, K.; Onorato, J.J.; Caron, M.G.; Lefkowitz, R.J. The receptor kinase family: Primary structure of rhodopsin kinase reveals similarities to the beta-adrenergic receptor kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 8715–8719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palczewski, K.; Buczylko, J.; Kaplan, M.W.; Polans, A.S.; Crabb, J.W. Mechanism of rhodopsin kinase activation. J. Biol. Chem. 1991, 266, 12949–12955. [Google Scholar] [PubMed]
- Chen, C.Y.; Dion, S.B.; Kim, C.M.; Benovic, J.L. Beta-adrenergic receptor kinase. Agonist-dependent receptor binding promotes kinase activation. J. Biol. Chem. 1993, 268, 7825–7831. [Google Scholar] [PubMed]
- Pack, T.F.; Orlen, M.I.; Ray, C.; Peterson, S.M.; Caron, M.G. The dopamine d2 receptor can directly recruit and activate grk2 without g protein activation. J. Biol. Chem. 2018, 293, 6161–6171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, H.; Hall, S.W.; Wilden, U. Light-induced binding of 48-kda protein to photoreceptor membranes is highly enhanced by phosphorylation of rhodopsin. FEBS Lett. 1984, 176, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Benovic, J.L.; Kühn, H.; Weyand, I.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Functional desensitization of the isolated β-adrenergic receptor by the β-adrenergic receptor kinase: Potential role of an analog of the retinal protein arrestin (48 kda protein). Proc. Natl. Acad. Sci. USA 1987, 84, 8879–8882. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, T.; Dietzschold, B.; Craft, C.M.; Wistow, G.; Early, J.J.; Donoso, L.A.; Horwitz, J.; Tao, R. Primary and secondary structure of bovine retinal s antigen (48 kda protein). Proc. Natl. Acad. Sci. USA 1987, 84, 6975–6979. [Google Scholar] [CrossRef] [Green Version]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Beta-arrestin: A protein that regulates beta-adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar] [CrossRef]
- Lohse, M.J.; Andexinger, S.; Pitcher, J.; Trukawinski, S.; Codina, J.; Faure, J.P.; Caron, M.G.; Lefkowitz, R.J. Receptor-specific desensitization with purified proteins. Kinase dependence and receptor specificity of beta-arrestin and arrestin in the beta 2-adrenergic receptor and rhodopsin systems. J. Biol. Chem. 1992, 267, 8558–8564. [Google Scholar]
- Chen, C.K.; Burns, M.E.; Spencer, M.; Niemi, G.A.; Chen, J.; Hurley, J.B.; Baylor, D.A.; Simon, M.I. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc. Natl. Acad. Sci. USA 1999, 96, 3718–3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Dodd, R.L.; Makino, C.L.; Simon, M.I.; Baylor, D.A.; Chen, J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature 1997, 389, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Mushegian, A.; Gurevich, V.V.; Gurevich, E.V. The origin and evolution of G protein-coupled receptor kinases. PLoS ONE 2012, 7, e33806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indrischek, H.; Prohaska, S.J.; Gurevich, V.V.; Gurevich, E.V.; Stadler, P.F. Uncovering missing pieces: Duplication and deletion history of arrestins in deuterostomes. BMC Evol. Biol. 2017, 17, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilden, U. Duration and amplitude of the light-induced cGMP hydrolysis in vertebrate photoreceptors are regulated by multiple phosphorylation of rhodopsin and by arrestin binding. Biochemistry 1995, 34, 1446–1454. [Google Scholar] [CrossRef]
- Krupnick, J.G.; Gurevich, V.V.; Benovic, J.L. Mechanism of quenching of phototransduction. Binding competition between arrestin and transducin for phosphorhodopsin. J. Biol. Chem. 1997, 272, 18125–18131. [Google Scholar] [CrossRef] [Green Version]
- Carman, C.V.; Benovic, J.L. G-protein-coupled receptors: Turn-ons and turn-offs. Curr. Opin. Neurobiol. 1998, 8, 335–344. [Google Scholar] [CrossRef]
- Goodman, O.B., Jr.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 1996, 383, 447–450. [Google Scholar] [CrossRef]
- Laporte, S.A.; Oakley, R.H.; Zhang, J.; Holt, J.A.; Ferguson, s.S.G.; Caron, M.G.; Barak, L.S. The 2-adrenergic receptor/arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. USA 1999, 96, 3712–3717. [Google Scholar] [CrossRef] [Green Version]
- Pals-Rylaarsdam, R.; Hosey, M.M. Two homologous phosphorylation domains differentially contribute to desensitization and internalization of the m2 muscarinic acetylcholine receptor. J. Biol. Chem. 1997, 272, 14152–14158. [Google Scholar] [CrossRef] [Green Version]
- Pals-Rylaarsdam, R.; Xu, Y.; Witt-Enderby, P.; Benovic, J.L.; Hosey, M.M. Desensitization and internalization of the m2 muscarinic acetylcholine receptor are directed by independent mechanisms. J. Biol. Chem. 1995, 270, 29004–29011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakata, H.; Kameyama, K.; Haga, K.; Haga, T. Location of agonist-dependent-phosphorylation sites in the third intracellular loop of muscarinic acetylcholine receptors (m2 subtype). Eur. J. Biochem. 1994, 220, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.J.; Gardner, B.R.; Williams, D.B.; Marinec, P.S.; Cabrera, D.M.; Peters, J.D.; Mak, C.C.; Kim, K.M.; Sibley, D.R. The role of phosphorylation in D1 dopamine receptor desensitization: Evidence for a novel mechanism of arrestin association. J. Biol. Chem. 2004, 279, 7999–8010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namkung, Y.; Dipace, C.; Javitch, J.A.; Sibley, D.R. G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J. Biol. Chem. 2009, 284, 15038–15051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharm. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausdorff, W.P.; Lohse, M.J.; Bouvier, M.; Liggett, S.B.; Caron, M.G.; Lefkowitz, R.J. Two kinases mediate agonist-dependent phosphorylation and desensitization of the beta 2-adrenergic receptor. Symp. Soc. Exp. Biol. 1990, 44, 225–240. [Google Scholar] [PubMed]
- Freedman, N.J.; Liggett, S.B.; Drachman, D.E.; Pei, G.; Caron, M.G.; Lefkowitz, R.J. Phosphorylation and desensitization of the human beta 1-adrenergic receptor. Involvement of g protein-coupled receptor kinases and cAMP-dependent protein kinase. J. Biol. Chem. 1995, 270, 17953–17961. [Google Scholar] [CrossRef] [Green Version]
- Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase a. Nature 1997, 390, 88–91. [Google Scholar] [CrossRef]
- Zamah, A.M.; Delahunty, M.; Luttrell, L.M.; Lefkowitz, R.J. Protein kinase A-mediated phosphorylation of the beta 2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. J. Biol. Chem. 2002, 277, 31249–31256. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.P.; Whalen, E.J.; Zamah, M.A.; Pierce, K.L.; Lefkowitz, R.J. PKA-mediated phosphorylation of the beta1-adrenergic receptor promotes Gs/Gi switching. Cell. Signal. 2004, 16, 1397–1403. [Google Scholar] [CrossRef]
- Molenaar, P.; Savarimuthu, S.M.; Sarsero, D.; Chen, L.; Semmler, A.B.; Carle, A.; Yang, I.; Bartel, S.; Vetter, D.; Beyerdörfer, I.; et al. (-)-adrenaline elicits positive inotropic, lusitropic, and biochemical effects through beta2-adrenoceptors in human atrial myocardium from nonfailing and failing hearts, consistent with Gs coupling but not with Gi coupling. Naunyn Schmiedebergs Arch. Pharm. 2007, 375, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Babu, B.; Clark, R.B. Beta(2)-adrenergic receptor lacking the cyclic amp-dependent protein kinase consensus sites fully activates extracellular signal-regulated kinase 1/2 in human embryonic kidney 293 cells: Lack of evidence for G(s)/G(i) switching. Mol. Pharm. 2002, 62, 1094–1102. [Google Scholar] [CrossRef]
- Liggett, S.B.; Freedman, N.J.; Schwinn, D.A.; Lefkowitz, R.J. Structural basis for receptor subtype-specific regulation revealed by a chimeric beta 3/beta 2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 3665–3669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell-Motz, E.A.; Small, K.M.; Theiss, C.T.; Liggett, S.B. Alpha 2A/alpha 2C-adrenergic receptor third loop chimera show that agonist interaction with receptor subtype backbone establishes g protein-coupled receptor kinase phosphorylation. J. Biol. Chem. 2000, 275, 28989–28993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, C.S.; Benovic, J.L. Structure/function analysis of α2A-adrenergic receptor interaction with G protein-coupled receptor kinase 2. J. Biol. Chem. 2005, 280, 11052–11058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell-Motz, E.A.; Liggett, S.B. G protein-coupled receptor kinase specificity for phosphorylation and desensitization of α2-adrenergic receptor subtypes. J. Biol. Chem. 1996, 271, 18082–18087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurose, H.; Lefkowitz, R.J. Differential desensitization and phosphorylation of three cloned and transfected alpha 2-adrenergic receptor subtypes. J. Biol. Chem. 1994, 269, 10093–10099. [Google Scholar]
- Gingrich, J.A.; Caron, M.G. Recent advances in the molecular biology of dopamine receptors. Annu. Rev. Neurosci. 1993, 16, 299–321. [Google Scholar] [CrossRef]
- Gimenez, L.E.; Vishnivetskiy, S.A.; Baameur, F.; Gurevich, V.V. Manipulation of very few receptor discriminator residues greatly enhances receptor specificity of non-visual arrestins. J. Biol. Chem. 2012, 287, 29495–29505. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, E.V.; Gainetdinov, R.R.; Gurevich, V.V. G protein-coupled receptor kinases as regulators of dopamine receptor functions. Pharm. Res. 2016, 111, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tiberi, M.; Nash, S.; Bertrand, L.; Lefkowitz, R.J.; Caron, M.G. Differential regulation of dopamine D1a receptor responsiveness by various g protein-coupled receptor kinases. J. Biol. Chem. 1996, 271, 3771–3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Homan, K.T.; Vishnivetskiy, S.A.; Manglik, A.; Tesmer, J.J.G.; Gurevich, V.V.; Gurevich, E.V. G protein-coupled receptor kinases of the GRK4 protein subfamily phosphorylate inactive G protein-coupled receptors (GPCRs). J. Biol. Chem. 2015, 290, 10775–10790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, J.L.; Free, R.B.; Sibley, D.R. Identification of G protein-biased agonists that fail to recruit β-arrestin or promote internalization of the D1 dopamine receptor. ACS Chem. Neurosci. 2015, 6, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Gray, D.L.; Allen, J.A.; Mente, S.; O’Connor, R.E.; DeMarco, G.J.; Efremov, I.; Tierney, P.; Volfson, D.; Davoren, J.; Guilmette, E.; et al. Impaired β-arrestin recruitment and reduced desensitization by non-catechol agonists of the D1 dopamine receptor. Nat. Commun. 2018, 9, 674. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-M.; Valenzano, K.J.; Robinson, S.R.; Yao, W.D.; Barak, L.S.; Caron, M.G. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and β-arrestins. J. Biol. Chem. 2001, 276, 37409–37414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, D.; Zheng, M.; Min, C.; Ma, L.; Kurose, H.; Park, J.H.; Kim, K.-M. Agonist-induced endocytosis and receptor phosphorylation mediate resensitization of dopamine D(2) receptors. Mol. Endocrinol. 2010, 24, 574–586. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.J.; Lachowicz, J.E.; Sibley, D.R. The D2s and D2l dopamine receptor isoforms are differentially regulated in chinese hamster ovary cells. Mol. Pharm. 1994, 45, 878–889. [Google Scholar]
- Namkung, Y.; Dipace, C.; Urizar, E.; Javitch, J.A.; Sibley, D.R. G protein-coupled receptor kinase-2 constitutively regulates D2 dopamine receptor expression and signaling independently of receptor phosphorylation. J. Biol. Chem. 2009, 284, 34103–34115. [Google Scholar] [CrossRef] [Green Version]
- Celver, J.; Sharma, M.; Thanawala, V.; Christopher Octeau, J.; Kovoor, A. Arrestin-dependent but G-protein coupled receptor kinase-independent uncoupling of D2-dopamine receptors. J. Neurochem. 2013, 127, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Haga, K.; Haga, T. Activation by G protein beta gamma subunits of agonist- or light-dependent phosphorylation of muscarinic acetylcholine receptors and rhodopsin. J. Biol. Chem. 1992, 267, 2222–2227. [Google Scholar]
- DebBurman, S.K.; Ptasienski, J.; Benovic, J.L.; Hosey, M.M. G protein-coupled receptor kinase GRK2 is a phospholipid-dependent enzyme that can be conditionally activated by G protein betagamma subunits. J. Biol. Chem. 1996, 271, 22552–22562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitcher, J.A.; Inglese, J.; Higgins, J.B.; Arriza, J.L.; Casey, P.J.; Kim, C.; Benovic, J.L.; Kwatra, M.M.; Caron, M.G.; Lefkowitz, R.J. Role of beta gamma subunits of G proteins in targeting the beta-adrenergic receptor kinase to membrane-bound receptors. Science 1992, 257, 1264–1267. [Google Scholar] [CrossRef] [PubMed]
- Gainetdinov, R.R.; Bohn, L.M.; Sotnikova, T.D.; Cyr, M.; Laakso, A.; Macrae, A.D.; Torres, G.E.; Kim, K.M.; Lefkowitz, R.J.; Caron, M.G.; et al. Dopaminergic supersensitivity in g protein-coupled receptor kinase 6-deficient mice. Neuron 2003, 38, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef]
- Murray, A.M.; Ryoo, H.L.; Gurevich, E.; Joyce, J.N. Localization of dopamine D3 receptors to mesolimbic and D2 receptors to mesostriatal regions of human forebrain. Proc. Natl. Acad. Sci. USA 1994, 91, 11271–11275. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, E.V.; Bordelon, Y.; Shapiro, R.M.; Arnold, S.E.; Gur, R.E.; Joyce, J.N. Mesolimbic dopamine D3 receptors and use of antipsychotics in patients with schizophrenia. A postmortem study. Arch. Gen. Psychiatry 1997, 54, 225–232. [Google Scholar] [CrossRef]
- Bouthenet, M.L.; Souil, E.; Martres, M.P.; Sokoloff, P.; Giros, B.; Schwartz, J.C. Localization of dopamine d3 receptor mrna in the rat brain using in situ hybridization histochemistry: Comparison with dopamine D2 receptor mRNA. Brain Res. 1991, 564, 203–219. [Google Scholar] [CrossRef]
- Min, C.; Zheng, M.; Zhang, X.; Caron, M.G.; Kim, K.M. Novel roles for β-arrestins in the regulation of pharmacological sequestration to predict agonist-induced desensitization of dopamine D3 receptors. Br. J. Pharm. 2013, 170, 1112–1129. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-M.; Gainetdinov, R.R.; Laporte, S.A.; Caron, M.G.; Barak, L.S. G protein-coupled receptor kinase regulates dopamine D3 receptor signaling by modulating the stability of a receptor-filamin-β-arrestin complex: A case of autoreceptor regulation. J. Biol Chem. 2005, 280, 12774–12780. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Reith, M.E.A.; Liu-Chen, L.-Y.; Kortagere, S. Biased signaling agonist of dopamine D3 receptor induces receptor internalization independent of β-arrestin recruitment. Pharm. Res. 2019, 143, 48–57. [Google Scholar] [CrossRef]
- Xu, W.; Wang, X.; Tocker, A.M.; Huang, P.; Reith, M.E.A.; Liu-Chen, L.-Y.; Smith, A.B.; Kortagere, S. Functional characterization of a novel series of biased signaling dopamine D3 receptor agonists. ACS Chem. Neurosci. 2017, 8, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Wess, J. Molecular basis of muscarinic acetylcholine receptor function. Trends Pharm. Sci. 1993, 14, 308–313. [Google Scholar] [CrossRef]
- Caulfield, M.P. Muscarinic receptors—characterization, coupling and function. Pharmacol. Ther. 1993, 58, 319–379. [Google Scholar] [CrossRef]
- Bünemann, M.; Hosey, M.M. G-protein coupled receptor kinases as modulators of g-protein signalling. J. Physiol. 1999, 517, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Pals-Rylaarsdam, R.; Gurevich, V.V.; Lee, K.B.; Ptasienski, J.; Benovic, J.L.; Hosey, M.M. Internalization of the m2 muscarinic acetylcholine receptor: Arrestin-independent and -dependent pathways. J. Biol. Chem. 1997, 272, 23682–23689. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.B.; Ptasienski, J.A.; Pals-Rylaarsdam, R.; Gurevich, V.V.; Hosey, M.M. Arrestin binding to the m2 muscarinic acetylcholine receptor is precluded by an inhibitory element in the third intracellular loop of the receptor. J. Biol. Chem. 2000, 275, 9284–9289. [Google Scholar] [CrossRef] [Green Version]
- Gainetdinov, R.R.; Bohn, L.M.; Walker, J.K.; Laporte, S.A.; Macrae, A.D.; Caron, M.G.; Lefkowitz, R.J.; Premont, R.T. Muscarinic supersensitivity and impaired receptor desensitization in G protein-coupled receptor kinase 5-deficient mice. Neuron 1999, 24, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Gomeza, J.; Shannon, H.; Kostenis, E.; Felder, C.; Zhang, L.; Brodkin, J.; Grinberg, A.; Sheng, H.; Wess, J. Pronounced pharmacologic deficits in m2 muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 1692–1697. [Google Scholar] [CrossRef] [Green Version]
- Gomeza, J.; Zhang, L.; Kostenis, E.; Felder, C.C.; Bymaster, F.P.; Brodkin, J.; Shannon, H.; Xia, B.; Duttaroy, A.; Deng, C.-X.; et al. Generation and pharmacological analysis of m2 and m4 muscarinic receptor knockout mice. Life Sci. 2001, 68, 2457–2466. [Google Scholar] [CrossRef]
- Walker, J.K.L.; Gainetdinov, R.R.; Feldman, D.S.; McFawn, P.K.; Caron, M.G.; Lefkowitz, R.J.; Premont, R.T.; Fisher, J.T. G protein-coupled receptor kinase 5 regulates airway responses induced by muscarinic receptor activation. Am. J. Physiol. 2004, 286, L312–L319. [Google Scholar] [CrossRef] [Green Version]
- Gainetdinov, R.R.; Premont, R.T.; Bohn, L.M.; Lefkowitz, R.J.; Caron, M.G. Desensitization of G protein-coupled receptors and neuronal function. Ann. Rev. Neurosci. 2004, 27, 107–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimenez, L.E.; Kook, S.; Vishnivetskiy, S.A.; Ahmed, M.R.; Gurevich, E.V.; Gurevich, V.V. Role of receptor-attached phosphates in binding of visual and non-visual arrestins to g protein-coupled receptors. J. Biol. Chem. 2012, 287, 9028–9040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, V.V.; Dion, S.B.; Onorato, J.J.; Ptasienski, J.; Kim, C.M.; Sterne-Marr, R.; Hosey, M.M.; Benovic, J.L. Arrestin interaction with g protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, b2-adrenergic, and m2 muscarinic cholinergic receptors. J. Biol. Chem. 1995, 270, 720–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, V.V.; Pals-Rylaarsdam, R.; Benovic, J.L.; Hosey, M.M.; Onorato, J.J. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J. Biol. Chem. 1997, 272, 28849–28852. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Richardson, R.M.; Kim, C.M.; Hosey, M.M.; Benovic, J.L. Binding of wild type and chimeric arrestins to the m2 muscarinic cholinergic receptor. J. Biol. Chem. 1993, 268, 16879–16882. [Google Scholar]
- Wan, M.; Zhang, W.; Tian, Y.; Xu, C.; Xu, T.; Liu, J.; Zhang, R. Unraveling a molecular determinant for clathrin-independent internalization of the m2 muscarinic acetylcholine receptor. Sci. Rep. 2015, 5, 11408. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, N.; Jojima, E.; Saito, H.; Haga, T. Role of the third intracellular loop in the subtype-specific internalization and recycling of muscarinic m2 and m4 receptors. Biomed. Res. 2014, 35, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Morisawa, K.; Saito, H.; Jojima, E.; Yoshida, N.; Haga, T. Muscarinic m4 receptor recycling requires a motif in the third intracellular loop. J. Pharm. Exp. 2008, 325, 947. [Google Scholar] [CrossRef] [Green Version]
- Lambert, L.; Dubayle, D.; Fafouri, A.; Herzog, E.; Csaba, Z.; Dournaud, P.; El Mestikawy, S.; Bernard, V. Endocytosis of activated muscarinic M2 receptor (M2R) in live mouse hippocampal neurons occurs via a clathrin-dependent pathway. Front. Cell Neurosci. 2018, 12, 450. [Google Scholar] [CrossRef]
- Haga, K.; Kameyama, K.; Haga, T.; Kikkawa, U.; Shiozaki, K.; Uchiyama, H. Phosphorylation of human m1 muscarinic acetylcholine receptors by G protein-coupled receptor kinase 2 and protein kinase C. J. Biol. Chem. 1996, 271, 2776–2782. [Google Scholar] [CrossRef] [Green Version]
- Lameh, J.; Philip, M.; Sharma, Y.K.; Moro, O.; Ramachandran, J.; Sadée, W. Hm1 muscarinic cholinergic receptor internalization requires a domain in the third cytoplasmic loop. J. Biol. Chem. 1992, 267, 13406–13412. [Google Scholar] [PubMed]
- Yeatman, H.R.; Lane, J.R.; Choy, K.; Ho, C.; Lambert, N.A.; Sexton, P.M.; Christopoulos, A.; Canals, M. Allosteric modulation of m1 muscarinic acetylcholine receptor internalization and subcellular trafficking. J. Biol. Chem. 2014, 289, 15856–15866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willets, J.M.; Nahorski, S.R.; Challiss, R.A.J. Roles of phosphorylation-dependent and -independent mechanisms in the regulation of m1 muscarinic acetylcholine receptors by G protein-coupled receptor kinase 2 in hippocampal neurons. J. Biol. Chem. 2005, 280, 18950–18958. [Google Scholar] [CrossRef] [Green Version]
- Willets, J.M.; Nash, M.S.; Challiss, R.A.J.; Nahorski, S.R. Imaging of muscarinic acetylcholine receptor signaling in hippocampal neurons: Evidence for phosphorylation-dependent and -independent regulation by G-protein-coupled receptor kinases. J. Neurosci. 2004, 24, 4157–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Bogatkevich, G.S.; Mukhin, Y.V.; Benovic, J.L.; Hildebrandt, J.D.; Lanier, S.M. Identification of Gβγ binding sites in the third intracellular loop of the m3-muscarinic receptor and their role in receptor regulation. J. Biol. Chem. 2000, 275, 9026–9034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, O.; Lameh, J.; Sadée, W. Serine- and threonine-rich domain regulates internalization of muscarinic cholinergic receptors. J. Biol. Chem. 1993, 268, 6862–6865. [Google Scholar]
- Debburman, S.K.; Kunapuli, P.; Benovic, J.L.; Hosey, M.M. Agonist-dependent phosphorylation of human muscarinic receptors in spodoptera frugiperda insect cell membranes by G protein-coupled receptor kinases. Mol. Pharm. 1995, 47, 224. [Google Scholar]
- Walker, J.K.L.; Peppel, K.; Lefkowitz, R.J.; Caron, M.G.; Fisher, J.T. Altered airway and cardiac responses in mice lacking G protein-coupled receptor kinase 3. Am. J. Physiol. 1999, 276, R1214–R1221. [Google Scholar] [CrossRef]
- Garssen, J.; Van Loveren, H.; Gierveld, C.M.; Van der Vliet, H.; Nijkamp, F.P. Functional characterization of muscarinic receptors in murine airways. Br. J. Pharm. 1993, 109, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Roffel, A.F.; Elzinga, C.R.; Zaagsma, J. Muscarinic m3 receptors mediate contraction of human central and peripheral airway smooth muscle. Pulm. Pharm. 1990, 3, 47–51. [Google Scholar] [CrossRef]
- Wolters, V.; Krasel, C.; Brockmann, J.; Bünemann, M. Influence of Gα on the dynamics of m3-acetylcholine receptor–G-protein–coupled receptor kinase 2 interaction. Mol. Pharm. 2015, 87, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, M.J.; Lee, K.A.; Niemi, G.A.; Craven, K.B.; Garwin, G.G.; Saari, J.C.; Hurley, J.B. Multiple phosphorylation of rhodopsin and the in vivo chemistry underlying rod photoreceptor dark adaptation. Neuron 2001, 31, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, A.W.; Doan, T.; Moaven, H.; Sokal, I.; Baameur, F.; Vishnivetskiy, S.A.; Homan, K.T.; Tesmer, J.J.; Gurevich, V.V.; Chen, J.; et al. C-terminal threonines and serines play distinct roles in the desensitization of rhodopsin, a G protein-coupled receptor. Elife 2015, 4. [Google Scholar] [CrossRef]
- Mendez, A.; Burns, M.E.; Roca, A.; Lem, J.; Wu, L.W.; Simon, M.I.; Baylor, D.A.; Chen, J. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron 2000, 28, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of phosphorylation codes for arrestin recruitment by g protein-coupled receptors. Cell 2017, 170, 457–469. [Google Scholar] [CrossRef] [Green Version]
- 1Onorato, J.J.; Palczewski, K.; Regan, J.W.; Caron, M.G.; Lefkowitz, R.J.; Benovic, J.L. Role of acidic amino acids in peptide substrates of the beta-adrenergic receptor kinase and rhodopsin kinase. Biochemistry 1991, 30, 5118–5125. [Google Scholar] [CrossRef]
- Allouche, S.; Noble, F.; Marie, N. Opioid receptor desensitization: Mechanisms and its link to tolerance. Front. Pharm. 2014, 5, 280. [Google Scholar] [CrossRef] [Green Version]
- Lemos Duarte, M.; Devi, L.A. Post-translational modifications of opioid receptors. Trends Neurosci. 2020, 43, 417–432. [Google Scholar] [CrossRef]
- Just, S.; Illing, S.; Trester-Zedlitz, M.; Lau, E.K.; Kotowski, S.J.; Miess, E.; Mann, A.; Doll, C.; Trinidad, J.C.; Burlingame, A.L.; et al. Differentiation of opioid drug effects by hierarchical multi-site phosphorylation. Mol. Pharm. 2013, 83, 633. [Google Scholar] [CrossRef] [Green Version]
- Doll, C.; Pöll, F.; Peuker, K.; Loktev, A.; Glück, L.; Schulz, S. Deciphering µ-opioid receptor phosphorylation and dephosphorylation in hek293 cells. Br. J. Pharm. 2012, 167, 1259–1270. [Google Scholar] [CrossRef] [Green Version]
- McPherson, J.; Rivero, G.; Baptist, M.; Llorente, J.; Al-Sabah, S.; Krasel, C.; Dewey, W.L.; Bailey, C.P.; Rosethorne, E.M.; Charlton, S.J.; et al. μ-opioid receptors: Correlation of agonist efficacy for signalling with ability to activate internalization. Mol. Pharm. 2010, 78, 756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grecksch, G.; Just, S.; Pierstorff, C.; Imhof, A.-K.; Glück, L.; Doll, C.; Lupp, A.; Becker, A.; Koch, T.; Stumm, R.; et al. Analgesic tolerance to high-efficacy agonists but not to morphine is diminished in phosphorylation-deficient s375a μ-opioid receptor knock-in mice. J. Neurosci. 2011, 31, 13890. [Google Scholar] [CrossRef] [Green Version]
- Miess, E.; Gondin, A.B.; Yousuf, A.; Steinborn, R.; Mösslein, N.; Yang, Y.; Göldner, M.; Ruland, J.G.; Bünemann, M.; Krasel, C.; et al. Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid μ-opioid receptor desensitization. Sci. Signal. 2018, 11, eaas9609. [Google Scholar] [CrossRef] [Green Version]
- Lau, E.K.; Trester-Zedlitz, M.; Trinidad, J.C.; Kotowski, S.J.; Krutchinsky, A.N.; Burlingame, A.L.; von Zastrow, M. Quantitative encoding of the effect of a partial agonist on individual opioid receptors by multisite phosphorylation and threshold detection. Sci. Signal. 2011, 4, ra52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glück, L.; Loktev, A.; Moulédous, L.; Mollereau, C.; Law, P.-Y.; Schulz, S. Loss of morphine reward and dependence in mice lacking g protein–coupled receptor kinase 5. Biol. Psychiatry 2014, 76, 767–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-J.; Oldfield, S.; Butcher, A.J.; Tobin, A.B.; Saxena, K.; Gurevich, V.V.; Benovic, J.L.; Henderson, G.; Kelly, E. Identification of phosphorylation sites in the cooh-terminal tail of the μ-opioid receptor. J. Neurochem. 2013, 124, 189–199. [Google Scholar] [CrossRef]
- Schulz, S.; Mayer, D.; Pfeiffer, M.; Stumm, R.; Koch, T.; Höllt, V. Morphine induces terminal μ-opioid receptor desensitization by sustained phosphorylation of serine-375. EMBO J. 2004, 23, 3282–3289. [Google Scholar] [CrossRef] [Green Version]
- Yousuf, A.; Miess, E.; Sianati, S.; Du, Y.-P.; Schulz, S.; Christie, M.J. Role of phosphorylation sites in desensitization of m-opioid receptor. Mol. Pharm. 2015, 88, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Møller, T.C.; Pedersen, M.F.; van Senten, J.R.; Seiersen, S.D.; Mathiesen, J.M.; Bouvier, M.; Bräuner-Osborne, H. Dissecting the roles of grk2 and grk3 in μ-opioid receptor internalization and β-arrestin2 recruitment using CRISPR/Cas9-edited hek293 cells. Sci. Rep. 2020, 10, 17395. [Google Scholar] [CrossRef]
- Lowe, J.D.; Sanderson, H.S.; Cooke, A.E.; Ostovar, M.; Tsisanova, E.; Withey, S.L.; Chavkin, C.; Husbands, S.M.; Kelly, E.; Henderson, G.; et al. Role of G protein–coupled receptor kinases 2 and 3 in m-opioid receptor desensitization and internalization. Mol. Pharm. 2015, 88, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Birdsong, W.T.; Arttamangkul, S.; Bunzow, J.R.; Williams, J.T. Agonist binding and desensitization of the m-opioid receptor is modulated by phosphorylation of the C-terminal tail domain. Mol. Pharm. 2015, 88, 816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.; Brown, S.; Derleth, C.; Mackie, K. Internalization and recycling of the cb1 cannabinoid receptor. J. Neurochem. 1999, 73, 493–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouhen, O.M.; Wang, G.; Solberg, J.; Erickson, L.J.; Law, P.Y.; Loh, H.H. Hierarchical phosphorylation of delta-opioid receptor regulates agonist-induced receptor desensitization and internalization. J. Biol. Chem. 2000, 275, 36659–36664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wang, F.; Chen, X.; Chen, Y.; Ma, L. Post-endocytic fates of δ-opioid receptor are regulated by GRK2-mediated receptor phosphorylation and distinct β-arrestin isoforms. J. Neurochem. 2008, 106, 781–792. [Google Scholar] [CrossRef]
- Guo, J.; Wu, Y.; Zhang, W.; Zhao, J.; Devi, L.A.; Pei, G.; Ma, L. Identification of G protein-coupled receptor kinase 2 phosphorylation sites responsible for agonist-stimulated δ-opioid receptor phosphorylation. Mol. Pharmacol. 2000, 58, 1050. [Google Scholar] [CrossRef]
- Mann, A.; Liebetrau, S.; Klima, M.; Dasgupta, P.; Massotte, D.; Schulz, S. Agonist-induced phosphorylation bar code and differential post-activation signaling of the delta opioid receptor revealed by phosphosite-specific antibodies. Sci. Rep. 2020, 10, 8585. [Google Scholar] [CrossRef]
- McLaughlin, J.P.; Xu, M.; Mackie, K.; Chavkin, C. Phosphorylation of a carboxyl-terminal serine within the κ-opioid receptor produces desensitization and internalization. J. Biol. Chem. 2003, 278, 34631–34640. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, J.P.; Myers, L.C.; Zarek, P.E.; Caron, M.G.; Lefkowitz, R.J.; Czyzyk, T.A.; Pintar, J.E.; Chavkin, C. Prolonged kappa opioid receptor phosphorylation mediated by g-protein receptor kinase underlies sustained analgesic tolerance. J. Biol. Chem. 2004, 279, 1810–1818. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chiu, Y.-T.; Wu, W.; Huang, P.; Mann, A.; Schulz, S.; Liu-Chen, L.-Y. Determination of sites of u50,488h-promoted phosphorylation of the mouse κ opioid receptor (kopr): Disconnect between kopr phosphorylation and internalization. Biochem. J. 2016, 473, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.R.; Planer, W.; Siuda, E.R.; Zhao, H.-C.; Stickler, L.; Chang, S.D.; Baird, M.A.; Cao, Y.-Q.; Bruchas, M.R. Serine 363 is required for nociceptin/orphanin FQ opioid receptor (NOPR) desensitization, internalization, and arrestin signaling. J. Biol. Chem. 2012, 287, 42019–42030. [Google Scholar] [CrossRef] [Green Version]
- Mann, A.; Moulédous, L.; Froment, C.; O’Neill, P.R.; Dasgupta, P.; Günther, T.; Brunori, G.; Kieffer, B.L.; Toll, L.; Bruchas, M.R.; et al. Agonist-selective nop receptor phosphorylation correlates in vitro and in vivo and reveals differential post-activation signaling by chemically diverse agonists. Sci. Signal. 2019, 12, eaau8072. [Google Scholar] [CrossRef] [PubMed]
- Al-Zoubi, R.; Morales, P.; Reggio, P.H. Structural insights into cb1 receptor biased signaling. Int. J. Mol. Sci. 2019, 20, 1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daigle, T.L.; Kwok, M.L.; Mackie, K. Regulation of CB1 cannabinoid receptor internalization by a promiscuous phosphorylation-dependent mechanism. J. Neurochem. 2008, 106, 70–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.; Brown, S.; Roche, J.P.; Hsieh, C.; Celver, J.P.; Kovoor, A.; Chavkin, C.; Mackie, K. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J. Neurosci. 1999, 19, 3773. [Google Scholar] [CrossRef] [Green Version]
- Daigle, T.L.; Kearn, C.S.; Mackie, K. Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacology 2008, 54, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Peraza, F.; Ahn, K.H.; Nogueras-Ortiz, C.; Mungrue, I.N.; Mackie, K.; Kendall, D.A.; Yudowski, G.A. Mechanisms of biased beta-arrestin-mediated signaling downstream from the cannabinoid 1 receptor. Mol. Pharm. 2016, 89, 618. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Gurevich, E.V. GPCR monomers and oligomers: It takes all kinds. Trends Neurosci. 2007, 31, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Gurevich, E.V. How and why do GPCRs dimerize? Trends Pharm. Sci. 2008, 29, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Gurevich, E.V. GPCRs and signal transducers: Interaction stoichiometry. Trends Pharm. Sci. 2018, 39, 672–684. [Google Scholar] [CrossRef]
- Tobin, A.B.; Butcher, A.J.; Kong, K.C. Location, location, location...Site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharm. Sci. 2008, 29, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.-Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal 2011, 4, ra51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zidar, D.A.; Violin, J.D.; Whalen, E.J.; Lefkowitz, R.J. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 9649–9654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohout, T.A.; Nicholas, S.L.; Perry, S.J.; Reinhart, G.; Junger, S.; Struthers, R.S. Differential desensitization, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J. Biol. Chem. 2004, 279, 23214–23222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Ahn, S.; Ren, X.-R.; Whalen, E.J.; Reiter, E.; Wei, H.; Lefkowitz, R.J. Functional antagonism of different G protein-coupled receptor kinases for β-arrestin-mediated angiotensin II receptor signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 1442–1447. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.R.; Reiter, E.; Ahn, S.; Kim, J.; Chen, W.; Lefkowitz, R.J. Different g protein-coupled receptor kinases govern G protein and beta-arrestin-mediated signaling of V2 vasopressin receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 1448–1453. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Staus, D.P.; Wingler, L.M.; Ahn, S.; Pani, B.; Capel, W.D.; Lefkowitz, R.J. G protein–coupled receptor kinases (GRKs) orchestrate biased agonism at the β2-adrenergic receptor. Sci. Signal. 2018, 11, eaar7084. [Google Scholar] [CrossRef] [Green Version]
- Kaya, A.I.; Perry, N.A.; Gurevich, V.V.; Iverson, T.M. Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 14139–14149. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. Biased GPCR signaling: Possible mechanisms and inherent limitations. Pharmacol. Ther. 2020, 211, 107540. [Google Scholar] [CrossRef]
- Inagaki, S.; Ghirlando, R.; Vishnivetskiy, S.A.; Homan, K.T.; White, J.F.; Tesmer, J.J.G.; Gurevich, V.V.; Grisshammer, R. G protein-coupled receptor kinase 2 (GRK2) and 5 (GRK5) exhibit selective phosphorylation of the neurotensin receptor in vitro. Biochemistry 2015, 54, 4320–4329. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.R.; Berthet, A.; Bychkov, E.; Porras, G.; Li, Q.; Bioulac, B.H.; Carl, Y.T.; Bloch, B.; Kook, S.; Aubert, I.; et al. Lentiviral overexpression of GRK6 alleviates l-dopa-induced dyskinesia in experimental Parkinson’s disease. Sci. Transl. Med. 2010, 2, 28ra28. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.R.; Bychkov, E.; Kook, S.; Zurkovsky, L.; Dalby, K.N.; Gurevich, E.V. Overexpression of GRK6 rescues l-dopa-induced signaling abnormalities in the dopamine-depleted striatum of hemiparkinsonian rats. Exp. Neurol. 2015, 266, 42–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daigle, T.L.; Ferris, M.J.; Gainetdinov, R.R.; Sotnikova, T.D.; Urs, N.M.; Jones, S.R.; Caron, M.G. Selective deletion of GRK2 alters psychostimulant-induced behaviors and dopamine neurotransmission. Neuropsychopharmacology 2014, 39, 2450–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daigle, T.L.; Caron, M.G. Elimination of GRK2 from cholinergic neurons reduces behavioral sensitivity to muscarinic receptor activation. J. Neurosci. 2012, 32, 11461–11466. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rasul, I.; Sun, Y.; Wu, G.; Li, L.; Premont, R.T.; Suo, W.Z. GRK5 deficiency leads to reduced hippocampal acetylcholine level via impaired presynaptic m2/m4 autoreceptor desensitization. J. Biol. Chem. 2009, 284, 19564–19571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Li, L.; He, S.; Liu, J.; Sun, Y.; He, M.; Grasing, K.; Premont, R.T.; Suo, W.Z. GRK5 deficiency accelerates β-amyloid accumulation in tg2576 mice via impaired cholinergic activity. J. Biol. Chem. 2010, 285, 41541–41548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomeza, J.; Zhang, L.; Kostenis, E.; Felder, C.; Bymaster, F.; Brodkin, J.; Shannon, H.; Xia, B.; Deng, C.; Wess, J. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in m(4) muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 10483–10488. [Google Scholar] [CrossRef] [Green Version]
- Gerber, D.J.; Sotnikova, T.D.; Gainetdinov, R.R.; Huang, S.Y.; Caron, M.G.; Tonegawa, S. Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in m1 muscarinic acetylcholine receptor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 15312–15317. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, A.; Schmiedel, F.; Sianati, S.; Bailey, A.; Bateman, J.T.; Levitt, E.S.; Williams, J.T.; Christie, M.J.; Schulz, S. Phosphorylation-deficient G-protein-biased μ-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun. 2019, 10, 367. [Google Scholar] [CrossRef]
- Terman, G.W.; Jin, W.; Cheong, Y.-P.; Lowe, J.; Caron, M.G.; Lefkowitz, R.J.; Chavkin, C. G-protein receptor kinase 3 (GRK3) influences opioid analgesic tolerance but not opioid withdrawal. Br. J. Pharm. 2004, 141, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Morgan, D.J.; Davis, B.J.; Kearn, C.S.; Marcus, D.; Cook, A.J.; Wager-Miller, J.; Straiker, A.; Myoga, M.H.; Karduck, J.; Leishman, E.; et al. Mutation of putative GRK phosphorylation sites in the cannabinoid receptor 1 (CB1R) confers resistance to cannabinoid tolerance and hypersensitivity to cannabinoids in mice. J. Neurosci. 2014, 34, 5152. [Google Scholar] [CrossRef]
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J. Regulation of m-opioid receptors: Desensitization, phosphorylation, internalization, and tolerance. Pharm. Rev. 2013, 65, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melief, E.J.; Miyatake, M.; Bruchas, M.R.; Chavkin, C. Ligand-directed C-jun N-terminal kinase activation disrupts opioid receptor signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 11608–11613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente-Sanchez, A.; Dripps, I.J.; Tipton, A.F.; Akbari, H.; Akbari, A.; Jutkiewicz, E.M.; Pradhan, A.A. Tolerance to high-internalizing δ opioid receptor agonist is critically mediated by arrestin 2. Br. J. Pharm. 2018, 175, 3050–3059. [Google Scholar] [CrossRef] [PubMed]
- Nealon, C.M.; Henderson-Redmond, A.N.; Hale, D.E.; Morgan, D.J. Tolerance to win55,212–2 is delayed in desensitization-resistant S426A/S430A mice. Neuropharmacology 2019, 148, 151–159. [Google Scholar] [CrossRef]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Rüttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K.; et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 2018, 9, 341. [Google Scholar] [CrossRef]
- Alvarez-Curto, E.; Inoue, A.; Jenkins, L.; Raihan, S.Z.; Prihandoko, R.; Tobin, A.B.; Milligan, G. Targeted elimination of G proteins and arrestins defines their specific contributions to both intensity and duration of G protein-coupled receptor signaling. J. Biol. Chem. 2016, 291, 27147–27159. [Google Scholar] [CrossRef] [Green Version]
- Luttrell, L.M.; Wang, J.; Plouffe, B.; Smith, J.S.; Yamani, L.; Kaur, S.; Jean-Charles, P.-Y.; Gauthier, C.; Lee, M.-H.; Pani, B.; et al. Manifold roles of beta-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 2018, 11, eaat7650. [Google Scholar] [CrossRef] [Green Version]
- O’Hayre, M.; Eichel, K.; Avino, S.; Zhao, X.; Steffen, D.J.; Feng, X.; Kawakami, K.; Aoki, J.; Messer, K.; Sunahara, R.; et al. Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci. Signal. 2017, 10, 484. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Gurevich, E.V. Arrestins and G proteins in cellular signaling: The coin has two sides. Sci. Signal. 2018, 11, eaav1646. [Google Scholar] [CrossRef]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [Green Version]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.-T.; Pitis, P.M.; Gotchev, D.; Yuan, C.; et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharm. Exp. 2013, 344, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Violin, J.D.; Crombie, A.L.; Soergel, D.G.; Lark, M.W. Biased ligands at G-protein-coupled receptors: Promise and progress. Trends Pharm. Sci. 2014, 35, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Raman, D.; Wei, J.; Kennedy, M.J.; Hurley, J.B.; Gurevich, V.V. Regulation of arrestin binding by rhodopsin phosphorylation level. J. Biol. Chem. 2007, 282, 32075–32083. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurevich, E.V.; Gurevich, V.V. GRKs as Modulators of Neurotransmitter Receptors. Cells 2021, 10, 52. https://doi.org/10.3390/cells10010052
Gurevich EV, Gurevich VV. GRKs as Modulators of Neurotransmitter Receptors. Cells. 2021; 10(1):52. https://doi.org/10.3390/cells10010052
Chicago/Turabian StyleGurevich, Eugenia V., and Vsevolod V. Gurevich. 2021. "GRKs as Modulators of Neurotransmitter Receptors" Cells 10, no. 1: 52. https://doi.org/10.3390/cells10010052
APA StyleGurevich, E. V., & Gurevich, V. V. (2021). GRKs as Modulators of Neurotransmitter Receptors. Cells, 10(1), 52. https://doi.org/10.3390/cells10010052