Master Regulators of Muscle Atrophy: Role of Costamere Components


Abstract
1. Introduction
2. Master Regulators of Muscle Atrophy
2.1. Transcriptional Regulators of Atrogenes
2.1.1. FoxOs
2.1.2. NF-κB
2.1.3. Smad3
2.1.4. ATF4
2.1.5. p53
2.1.6. Hippo Pathway
2.2. Oxidative and Nitrosative Stress
2.2.1. Reactive Oxygen Species (ROS)
2.2.2. Reactive Nitrogen Species (RNS)
2.3. Mechanotransduction
2.3.1. Dystrophin Glycoprotein Complex (DGC)
2.3.2. Integrins and Integrin-Associated Signaling
2.3.3. IR/IGF-R
2.3.4. Na+/K+ ATPase and Ion Channels
3. Involvement of Costamere Components in Different Muscle Atrophy Types
3.1. Unloading/Bed Rest/Immobilization
3.2. Denervation/Spinal Cord Injury
3.3. Cachexia
3.4. Sarcopenia
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 4-PBA | 4-phenyl butyric acid |
| AAV-9 | Adeno-associated virus serotype 9 |
| Ac | Acetylation |
| ActRIIB | Activin receptor type IIB |
| AICAR | 5-amino-imidazole-4-carboxamide ribonucleotide (AMPK activator) |
| Akt | Protein kinase B |
| AMP | Adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| ATF4 | Activating transcription factor 4 |
| ATP | Adenosine triphosphate |
| BAFFR | Tumor necrosis factor receptor superfamily member 13C |
| cAMP | Cyclic adenosine monophosphate |
| Cbl-b | Casitas B-lineage lymphoma-b ubiquitin ligase |
| CD40 | Cluster of differentiation 40 |
| CHORDS | Cysteine and Histidine-Rich Domains |
| c-Raf | RAF proto-oncogene serine/threonine-protein kinase |
| c-Rel | Proto-oncogene c-Rel |
| CS | CHORD and Sgt1 domain |
| DGC | Dystrophin glycoprotein complex |
| ECM | Extracellular matrix |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| FAK | Focal adhesion kinase |
| FOXO1/3/4 | Forkhead box O1/3/4 |
| GLUT-4 | Glucose transporter 4 |
| Grp94/gp96 | Glucose-regulated protein 94/glycoprotein 96 |
| GSK-3β | Glycogen synthase kinase-3β |
| HDAC1/4 | Histone deacetylase 1/4 |
| HER2 | Receptor tyrosine-protein kinase erbB-2 |
| Hsp90 | Heat shock protein 90 |
| IGF1R | Insulin-like growth factor 1 receptor |
| IGF-I and –II | Insulin-like growth factor I and II |
| IKKα/β | Inhibitor of nuclear factor kappa-B kinase subunit α/β |
| IL-1 | Interleukin 1 |
| ILK | Integrin-linked kinase |
| IQGAP-1 | IQ Motif Containing GTPase Activating Protein 1 |
| IR | Insulin receptor |
| IRS-1 | Insulin receptor substrate-1 |
| ITGB1BP2 | Integrin beta-1-binding protein 2 |
| IκB | Inhibitor of nuclear factor kappa B |
| JNK | c-jun N-terminal kinase |
| LGMD2A | Limb-girdle muscular dystrophy type 2A |
| LTβR | Lymphotoxin-β receptor |
| MAFbx | Muscle atrophy F-box ubiquitin ligase |
| MAPK | Mitogen-activated protein kinase |
| MEK1/2 | Dual specificity mitogen-activated protein kinase kinase 1/2 |
| MLP | Muscle LIM protein |
| MRF4 | Myogenic regulatory factor 4 |
| MST1 | Macrophage stimulating 1 |
| mTORC1 | Mammalian/mechanistic target of rapamycin |
| MuRF-1 | Muscle RING finger—1 ubiquitin ligase |
| MyoD | Gene coding for Myoblast determination protein 1 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NF-κB1 | Nuclear factor NF-kappa-B p105 subunit |
| NF-κB2 | Nuclear factor NF-kappa-B p100 subunit |
| NIK | NF-κB inducing kinase |
| NMJ | Neuromuscular junction |
| nNOS | Neuronal nitric oxide synthase |
| NO | Nitric oxide |
| p38 MAPK | p38 mitogen-activated protein kinase |
| p70S6K | Ribosomal protein S6 kinase p70 |
| PCG-1α and β | Peroxisome proliferator-activated receptor-gamma coactivator 1α and β |
| PDZ domain | PSD-95/Dlg/ZO-1 domain |
| PI3K | Phosphoinositide 3-kinase |
| PINCH | Particularly interesting new Cys-His protein 1 |
| PKC | Protein kinase C |
| PLA2 | Phospholipase A2 |
| PW1 | Gene coding for Paternally-expressed gene 3 protein |
| RANK | Tumor necrosis factor receptor superfamily member 11A |
| RelA/p65 | Transcription factor p65 |
| RelB | Transcription factor RelB |
| ROS | Reactive oxygen species |
| RYR1 | Ryanodine receptor 1 |
| SAC | Stretch-activated ion channel |
| SCN4A | Gene coding for Sodium channel protein type 4 subunit alpha |
| Smad | Small mother against decapentaplegic |
| SR | Sarcoplasmic reticulum |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| TLR | Toll-like receptor |
| TNFR | Tumor necrosis factor receptor |
| TNF-α | Tumor necrosis factor a |
| Trim 32 | Tripartite motif-containing protein 32 |
| TRPC1 | Transient receptor potential channel 1 |
| TWEAK | TNF-related weak inducer of apoptosis |
| YAP | Yes-associated protein |
References
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, K.M.; Haddad, F.; Pandorf, C.E.; Roy, R.R.; Edgerton, V.R. Alterations in muscle mass and contractile phenotype in response to unloading models: Role of transcriptional/pretranslational mechanisms. Front. Physiol. 2013, 4, 284. [Google Scholar] [CrossRef] [PubMed]
- Pisot, R.; Marusic, U.; Biolo, G.; Mazzucco, S.; Lazzer, S.; Grassi, B.; Reggiani, C.; Toniolo, L.; di Prampero, P.E.; Passaro, A.; et al. Greater loss in muscle mass and function but smaller metabolic alterations in older compared with younger men following 2 wk of bed rest and recovery. J. Appl. Physiol. 2016, 120, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Vikne, H.; Strom, V.; Pripp, A.H.; Gjovaag, T. Human skeletal muscle fiber type percentage and area after reduced muscle use: A systematic review and meta-analysis. Scand. J. Med. Sci. Sports 2020, 30, 1298–1317. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Khaghani, S.; Turk, C.; Wiederstein, J.L.; Holper, S.; Piller, T.; Nogara, L.; Blaauw, B.; Gunther, S.; Muller, S.; et al. Single Muscle Fiber Proteomics Reveals Distinct Protein Changes in Slow and Fast Fibers during Muscle Atrophy. J. Proteome Res. 2018, 17, 3333–3347. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.E.; von Allmen, M.T.; Devries, M.C.; Phillips, S.M. Muscle Disuse as a Pivotal Problem in Sarcopenia-related Muscle Loss and Dysfunction. J. Frailty Aging 2016, 5, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Ballak, S.B.; Degens, H.; de Haan, A.; Jaspers, R.T. Aging related changes in determinants of muscle force generating capacity: A comparison of muscle aging in men and male rodents. Ageing Res. Rev. 2014, 14, 43–55. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyere, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef]
- Narici, M.V.; Maffulli, N. Sarcopenia: Characteristics, mechanisms and functional significance. Br. Med. Bull. 2010, 95, 139–159. [Google Scholar] [CrossRef]
- Bielecka-Dabrowa, A.; Ebner, N.; Dos Santos, M.R.; Ishida, J.; Hasenfuss, G.; von Haehling, S. Cachexia, muscle wasting, and frailty in cardiovascular disease. Eur. J. Heart Fail. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.F.; Rohm, M.; Herzig, S.; Berriel Diaz, M. Cancer Cachexia: More Than Skeletal Muscle Wasting. Trends Cancer 2018, 4, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Peixoto da Silva, S.; Santos, J.M.O.; Costa, E.S.M.P.; Gil da Costa, R.M.; Medeiros, R. Cancer cachexia and its pathophysiology: Links with sarcopenia, anorexia and asthenia. J. Cachexia Sarcopenia Muscle 2020, 11, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.T.; Ma, J.F.; Marco, S.D.; Gallouzi, I.E. Inducible nitric oxide synthase (iNOS) in muscle wasting syndrome, sarcopenia, and cachexia. Aging 2011, 3, 702–715. [Google Scholar] [CrossRef]
- Hughes, D.C.; Wallace, M.A.; Baar, K. Effects of aging, exercise, and disease on force transfer in skeletal muscle. Am. J. Physiol.. Endocrinol. Metab. 2015, 309, E1–E10. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef]
- Calura, E.; Cagnin, S.; Raffaello, A.; Laveder, P.; Lanfranchi, G.; Romualdi, C. Meta-analysis of expression signatures of muscle atrophy: Gene interaction networks in early and late stages. BMC Genom. 2008, 9, 630. [Google Scholar] [CrossRef]
- Vainshtein, A.; Sandri, M. Signaling Pathways That Control Muscle Mass. Int. J. Mol. Sci. 2020, 21, 4759. [Google Scholar] [CrossRef]
- Shenkman, B.S. How Postural Muscle Senses Disuse? Early Signs and Signals. Int. J. Mol. Sci. 2020, 21, 5037. [Google Scholar] [CrossRef]
- Bohnert, K.R.; McMillan, J.D.; Kumar, A. Emerging roles of ER stress and unfolded protein response pathways in skeletal muscle health and disease. J. Cell. Physiol. 2018, 233, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Watt, K.I.; Goodman, C.A.; Hornberger, T.A.; Gregorevic, P. The Hippo Signaling Pathway in the Regulation of Skeletal Muscle Mass and Function. Exerc. Sport Sci. Rev. 2018, 46, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Brocca, L.; Toniolo, L.; Reggiani, C.; Bottinelli, R.; Sandri, M.; Pellegrino, M.A. FoxO-dependent atrogenes vary among catabolic conditions and play a key role in muscle atrophy induced by hindlimb suspension. J. Physiol. 2017, 595, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Barberi, L.; Bijlsma, A.Y.; Blaauw, B.; Dyar, K.A.; Milan, G.; Mammucari, C.; Meskers, C.G.; Pallafacchina, G.; Paoli, A.; et al. Signalling pathways regulating muscle mass in ageing skeletal muscle: The role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology 2013, 14, 303–323. [Google Scholar] [CrossRef] [PubMed]
- Hwee, D.T.; Baehr, L.M.; Philp, A.; Baar, K.; Bodine, S.C. Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell 2014, 13, 92–101. [Google Scholar] [CrossRef]
- Suzuki, N.; Motohashi, N.; Uezumi, A.; Fukada, S.; Yoshimura, T.; Itoyama, Y.; Aoki, M.; Miyagoe-Suzuki, Y.; Takeda, S. NO production results in suspension-induced muscle atrophy through dislocation of neuronal NOS. J. Clin. Investig. 2007, 117, 2468–2476. [Google Scholar] [CrossRef]
- Vitadello, M.; Gherardini, J.; Gorza, L. The stress protein/chaperone Grp94 counteracts muscle disuse atrophy by stabilizing subsarcolemmal neuronal nitric oxide synthase. Antioxid. Redox Signal. 2014, 20, 2479–2496. [Google Scholar] [CrossRef]
- Vitadello, M.; Germinario, E.; Ravara, B.; Libera, L.D.; Danieli-Betto, D.; Gorza, L. Curcumin counteracts loss of force and atrophy of hindlimb unloaded rat soleus by hampering neuronal nitric oxide synthase untethering from sarcolemma. J. Physiol. 2014, 592, 2637–2652. [Google Scholar] [CrossRef]
- Lechado i Terradas, A.; Vitadello, M.; Traini, L.; Namuduri, A.V.; Gastaldello, S.; Gorza, L. Sarcolemmal loss of active nNOS (Nos1) is an oxidative stress-dependent, early event driving disuse atrophy. J. Pathol. 2018, 246, 433–446. [Google Scholar] [CrossRef]
- Belova, S.P.; Vilchinskaya, N.A.; Mochalova, E.P.; Mirzoev, T.M.; Nemirovskaya, T.L.; Shenkman, B.S. Elevated p70S6K phosphorylation in rat soleus muscle during the early stage of unloading: Causes and consequences. Arch. Biochem. Biophys. 2019, 674, 108105. [Google Scholar] [CrossRef] [PubMed]
- Bertaggia, E.; Coletto, L.; Sandri, M. Posttranslational modifications control FoxO3 activity during denervation. Am. J. Physiol. Cell Physiol. 2012, 302, C587–C596. [Google Scholar] [CrossRef] [PubMed]
- Beharry, A.W.; Sandesara, P.B.; Roberts, B.M.; Ferreira, L.F.; Senf, S.M.; Judge, A.R. HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J. Cell Sci. 2014, 127, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Vernus, B.; Chelh, I.; Cassar-Malek, I.; Gabillard, J.C.; Hadj Sassi, A.; Seiliez, I.; Picard, B.; Bonnieu, A. Myostatin and the skeletal muscle atrophy and hypertrophy signaling pathways. Cell. Mol. Life Sci. Cmls 2014, 71, 4361–4371. [Google Scholar] [CrossRef] [PubMed]
- Fusella, F.; Secli, L.; Cannata, C.; Brancaccio, M. The one thousand and one chaperones of the NF-kappaB pathway. Cell. Mol. Life Sci. CMLS 2020, 77, 2275–2288. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, D.; Smith, K.; Babraj, J.; Leese, G.; Waddell, T.; Atherton, P.; Wackerhage, H.; Taylor, P.M.; Rennie, M.J. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shai, M.; Carmeli, E.; Reznick, A.Z. The role of NF-kappaB in protein breakdown in immobilization, aging, and exercise: From basic processes to promotion of health. Ann. N. Y. Acad. Sci. 2005, 1057, 431–447. [Google Scholar] [CrossRef]
- Hunter, R.B.; Stevenson, E.; Koncarevic, A.; Mitchell-Felton, H.; Essig, D.A.; Kandarian, S.C. Activation of an alternative NF-kappaB pathway in skeletal muscle during disuse atrophy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 529–538. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.Y.; Baldwin, A.S., Jr. NF-kappaB-induced loss of MyoD messenger RNA: Possible role in muscle decay and cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef]
- Cai, D.; Frantz, J.D.; Tawa, N.E., Jr.; Melendez, P.A.; Oh, B.C.; Lidov, H.G.; Hasselgren, P.O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef]
- Mourkioti, F.; Kratsios, P.; Luedde, T.; Song, Y.H.; Delafontaine, P.; Adami, R.; Parente, V.; Bottinelli, R.; Pasparakis, M.; Rosenthal, N. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J. Clin. Investig. 2006, 116, 2945–2954. [Google Scholar] [CrossRef] [PubMed]
- Thoma, A.; Lightfoot, A.P. NF-kB and Inflammatory Cytokine Signalling: Role in Skeletal Muscle Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.B.; Kandarian, S.C. Disruption of either the Nfkb1 or the Bcl3 gene inhibits skeletal muscle atrophy. J. Clin. Investig. 2004, 114, 1504–1511. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.R.; Koncarevic, A.; Hunter, R.B.; Liou, H.C.; Jackman, R.W.; Kandarian, S.C. Role for IkappaBalpha, but not c-Rel, in skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 2007, 292, C372–C382. [Google Scholar] [CrossRef] [PubMed]
- Van Gammeren, D.; Damrauer, J.S.; Jackman, R.W.; Kandarian, S.C. The IkappaB kinases IKKalpha and IKKbeta are necessary and sufficient for skeletal muscle atrophy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 362–370. [Google Scholar] [CrossRef]
- Reed, S.A.; Senf, S.M.; Cornwell, E.W.; Kandarian, S.C.; Judge, A.R. Inhibition of IkappaB kinase alpha (IKKalpha) or IKKbeta (IKKbeta) plus forkhead box O (Foxo) abolishes skeletal muscle atrophy. Biochem. Biophys. Res. Commun. 2011, 405, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Belova, S.P.; Shenkman, B.S.; Kostrominova, T.Y.; Nemirovskaya, T.L. Paradoxical effect of IKKbeta inhibition on the expression of E3 ubiquitin ligases and unloading-induced skeletal muscle atrophy. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Langen, R.C.; Schols, A.M.; Kelders, M.C.; Wouters, E.F.; Janssen-Heininger, Y.M. Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor-kappaB. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 1169–1180. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Kumar, A. The TWEAK-Fn14 system: Breaking the silence of cytokine-induced skeletal muscle wasting. Curr. Mol. Med. 2012, 12, 3–13. [Google Scholar] [CrossRef]
- Brancaccio, M.; Pirozzi, F.; Hirsch, E.; Ghigo, A. Mechanisms underlying the cross-talk between heart and cancer. J. Physiol. 2020, 598, 3015–3027. [Google Scholar] [CrossRef]
- Mittal, A.; Bhatnagar, S.; Kumar, A.; Lach-Trifilieff, E.; Wauters, S.; Li, H.; Makonchuk, D.Y.; Glass, D.J.; Kumar, A. The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J. Cell Biol. 2010, 188, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Dogra, C.; Changotra, H.; Wedhas, N.; Qin, X.; Wergedal, J.E.; Kumar, A. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 1857–1869. [Google Scholar] [CrossRef] [PubMed]
- Elkina, Y.; von Haehling, S.; Anker, S.D.; Springer, J. The role of myostatin in muscle wasting: An overview. J. Cachexia Sarcopenia Muscle 2011, 2, 143–151. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.D.; Popovic, L.; Oldham, J.M.; Jeanplong, F.; Smith, H.K.; Kambadur, R.; Sharma, M.; Maxwell, L.; Bass, J.J. Myostatin-deficient mice lose more skeletal muscle mass than wild-type controls during hindlimb suspension. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E82–E87. [Google Scholar] [CrossRef] [PubMed]
- Joulia, D.; Bernardi, H.; Garandel, V.; Rabenoelina, F.; Vernus, B.; Cabello, G. Mechanisms involved in the inhibition of myoblast proliferation and differentiation by myostatin. Exp. Cell Res. 2003, 286, 263–275. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Fox, D.K.; Ebert, S.M.; Bongers, K.S.; Dyle, M.C.; Bullard, S.A.; Dierdorff, J.M.; Kunkel, S.D.; Adams, C.M. p53 and ATF4 mediate distinct and additive pathways to skeletal muscle atrophy during limb immobilization. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E245–E261. [Google Scholar] [CrossRef]
- Schmalbruch, H.; Lewis, D.M. Dynamics of nuclei of muscle fibers and connective tissue cells in normal and denervated rat muscles. Muscle Nerve 2000, 23, 617–626. [Google Scholar] [CrossRef]
- Pharaoh, G.; Brown, J.L.; Sataranatarajan, K.; Kneis, P.; Bian, J.; Ranjit, R.; Hadad, N.; Georgescu, C.; Rabinovitch, P.; Ran, Q.; et al. Targeting cPLA2 derived lipid hydroperoxides as a potential intervention for sarcopenia. Sci. Rep. 2020, 10, 13968. [Google Scholar] [CrossRef]
- Mensch, A.; Zierz, S. Cellular Stress in the Pathogenesis of Muscular Disorders-From Cause to Consequence. Int. J. Mol. Sci. 2020, 21, 5830. [Google Scholar] [CrossRef]
- Vitadello, M.; Doria, A.; Tarricone, E.; Ghirardello, A.; Gorza, L. Myofiber stress-response in myositis: Parallel investigations on patients and experimental animal models of muscle regeneration and systemic inflammation. Arthritis Res. Ther. 2010, 12, R52. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, S.M.; Dahlby, T.; Hede Andersen, C.; Haataja, L.; Petersen, S.; Omar-Hmeadi, M.; Yang, M.; Pihl, C.; Bresson, S.E.; Khilji, M.S.; et al. Endoplasmic Reticulum Chaperone Glucose-Regulated Protein 94 Is Essential for Proinsulin Handling. Diabetes 2019, 68, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Gorza, L.; Vitadello, M. Grp94 (HSP90B1). In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 2276–2287. [Google Scholar]
- Edwards, M.G.; Anderson, R.M.; Yuan, M.; Kendziorski, C.M.; Weindruch, R.; Prolla, T.A. Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC Genom. 2007, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Ehrnhoefer, D.E.; Skotte, N.H.; Ladha, S.; Nguyen, Y.T.; Qiu, X.; Deng, Y.; Huynh, K.T.; Engemann, S.; Nielsen, S.M.; Becanovic, K.; et al. p53 increases caspase-6 expression and activation in muscle tissue expressing mutant huntingtin. Hum. Mol. Genet. 2014, 23, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Siu, P.M.; Alway, S.E. Id2 and p53 participate in apoptosis during unloading-induced muscle atrophy. Am. J. Physiol. Cell Physiol. 2005, 288, C1058–C1073. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Siu, P.M.; Alway, S.E. Mitochondria-associated apoptotic signalling in denervated rat skeletal muscle. J. Physiol. 2005, 565, 309–323. [Google Scholar] [CrossRef]
- Stevenson, E.J.; Giresi, P.G.; Koncarevic, A.; Kandarian, S.C. Global analysis of gene expression patterns during disuse atrophy in rat skeletal muscle. J. Physiol. 2003, 551, 33–48. [Google Scholar] [CrossRef]
- Welle, S.; Brooks, A.I.; Delehanty, J.M.; Needler, N.; Bhatt, K.; Shah, B.; Thornton, C.A. Skeletal muscle gene expression profiles in 20-29 year old and 65-71 year old women. Exp. Gerontol. 2004, 39, 369–377. [Google Scholar] [CrossRef]
- Welle, S.; Brooks, A.I.; Delehanty, J.M.; Needler, N.; Thornton, C.A. Gene expression profile of aging in human muscle. Physiol. Genom. 2003, 14, 149–159. [Google Scholar] [CrossRef]
- Yang, Z.J.; Broz, D.K.; Noderer, W.L.; Ferreira, J.P.; Overton, K.W.; Spencer, S.L.; Meyer, T.; Tapscott, S.J.; Attardi, L.D.; Wang, C.L. p53 suppresses muscle differentiation at the myogenin step in response to genotoxic stress. Cell Death Differ. 2015, 22, 560–573. [Google Scholar] [CrossRef]
- Atherton, P.J.; Greenhaff, P.L.; Phillips, S.M.; Bodine, S.C.; Adams, C.M.; Lang, C.H. Control of skeletal muscle atrophy in response to disuse: Clinical/preclinical contentions and fallacies of evidence. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E594–E604. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Neuparth, M.J.; Vitorino, R.; Appell, H.J.; Amado, F.; Duarte, J.A. Evidences of apoptosis during the early phases of soleus muscle atrophy in hindlimb suspended mice. Physiol. Res. 2008, 57, 601–611. [Google Scholar] [PubMed]
- Schwarzkopf, M.; Coletti, D.; Sassoon, D.; Marazzi, G. Muscle cachexia is regulated by a p53-PW1/Peg3-dependent pathway. Genes Dev. 2006, 20, 3440–3452. [Google Scholar] [CrossRef] [PubMed]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar] [CrossRef]
- Rodriguez-Munoz, R.; Cardenas-Aguayo Mdel, C.; Aleman, V.; Osorio, B.; Chavez-Gonzalez, O.; Rendon, A.; Martinez-Rojas, D.; Meraz-Rios, M.A. Novel Nuclear Protein Complexes of Dystrophin 71 Isoforms in Rat Cultured Hippocampal GABAergic and Glutamatergic Neurons. PLoS ONE 2015, 10, e0137328. [Google Scholar] [CrossRef]
- Baldelli, S.; Ciriolo, M.R. Altered S-nitrosylation of p53 is responsible for impaired antioxidant response in skeletal muscle during aging. Aging 2016, 8, 3450–3467. [Google Scholar] [CrossRef]
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef]
- Wei, B.; Dui, W.; Liu, D.; Xing, Y.; Yuan, Z.; Ji, G. MST1, a key player, in enhancing fast skeletal muscle atrophy. BMC Biol. 2013, 11, 12. [Google Scholar] [CrossRef]
- Watt, K.I.; Turner, B.J.; Hagg, A.; Zhang, X.; Davey, J.R.; Qian, H.; Beyer, C.; Winbanks, C.E.; Harvey, K.F.; Gregorevic, P. The Hippo pathway effector YAP is a critical regulator of skeletal muscle fibre size. Nat. Commun. 2015, 6, 6048. [Google Scholar] [CrossRef]
- McKenna, C.F.; Fry, C.S. Altered satellite cell dynamics accompany skeletal muscle atrophy during chronic illness, disuse, and aging. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 447–452. [Google Scholar] [CrossRef]
- Yoshida, N.; Endo, J.; Kinouchi, K.; Kitakata, H.; Moriyama, H.; Kataoka, M.; Yamamoto, T.; Shirakawa, K.; Morimoto, S.; Nishiyama, A.; et al. (Pro)renin receptor accelerates development of sarcopenia via activation of Wnt/YAP signaling axis. Aging Cell 2019, 18, e12991. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.; Kabayo, T.; Ng, R.; Chamberlain, J.; McArdle, A.; Jackson, M.J. Skeletal muscle contractions induce acute changes in cytosolic superoxide, but slower responses in mitochondrial superoxide and cellular hydrogen peroxide. PLoS ONE 2014, 9, e96378. [Google Scholar] [CrossRef] [PubMed]
- Henriquez-Olguin, C.; Meneses-Valdes, R.; Jensen, T.E. Compartmentalized muscle redox signals controlling exercise metabolism - Current state, future challenges. Redox Biol. 2020, 35, 101473. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Morton, A.B.; Ahn, B.; Smuder, A.J. Redox control of skeletal muscle atrophy. Free Radic. Biol. Med. 2016, 98, 208–217. [Google Scholar] [CrossRef]
- Dalla Libera, L.; Ravara, B.; Gobbo, V.; Tarricone, E.; Vitadello, M.; Biolo, G.; Vescovo, G.; Gorza, L. A transient antioxidant stress response accompanies the onset of disuse atrophy in human skeletal muscle. J. Appl. Physiol. 2009, 107, 549–557. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, R.; Xu, L.; Wan, Q.; Zhu, J.; Gu, J.; Huang, Z.; Ma, W.; Shen, M.; Ding, F.; et al. Microarray Analysis of Gene Expression Provides New Insights Into Denervation-Induced Skeletal Muscle Atrophy. Front. Physiol. 2019, 10, 1298. [Google Scholar] [CrossRef]
- Powers, S.K.; Hudson, M.B.; Nelson, W.B.; Talbert, E.E.; Min, K.; Szeto, H.H.; Kavazis, A.N.; Smuder, A.J. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med. 2011, 39, 1749–1759. [Google Scholar] [CrossRef]
- Lawler, J.M.; Kunst, M.; Hord, J.M.; Lee, Y.; Joshi, K.; Botchlett, R.E.; Ramirez, A.; Martinez, D.A. EUK-134 ameliorates nNOSmu translocation and skeletal muscle fiber atrophy during short-term mechanical unloading. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R470–R482. [Google Scholar] [CrossRef]
- Fernando, R.; Castro, J.P.; Flore, T.; Deubel, S.; Grune, T.; Ott, C. Age-Related Maintenance of the Autophagy-Lysosomal System Is Dependent on Skeletal Muscle Type. Oxidative Med. Cell. Longev. 2020, 2020, 4908162. [Google Scholar] [CrossRef]
- Pearson, T.; McArdle, A.; Jackson, M.J. Nitric oxide availability is increased in contracting skeletal muscle from aged mice, but does not differentially decrease muscle superoxide. Free Radic. Biol. Med. 2015, 78, 82–88. [Google Scholar] [CrossRef][Green Version]
- Jackson, M.J. Recent advances and long-standing problems in detecting oxidative damage and reactive oxygen species in skeletal muscle. J. Physiol. 2016, 594, 5185–5193. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, H.; Deminice, R.; Yoshihara, T.; Powers, S.K. Mitochondrial dysfunction induces muscle atrophy during prolonged inactivity: A review of the causes and effects. Arch. Biochem. Biophys. 2019, 662, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Sandri, M. The connection between the dynamic remodeling of the mitochondrial network and the regulation of muscle mass. Cell. Mol. Life Sci. CMLS 2020. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Arc-Chagnaud, C.; Salvador-Pascual, A.; Brioche, T.; Chopard, A.; Olaso-Gonzalez, G.; Vina, J. Redox modulation of muscle mass and function. Redox Biol. 2020, 35, 101531. [Google Scholar] [CrossRef]
- Ji, L.L.; Yeo, D.; Kang, C. Muscle Disuse Atrophy Caused by Discord of Intracellular Signaling. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Mikhaeil, J.S.; Leveille, C.F.; Saint, C.; LeBlanc, P.J. Cardiolipin content, linoleic acid composition, and tafazzin expression in response to skeletal muscle overload and unload stimuli. Sci. Rep. 2017, 7, 2060. [Google Scholar] [CrossRef]
- Matecki, S.; Dridi, H.; Jung, B.; Saint, N.; Reiken, S.R.; Scheuermann, V.; Mrozek, S.; Santulli, G.; Umanskaya, A.; Petrof, B.J.; et al. Leaky ryanodine receptors contribute to diaphragmatic weakness during mechanical ventilation. Proc. Natl. Acad. Sci. USA 2016, 113, 9069–9074. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef]
- Eshima, H.; Siripoksup, P.; Mahmassani, Z.S.; Johnson, J.M.; Ferrara, P.J.; Verkerke, A.R.P.; Salcedo, A.; Drummond, M.J.; Funai, K. Neutralizing mitochondrial ROS does not rescue muscle atrophy induced by hindlimb unloading in female mice. J. Appl. Physiol. 2020, 129, 124–132. [Google Scholar] [CrossRef]
- Dodd, S.L.; Gagnon, B.J.; Senf, S.M.; Hain, B.A.; Judge, A.R. Ros-mediated activation of NF-kappaB and Foxo during muscle disuse. Muscle Nerve 2010, 41, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef] [PubMed]
- Pollock, N.; Staunton, C.A.; Vasilaki, A.; McArdle, A.; Jackson, M.J. Denervated muscle fibers induce mitochondrial peroxide generation in neighboring innervated fibers: Role in muscle aging. Free Radic. Biol. Med. 2017, 112, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Percival, J.M. nNOS regulation of skeletal muscle fatigue and exercise performance. Biophys. Rev. 2011, 3, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Uchida, H.; Kofuji, A.; Ito, J.; Shimizu, M.; Kim, H.; Sekiguchi, Y.; Kushibe, S. Molecular regulation of skeletal muscle mass and the contribution of nitric oxide: A review. FASEB Bioadv. 2019, 1, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Lomonosova, Y.N.; Kalamkarov, G.R.; Bugrova, A.E.; Shevchenko, T.F.; Kartashkina, N.L.; Lysenko, E.A.; Shvets, V.I.; Nemirovskaya, T.L. Protective effect of L-Arginine administration on proteins of unloaded m. soleus. Biochem. Biokhimiia 2011, 76, 571–580. [Google Scholar] [CrossRef]
- Lomonosova, Y.N.; Kalamkarov, G.R.; Bugrova, A.E.; Shevchenko, T.F.; Kartashkina, N.L.; Lysenko, E.A.; Shenkman, B.S.; Nemirovskaya, T.L. Role of NO-synthase in regulation of protein metabolism of stretched rat m. soleus muscle during functional unloading. Biochem. Biokhimiia 2012, 77, 208–216. [Google Scholar] [CrossRef]
- Sharlo, K.A.; Paramonova, I.I.; Lvova, I.D.; Vilchinskaya, N.A.; Bugrova, A.E.; Shevchenko, T.F.; Kalamkarov, G.R.; Shenkman, B.S. NO-Dependent Mechanisms of Myosin Heavy Chain Transcription Regulation in Rat Soleus Muscle After 7-Days Hindlimb Unloading. Front. Physiol. 2020, 11, 814. [Google Scholar] [CrossRef]
- Tews, D.S. Role of nitric oxide and nitric oxide synthases in experimental models of denervation and reinnervation. Microsc. Res. Tech. 2001, 55, 181–186. [Google Scholar] [CrossRef]
- Yamada, T.; Ashida, Y.; Tatebayashi, D.; Himori, K. Myofibrillar function differs markedly between denervated and dexamethasone-treated rat skeletal muscles: Role of mechanical load. PLoS ONE 2019, 14, e0223551. [Google Scholar] [CrossRef]
- Tidball, J.G.; Lavergne, E.; Lau, K.S.; Spencer, M.J.; Stull, J.T.; Wehling, M. Mechanical loading regulates NOS expression and activity in developing and adult skeletal muscle. Am. J. Physiol. 1998, 275, C260–C266. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.; Takeda, S.; Yokota, T. Nonmechanical Roles of Dystrophin and Associated Proteins in Exercise, Neuromuscular Junctions, and Brains. Brain Sci. 2015, 5, 275–298. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.E.; Zhu, A.; Mizuno, T.M. Nitric oxide treatment attenuates muscle atrophy during hind limb suspension in mice. Free Radic. Biol. Med. 2018, 115, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Mizunoya, W.; Upadhaya, R.; Burczynski, F.J.; Wang, G.; Anderson, J.E. Nitric oxide donors improve prednisone effects on muscular dystrophy in the mdx mouse diaphragm. Am. J. Physiol. Cell Physiol. 2011, 300, C1065–C1077. [Google Scholar] [CrossRef]
- Li, D.; Yue, Y.; Lai, Y.; Hakim, C.H.; Duan, D. Nitrosative stress elicited by nNOSmicro delocalization inhibits muscle force in dystrophin-null mice. J. Pathol. 2011, 223, 88–98. [Google Scholar] [CrossRef]
- Rebolledo, D.L.; Kim, M.J.; Whitehead, N.P.; Adams, M.E.; Froehner, S.C. Sarcolemmal targeting of nNOSmu improves contractile function of mdx muscle. Hum. Mol. Genet. 2016, 25, 158–166. [Google Scholar] [CrossRef]
- Wehling-Henricks, M.; Tidball, J.G. Neuronal nitric oxide synthase-rescue of dystrophin/utrophin double knockout mice does not require nNOS localization to the cell membrane. PLoS ONE 2011, 6, e25071. [Google Scholar] [CrossRef]
- Ansa-Addo, E.A.; Thaxton, J.; Hong, F.; Wu, B.X.; Zhang, Y.; Fugle, C.W.; Metelli, A.; Riesenberg, B.; Williams, K.; Gewirth, D.T.; et al. Clients and Oncogenic Roles of Molecular Chaperone gp96/grp94. Curr. Top. Med. Chem. 2016, 16, 2765–2778. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Rothe, F.; Langnaese, K.; Wolf, G. New aspects of the location of neuronal nitric oxide synthase in the skeletal muscle: A light and electron microscopic study. Nitric Oxide Biol. Chem. 2005, 13, 21–35. [Google Scholar] [CrossRef]
- Ervasti, J.M. Costameres: The Achilles’ heel of Herculean muscle. J. Biol. Chem. 2003, 278, 13591–13594. [Google Scholar] [CrossRef] [PubMed]
- Bloch, R.J.; Gonzalez-Serratos, H. Lateral force transmission across costameres in skeletal muscle. Exerc. Sport Sci. Rev. 2003, 31, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.A.; Gomez, C.G.; Novak, S.M.; Mi-Mi, L.; Gregorio, C.C. Overview of the Muscle Cytoskeleton. Compr. Physiol. 2017, 7, 891–944. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Mathes, S.; Vanmunster, M.; Bloch, W.; Suhr, F. Evidence for skeletal muscle fiber type-specific expressions of mechanosensors. Cell. Mol. Life Sci. CMLS 2019, 76, 2987–3004. [Google Scholar] [CrossRef]
- Fluck, M.; Li, R.; Valdivieso, P.; Linnehan, R.M.; Castells, J.; Tesch, P.; Gustafsson, T. Early changes in costameric and mitochondrial protein expression with unloading are muscle specific. Biomed. Res. Int. 2014, 2014, 519310. [Google Scholar] [CrossRef]
- Vitadello, M.; Sorge, M.; Percivalle, E.; Germinario, E.; Danieli-Betto, D.; Turco, E.; Tarone, G.; Brancaccio, M.; Gorza, L. Loss of melusin is a novel, neuronal NO synthase/FoxO3-independent master switch of unloading-induced muscle atrophy. J. Cachexia Sarcopenia Muscle 2020, 11, 802–819. [Google Scholar] [CrossRef]
- Eid Mutlak, Y.; Aweida, D.; Volodin, A.; Ayalon, B.; Dahan, N.; Parnis, A.; Cohen, S. A signaling hub of insulin receptor, dystrophin glycoprotein complex and plakoglobin regulates muscle size. Nat. Commun. 2020, 11, 1381. [Google Scholar] [CrossRef]
- Gawlik, K.I.; Durbeej, M. Skeletal muscle laminin and MDC1A: Pathogenesis and treatment strategies. Skelet. Muscle 2011, 1, 9. [Google Scholar] [CrossRef]
- Talts, J.F.; Andac, Z.; Gohring, W.; Brancaccio, A.; Timpl, R. Binding of the G domains of laminin alpha1 and alpha2 chains and perlecan to heparin, sulfatides, alpha-dystroglycan and several extracellular matrix proteins. EMBO J. 1999, 18, 863–870. [Google Scholar] [CrossRef]
- Hohenester, E.; Tisi, D.; Talts, J.F.; Timpl, R. The crystal structure of a laminin G-like module reveals the molecular basis of alpha-dystroglycan binding to laminins, perlecan, and agrin. Mol. Cell 1999, 4, 783–792. [Google Scholar] [CrossRef]
- Peng, H.B.; Ali, A.A.; Daggett, D.F.; Rauvala, H.; Hassell, J.R.; Smalheiser, N.R. The relationship between perlecan and dystroglycan and its implication in the formation of the neuromuscular junction. Cell Adhes. Commun. 1998, 5, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Ghahramani Seno, M.M.; Graham, I.R.; Athanasopoulos, T.; Trollet, C.; Pohlschmidt, M.; Crompton, M.R.; Dickson, G. RNAi-mediated knockdown of dystrophin expression in adult mice does not lead to overt muscular dystrophy pathology. Hum. Mol. Genet. 2008, 17, 2622–2632. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.C.; Marcotte, G.R.; Baehr, L.M.; West, D.W.D.; Marshall, A.G.; Ebert, S.M.; Davidyan, A.; Adams, C.M.; Bodine, S.C.; Baar, K. Alterations in the muscle force transfer apparatus in aged rats during unloading and reloading: Impact of microRNA-31. J. Physiol. 2018, 596, 2883–2900. [Google Scholar] [CrossRef]
- Acharyya, S.; Butchbach, M.E.; Sahenk, Z.; Wang, H.; Saji, M.; Carathers, M.; Ringel, M.D.; Skipworth, R.J.; Fearon, K.C.; Hollingsworth, M.A.; et al. Dystrophin glycoprotein complex dysfunction: A regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005, 8, 421–432. [Google Scholar] [CrossRef]
- Molocea, C.E.; Tsokanos, F.F.; Herzig, S. Exploiting common aspects of obesity and cancer cachexia for future therapeutic strategies. Curr. Opin. Pharmacol. 2020, 53, 101–116. [Google Scholar] [CrossRef]
- Molza, A.E.; Mangat, K.; Le Rumeur, E.; Hubert, J.F.; Menhart, N.; Delalande, O. Structural Basis of Neuronal Nitric-oxide Synthase Interaction with Dystrophin Repeats 16 and 17. J. Biol. Chem. 2015, 290, 29531–29541. [Google Scholar] [CrossRef]
- Harris, M.B.; Mitchell, B.M.; Sood, S.G.; Webb, R.C.; Venema, R.C. Increased nitric oxide synthase activity and Hsp90 association in skeletal muscle following chronic exercise. Eur. J. Appl. Physiol. 2008, 104, 795–802. [Google Scholar] [CrossRef]
- Seo, Y.; Lee, K.; Park, K.; Bae, K.; Choi, I. A proteomic assessment of muscle contractile alterations during unloading and reloading. J. Biochem. 2006, 139, 71–80. [Google Scholar] [CrossRef]
- Brancaccio, M.; Hirsch, E.; Notte, A.; Selvetella, G.; Lembo, G.; Tarone, G. Integrin signalling: The tug-of-war in heart hypertrophy. Cardiovasc. Res. 2006, 70, 422–433. [Google Scholar] [CrossRef]
- Belkin, A.M.; Zhidkova, N.I.; Balzac, F.; Altruda, F.; Tomatis, D.; Maier, A.; Tarone, G.; Koteliansky, V.E.; Burridge, K. Beta 1D integrin displaces the beta 1A isoform in striated muscles: Localization at junctional structures and signaling potential in nonmuscle cells. J. Cell Biol. 1996, 132, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Jaka, O.; Casas-Fraile, L.; Lopez de Munain, A.; Saenz, A. Costamere proteins and their involvement in myopathic processes. Expert Rev. Mol. Med. 2015, 17, e12. [Google Scholar] [CrossRef] [PubMed]
- Boppart, M.D.; Mahmassani, Z.S. Integrin signaling: Linking mechanical stimulation to skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 2019, 317, C629–C641. [Google Scholar] [CrossRef] [PubMed]
- Martino, F.; Perestrelo, A.R.; Vinarsky, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef] [PubMed]
- VanWinkle, W.B.; Snuggs, M.B.; De Hostos, E.L.; Buja, L.M.; Woods, A.; Couchman, J.R. Localization of the transmembrane proteoglycan syndecan-4 and its regulatory kinases in costameres of rat cardiomyocytes: A deconvolution microscopic study. Anat. Rec. 2002, 268, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Ugarte, G.; Santander, C.; Brandan, E. Syndecan-4 and beta1 integrin are regulated by electrical activity in skeletal muscle: Implications for cell adhesion. Matrix Biol. J. Int. Soc. Matrix Biol. 2010, 29, 383–392. [Google Scholar] [CrossRef]
- Saoncella, S.; Echtermeyer, F.; Denhez, F.; Nowlen, J.K.; Mosher, D.F.; Robinson, S.D.; Hynes, R.O.; Goetinck, P.F. Syndecan-4 signals cooperatively with integrins in a Rho-dependent manner in the assembly of focal adhesions and actin stress fibers. Proc. Natl. Acad. Sci. USA 1999, 96, 2805–2810. [Google Scholar] [CrossRef]
- Fluck, M.; Carson, J.A.; Gordon, S.E.; Ziemiecki, A.; Booth, F.W. Focal adhesion proteins FAK and paxillin increase in hypertrophied skeletal muscle. Am. J. Physiol. 1999, 277, C152–C162. [Google Scholar] [CrossRef]
- Gordon, S.E.; Fluck, M.; Booth, F.W. Selected Contribution: Skeletal muscle focal adhesion kinase, paxillin, and serum response factor are loading dependent. J. Appl. Physiol. 2001, 90, 1174–1183, discussion 1165. [Google Scholar] [CrossRef]
- Durieux, A.C.; D’Antona, G.; Desplanches, D.; Freyssenet, D.; Klossner, S.; Bottinelli, R.; Fluck, M. Focal adhesion kinase is a load-dependent governor of the slow contractile and oxidative muscle phenotype. J. Physiol. 2009, 587, 3703–3717. [Google Scholar] [CrossRef]
- de Boer, M.D.; Selby, A.; Atherton, P.; Smith, K.; Seynnes, O.R.; Maganaris, C.N.; Maffulli, N.; Movin, T.; Narici, M.V.; Rennie, M.J. The temporal responses of protein synthesis, gene expression and cell signalling in human quadriceps muscle and patellar tendon to disuse. J. Physiol. 2007, 585, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Glover, E.I.; Phillips, S.M.; Oates, B.R.; Tang, J.E.; Tarnopolsky, M.A.; Selby, A.; Smith, K.; Rennie, M.J. Immobilization induces anabolic resistance in human myofibrillar protein synthesis with low and high dose amino acid infusion. J. Physiol. 2008, 586, 6049–6061. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.B.; Phillips, S.M.; Atherton, P.J.; Patel, R.; Yarasheski, K.E.; Tarnopolsky, M.A.; Rennie, M.J. Differential effects of resistance and endurance exercise in the fed state on signalling molecule phosphorylation and protein synthesis in human muscle. J. Physiol. 2008, 586, 3701–3717. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Narici, M.V.; Erskine, R.M.; Seynnes, O.R.; Rittweger, J.; Pisot, R.; Simunic, B.; Fluck, M. Costamere remodeling with muscle loading and unloading in healthy young men. J. Anat. 2013, 223, 525–536. [Google Scholar] [CrossRef]
- Graham, Z.A.; Qin, W.; Harlow, L.C.; Ross, N.H.; Bauman, W.A.; Gallagher, P.M.; Cardozo, C.P. Focal adhesion kinase signaling is decreased 56 days following spinal cord injury in rat gastrocnemius. Spinal Cord 2016, 54, 502–509. [Google Scholar] [CrossRef][Green Version]
- Graham, Z.A.; Gallagher, P.M.; Cardozo, C.P. Focal adhesion kinase and its role in skeletal muscle. J. Muscle Res. Cell Motil. 2015, 36, 305–315. [Google Scholar] [CrossRef]
- Wang, H.V.; Chang, L.W.; Brixius, K.; Wickstrom, S.A.; Montanez, E.; Thievessen, I.; Schwander, M.; Muller, U.; Bloch, W.; Mayer, U.; et al. Integrin-linked kinase stabilizes myotendinous junctions and protects muscle from stress-induced damage. J. Cell Biol. 2008, 180, 1037–1049. [Google Scholar] [CrossRef]
- Legate, K.R.; Montanez, E.; Kudlacek, O.; Fassler, R. ILK, PINCH and parvin: The tIPP of integrin signalling. Nat. Rev. Mol. Cell Biol. 2006, 7, 20–31. [Google Scholar] [CrossRef]
- Gheyara, A.L.; Vallejo-Illarramendi, A.; Zang, K.; Mei, L.; St-Arnaud, R.; Dedhar, S.; Reichardt, L.F. Deletion of integrin-linked kinase from skeletal muscles of mice resembles muscular dystrophy due to alpha 7 beta 1-integrin deficiency. Am. J. Pathol. 2007, 171, 1966–1977. [Google Scholar] [CrossRef]
- Slimani, L.; Vazeille, E.; Deval, C.; Meunier, B.; Polge, C.; Dardevet, D.; Bechet, D.; Taillandier, D.; Micol, D.; Listrat, A.; et al. The delayed recovery of the remobilized rat tibialis anterior muscle reflects a defect in proliferative and terminal differentiation that impairs early regenerative processes. J. Cachexia Sarcopenia Muscle 2015, 6, 73–83. [Google Scholar] [CrossRef]
- Chen, H.; Huang, X.N.; Yan, W.; Chen, K.; Guo, L.; Tummalapali, L.; Dedhar, S.; St-Arnaud, R.; Wu, C.; Sepulveda, J.L. Role of the integrin-linked kinase/PINCH1/alpha-parvin complex in cardiac myocyte hypertrophy. Lab. Investig. A J. Tech. Methods Pathol. 2005, 85, 1342–1356. [Google Scholar] [CrossRef] [PubMed][Green Version]
- White, D.E.; Coutu, P.; Shi, Y.F.; Tardif, J.C.; Nattel, S.; St Arnaud, R.; Dedhar, S.; Muller, W.J. Targeted ablation of ILK from the murine heart results in dilated cardiomyopathy and spontaneous heart failure. Genes Dev. 2006, 20, 2355–2360. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Fedak, P.W.; Dai, X.; Du, C.; Zhou, Y.Q.; Henkelman, M.; Mongroo, P.S.; Lau, A.; Yamabi, H.; Hinek, A.; et al. Integrin-linked kinase expression is elevated in human cardiac hypertrophy and induces hypertrophy in transgenic mice. Circulation 2006, 114, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Mahmassani, Z.S.; Reidy, P.T.; McKenzie, A.I.; Stubben, C.; Howard, M.T.; Drummond, M.J. Age-dependent skeletal muscle transcriptome response to bed rest-induced atrophy. J. Appl. Physiol. 2019, 126, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Shirasu, K.; Lahaye, T.; Tan, M.W.; Zhou, F.; Azevedo, C.; Schulze-Lefert, P. A novel class of eukaryotic zinc-binding proteins is required for disease resistance signaling in barley and development in C. elegans. Cell 1999, 99, 355–366. [Google Scholar] [CrossRef]
- Brancaccio, M.; Guazzone, S.; Menini, N.; Sibona, E.; Hirsch, E.; De Andrea, M.; Rocchi, M.; Altruda, F.; Tarone, G.; Silengo, L. Melusin is a new muscle-specific interactor for beta(1) integrin cytoplasmic domain. J. Biol. Chem. 1999, 274, 29282–29288. [Google Scholar] [CrossRef]
- Sbroggio, M.; Ferretti, R.; Percivalle, E.; Gutkowska, M.; Zylicz, A.; Michowski, W.; Kuznicki, J.; Accornero, F.; Pacchioni, B.; Lanfranchi, G.; et al. The mammalian CHORD-containing protein melusin is a stress response protein interacting with Hsp90 and Sgt1. FEBS Lett. 2008, 582, 1788–1794. [Google Scholar] [CrossRef]
- Gano, J.J.; Simon, J.A. A proteomic investigation of ligand-dependent HSP90 complexes reveals CHORDC1 as a novel ADP-dependent HSP90-interacting protein. Mol. Cell. Proteom. MCP 2010, 9, 255–270. [Google Scholar] [CrossRef]
- Zhang, M.; Kadota, Y.; Prodromou, C.; Shirasu, K.; Pearl, L.H. Structural basis for assembly of Hsp90-Sgt1-CHORD protein complexes: Implications for chaperoning of NLR innate immunity receptors. Mol. Cell 2010, 39, 269–281. [Google Scholar] [CrossRef]
- Hong, T.J.; Kim, S.; Wi, A.R.; Lee, P.; Kang, M.; Jeong, J.H.; Hahn, J.S. Dynamic nucleotide-dependent interactions of cysteine- and histidine-rich domain (CHORD)-containing Hsp90 cochaperones Chp-1 and melusin with cochaperones PP5 and Sgt1. J. Biol. Chem. 2013, 288, 215–222. [Google Scholar] [CrossRef]
- Garcia-Ranea, J.A.; Mirey, G.; Camonis, J.; Valencia, A. p23 and HSP20/alpha-crystallin proteins define a conserved sequence domain present in other eukaryotic protein families. FEBS Lett. 2002, 529, 162–167. [Google Scholar] [CrossRef]
- Lee, Y.T.; Jacob, J.; Michowski, W.; Nowotny, M.; Kuznicki, J.; Chazin, W.J. Human Sgt1 binds HSP90 through the CHORD-Sgt1 domain and not the tetratricopeptide repeat domain. J. Biol. Chem. 2004, 279, 16511–16517. [Google Scholar] [CrossRef] [PubMed]
- Sbroggio, M.; Bertero, A.; Velasco, S.; Fusella, F.; De Blasio, E.; Bahou, W.F.; Silengo, L.; Turco, E.; Brancaccio, M.; Tarone, G. ERK1/2 activation in heart is controlled by melusin, focal adhesion kinase and the scaffold protein IQGAP1. J. Cell Sci. 2011, 124, 3515–3524. [Google Scholar] [CrossRef] [PubMed]
- Tarone, G.; Sbroggio, M.; Brancaccio, M. Key role of ERK1/2 molecular scaffolds in heart pathology. Cell. Mol. Life Sci. CMLS 2013, 70, 4047–4054. [Google Scholar] [CrossRef] [PubMed]
- Waardenberg, A.J.; Bernardo, B.C.; Ng, D.C.; Shepherd, P.R.; Cemerlang, N.; Sbroggio, M.; Wells, C.A.; Dalrymple, B.P.; Brancaccio, M.; Lin, R.C.; et al. Phosphoinositide 3-kinase (PI3K(p110alpha)) directly regulates key components of the Z-disc and cardiac structure. J. Biol. Chem. 2011, 286, 30837–30846. [Google Scholar] [CrossRef]
- Tarone, G.; Lembo, G. Molecular interplay between mechanical and humoral signalling in cardiac hypertrophy. Trends Mol. Med. 2003, 9, 376–382. [Google Scholar] [CrossRef]
- De Acetis, M.; Notte, A.; Accornero, F.; Selvetella, G.; Brancaccio, M.; Vecchione, C.; Sbroggio, M.; Collino, F.; Pacchioni, B.; Lanfranchi, G.; et al. Cardiac overexpression of melusin protects from dilated cardiomyopathy due to long-standing pressure overload. Circ. Res. 2005, 96, 1087–1094. [Google Scholar] [CrossRef]
- Donker, D.W.; Maessen, J.G.; Verheyen, F.; Ramaekers, F.C.; Spatjens, R.L.; Kuijpers, H.; Ramakers, C.; Schiffers, P.M.; Vos, M.A.; Crijns, H.J.; et al. Impact of acute and enduring volume overload on mechanotransduction and cytoskeletal integrity of canine left ventricular myocardium. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2324–H2332. [Google Scholar] [CrossRef]
- Aro, J.; Tokola, H.; Ronkainen, V.P.; Koivisto, E.; Tenhunen, O.; Ilves, M.; Szokodi, I.; Ruskoaho, H.; Rysa, J. Regulation of cardiac melusin gene expression by hypertrophic stimuli in the rat. Acta Physiol. 2013, 207, 470–484. [Google Scholar] [CrossRef]
- Brancaccio, M.; Menini, N.; Bongioanni, D.; Ferretti, R.; De Acetis, M.; Silengo, L.; Tarone, G. Chp-1 and melusin, two CHORD containing proteins in vertebrates. FEBS Lett. 2003, 551, 47–52. [Google Scholar] [CrossRef]
- Unsold, B.; Kaul, A.; Sbroggio, M.; Schubert, C.; Regitz-Zagrosek, V.; Brancaccio, M.; Damilano, F.; Hirsch, E.; Van Bilsen, M.; Munts, C.; et al. Melusin protects from cardiac rupture and improves functional remodelling after myocardial infarction. Cardiovasc. Res. 2014, 101, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Zheng, D.; Bai, J.; Xie, J.; Dai, Q.; Xu, B. Altered melusin pathways involved in cardiac remodeling following acute myocardial infarction. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2012, 21, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Brancaccio, M.; Tullio, F.; Rubinetto, C.; Perrelli, M.G.; Angotti, C.; Pagliaro, P.; Tarone, G. Overexpression of the muscle-specific protein, melusin, protects from cardiac ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 418. [Google Scholar] [CrossRef] [PubMed]
- Brokat, S.; Thomas, J.; Herda, L.R.; Knosalla, C.; Pregla, R.; Brancaccio, M.; Accornero, F.; Tarone, G.; Hetzer, R.; Regitz-Zagrosek, V. Altered melusin expression in the hearts of aortic stenosis patients. Eur. J. Heart Fail. 2007, 9, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Jaka, O.; Casas-Fraile, L.; Azpitarte, M.; Aiastui, A.; Lopez de Munain, A.; Saenz, A. FRZB and melusin, overexpressed in LGMD2A, regulate integrin beta1D isoform replacement altering myoblast fusion and the integrin-signalling pathway. Expert Rev. Mol. Med. 2017, 19, e2. [Google Scholar] [CrossRef] [PubMed]
- Vafiadaki, E.; Arvanitis, D.A.; Sanoudou, D. Muscle LIM Protein: Master regulator of cardiac and skeletal muscle functions. Gene 2015, 566, 1–7. [Google Scholar] [CrossRef]
- Arber, S.; Hunter, J.J.; Ross, J., Jr.; Hongo, M.; Sansig, G.; Borg, J.; Perriard, J.C.; Chien, K.R.; Caroni, P. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 1997, 88, 393–403. [Google Scholar] [CrossRef]
- Knoll, R.; Hoshijima, M.; Hoffman, H.M.; Person, V.; Lorenzen-Schmidt, I.; Bang, M.L.; Hayashi, T.; Shiga, N.; Yasukawa, H.; Schaper, W.; et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 2002, 111, 943–955. [Google Scholar] [CrossRef]
- Knoll, R.; Kostin, S.; Klede, S.; Savvatis, K.; Klinge, L.; Stehle, I.; Gunkel, S.; Kotter, S.; Babicz, K.; Sohns, M.; et al. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ. Res. 2010, 106, 695–704. [Google Scholar] [CrossRef]
- Barash, I.A.; Mathew, L.; Lahey, M.; Greaser, M.L.; Lieber, R.L. Muscle LIM protein plays both structural and functional roles in skeletal muscle. Am. J. Physiol. Cell Physiol. 2005, 289, C1312–C1320. [Google Scholar] [CrossRef]
- Von der Hagen, M.; Laval, S.H.; Cree, L.M.; Haldane, F.; Pocock, M.; Wappler, I.; Peters, H.; Reitsamer, H.A.; Hoger, H.; Wiedner, M.; et al. The differential gene expression profiles of proximal and distal muscle groups are altered in pre-pathological dysferlin-deficient mice. Neuromuscul. Disord. NMD 2005, 15, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Winokur, S.T.; Chen, Y.W.; Masny, P.S.; Martin, J.H.; Ehmsen, J.T.; Tapscott, S.J.; van der Maarel, S.M.; Hayashi, Y.; Flanigan, K.M. Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum. Mol. Genet. 2003, 12, 2895–2907. [Google Scholar] [CrossRef] [PubMed]
- Sanoudou, D.; Corbett, M.A.; Han, M.; Ghoddusi, M.; Nguyen, M.A.; Vlahovich, N.; Hardeman, E.C.; Beggs, A.H. Skeletal muscle repair in a mouse model of nemaline myopathy. Hum. Mol. Genet. 2006, 15, 2603–2612. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Han, S.; Tang, S.; He, H.; Shen, X.; Zhao, J.; Chen, Y.; Wei, Y.; Wang, Y.; Zhu, Q.; et al. The Autophagy Regulatory Molecule CSRP3 Interacts with LC3 and Protects Against Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 749. [Google Scholar] [CrossRef] [PubMed]
- Louis, H.A.; Pino, J.D.; Schmeichel, K.L.; Pomies, P.; Beckerle, M.C. Comparison of three members of the cysteine-rich protein family reveals functional conservation and divergent patterns of gene expression. J. Biol. Chem. 1997, 272, 27484–27491. [Google Scholar] [CrossRef] [PubMed]
- Flick, M.J.; Konieczny, S.F. The muscle regulatory and structural protein MLP is a cytoskeletal binding partner of betaI-spectrin. J. Cell Sci. 2000, 113 Pt 9, 1553–1564. [Google Scholar]
- Postel, R.; Vakeel, P.; Topczewski, J.; Knoll, R.; Bakkers, J. Zebrafish integrin-linked kinase is required in skeletal muscles for strengthening the integrin-ECM adhesion complex. Dev. Biol. 2008, 318, 92–101. [Google Scholar] [CrossRef]
- Moretti, I.; Ciciliot, S.; Dyar, K.A.; Abraham, R.; Murgia, M.; Agatea, L.; Akimoto, T.; Bicciato, S.; Forcato, M.; Pierre, P.; et al. MRF4 negatively regulates adult skeletal muscle growth by repressing MEF2 activity. Nat. Commun. 2016, 7, 12397. [Google Scholar] [CrossRef]
- Tu, Y.; Li, F.; Wu, C. Nck-2, a novel Src homology2/3-containing adaptor protein that interacts with the LIM-only protein PINCH and components of growth factor receptor kinase-signaling pathways. Mol. Biol. Cell 1998, 9, 3367–3382. [Google Scholar] [CrossRef]
- Price, S.R.; Bailey, J.L.; Wang, X.; Jurkovitz, C.; England, B.K.; Ding, X.; Phillips, L.S.; Mitch, W.E. Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J. Clin. Investig. 1996, 98, 1703–1708. [Google Scholar] [CrossRef]
- Wang, X.; Hu, Z.; Hu, J.; Du, J.; Mitch, W.E. Insulin resistance accelerates muscle protein degradation: Activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 2006, 147, 4160–4168. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.M.; Kim, J.K.; Yakar, S.; Dupont, J.; Hernandez-Sanchez, C.; Castle, A.L.; Filmore, J.; Shulman, G.I.; Le Roith, D. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev. 2001, 15, 1926–1934. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Lee, D.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Trim32 reduces PI3K-Akt-FoxO signaling in muscle atrophy by promoting plakoglobin-PI3K dissociation. J. Cell Biol. 2014, 204, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Hinchee-Rodriguez, K.; Garg, N.; Venkatakrishnan, P.; Roman, M.G.; Adamo, M.L.; Masters, B.S.; Roman, L.J. Neuronal nitric oxide synthase is phosphorylated in response to insulin stimulation in skeletal muscle. Biochem. Biophys. Res. Commun. 2013, 435, 501–505. [Google Scholar] [CrossRef]
- Ayalon, G.; Hostettler, J.D.; Hoffman, J.; Kizhatil, K.; Davis, J.Q.; Bennett, V. Ankyrin-B interactions with spectrin and dynactin-4 are required for dystrophin-based protection of skeletal muscle from exercise injury. J. Biol. Chem. 2011, 286, 7370–7378. [Google Scholar] [CrossRef]
- Pratt, R.D.; Brickman, C.R.; Cottrill, C.L.; Shapiro, J.I.; Liu, J. The Na/K-ATPase Signaling: From Specific Ligands to General Reactive Oxygen Species. Int. J. Mol. Sci. 2018, 19, 2600. [Google Scholar] [CrossRef]
- Radzyukevich, T.L.; Neumann, J.C.; Rindler, T.N.; Oshiro, N.; Goldhamer, D.J.; Lingrel, J.B.; Heiny, J.A. Tissue-specific role of the Na,K-ATPase alpha2 isozyme in skeletal muscle. J. Biol. Chem. 2013, 288, 1226–1237. [Google Scholar] [CrossRef]
- Altarawneh, M.M.; Petersen, A.C.; Farr, T.; Garnham, A.; Broatch, J.R.; Halson, S.; Bishop, D.J.; McKenna, M.J. Resistance training upregulates skeletal muscle Na(+), K(+)-ATPase content, with elevations in both alpha1 and alpha2, but not beta isoforms. Eur. J. Appl. Physiol. 2020, 120, 1777–1785. [Google Scholar] [CrossRef]
- Petrov, A.M.; Shalagina, M.N.; Protopopov, V.A.; Sergeev, V.G.; Ovechkin, S.V.; Ovchinina, N.G.; Sekunov, A.V.; Zefirov, A.L.; Zakirjanova, G.F.; Bryndina, I.G. Changes in Membrane Ceramide Pools in Rat Soleus Muscle in Response to Short-Term Disuse. Int. J. Mol. Sci. 2019, 20, 4860. [Google Scholar] [CrossRef]
- Kravtsova, V.V.; Vilchinskaya, N.A.; Rozlomii, V.L.; Shenkman, B.S.; Krivoi, I.I. Low Ouabain Doses and AMP-Activated Protein Kinase as Factors Supporting Electrogenesis in Skeletal Muscle. Biochem. Biokhimiia 2019, 84, 1085–1092. [Google Scholar] [CrossRef]
- Desaphy, J.F.; Pierno, S.; Leoty, C.; George, A.L., Jr.; De Luca, A.; Camerino, D.C. Skeletal muscle disuse induces fibre type-dependent enhancement of Na(+) channel expression. Brain A J. Neurol. 2001, 124, 1100–1113. [Google Scholar] [CrossRef] [PubMed]
- Cisterna, B.A.; Cardozo, C.; Saez, J.C. Neuronal involvement in muscular atrophy. Front. Cell. Neurosci. 2014, 8, 405. [Google Scholar] [CrossRef] [PubMed]
- Nourse, J.L.; Pathak, M.M. How cells channel their stress: Interplay between Piezo1 and the cytoskeleton. Semin. Cell Dev. Biol. 2017, 71, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Stiber, J.A.; Seth, M.; Rosenberg, P.B. Mechanosensitive channels in striated muscle and the cardiovascular system: Not quite a stretch anymore. J. Cardiovasc. Pharmacol. 2009, 54, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.D.; Coon, B.G.; Yun, S.; Baeyens, N.; Tanaka, K.; Ouyang, M.; Schwartz, M.A. Integrins in mechanotransduction. Curr. Opin. Cell Biol. 2013, 25, 613–618. [Google Scholar] [CrossRef]
- Spangenburg, E.E.; McBride, T.A. Inhibition of stretch-activated channels during eccentric muscle contraction attenuates p70S6K activation. J. Appl. Physiol. 2006, 100, 129–135. [Google Scholar] [CrossRef]
- Tyganov, S.; Mirzoev, T.; Shenkman, B. An Anabolic Signaling Response of Rat Soleus Muscle to Eccentric Contractions Following Hindlimb Unloading: A Potential Role of Stretch-Activated Ion Channels. Int. J. Mol. Sci. 2019, 20, 1165. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Yeung, E.W. Mechanisms of stretch-induced muscle damage in normal and dystrophic muscle: Role of ionic changes. J. Physiol. 2005, 567, 723–735. [Google Scholar] [CrossRef]
- Vandebrouck, C.; Martin, D.; Colson-Van Schoor, M.; Debaix, H.; Gailly, P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [CrossRef]
- Vandebrouck, A.; Sabourin, J.; Rivet, J.; Balghi, H.; Sebille, S.; Kitzis, A.; Raymond, G.; Cognard, C.; Bourmeyster, N.; Constantin, B. Regulation of capacitative calcium entries by alpha1-syntrophin: Association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 608–617. [Google Scholar] [CrossRef]
- Gervasio, O.L.; Whitehead, N.P.; Yeung, E.W.; Phillips, W.D.; Allen, D.G. TRPC1 binds to caveolin-3 and is regulated by Src kinase - role in Duchenne muscular dystrophy. J. Cell Sci. 2008, 121, 2246–2255. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Jeong, S.Y.; Oh, M.R.; Allen, P.D.; Lee, E.H. TRPCs: Influential Mediators in Skeletal Muscle. Cells 2020, 9, 850. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Cheung, K.K.; Yeung, S.S.; Yeung, E.W. The involvement of transient receptor potential canonical type 1 in skeletal muscle regrowth after unloading-induced atrophy. J. Physiol. 2016, 594, 3111–3126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.T.; Yeung, S.S.; Cheung, K.K.; Chai, Z.Y.; Yeung, E.W. Adaptive responses of TRPC1 and TRPC3 during skeletal muscle atrophy and regrowth. Muscle Nerve 2014, 49, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, C. Not all disuse protocols are equal: New insight into the signalling pathways to muscle atrophy. J. Physiol. 2015, 593, 5227–5228. [Google Scholar] [CrossRef] [PubMed]
- Ryder, D.J.; Judge, S.M.; Beharry, A.W.; Farnsworth, C.L.; Silva, J.C.; Judge, A.R. Identification of the Acetylation and Ubiquitin-Modified Proteome during the Progression of Skeletal Muscle Atrophy. PLoS ONE 2015, 10, e0136247. [Google Scholar] [CrossRef]
- Nakao, R.; Hirasaka, K.; Goto, J.; Ishidoh, K.; Yamada, C.; Ohno, A.; Okumura, Y.; Nonaka, I.; Yasutomo, K.; Baldwin, K.M.; et al. Ubiquitin ligase Cbl-b is a negative regulator for insulin-like growth factor 1 signaling during muscle atrophy caused by unloading. Mol. Cell. Biol. 2009, 29, 4798–4811. [Google Scholar] [CrossRef]
- Ogneva, I.V.; Biryukov, N.S.; Leinsoo, T.A.; Larina, I.M. Possible role of non-muscle alpha-actinins in muscle cell mechanosensitivity. PLoS ONE 2014, 9, e96395. [Google Scholar] [CrossRef]
- Ogneva, I.V.; Biryukov, N.S. Lecithin Prevents Cortical Cytoskeleton Reorganization in Rat Soleus Muscle Fibers under Short-Term Gravitational Disuse. PLoS ONE 2016, 11, e0153650. [Google Scholar] [CrossRef]
- Petrov, A.M.; Kravtsova, V.V.; Matchkov, V.V.; Vasiliev, A.N.; Zefirov, A.L.; Chibalin, A.V.; Heiny, J.A.; Krivoi, I.I. Membrane lipid rafts are disturbed in the response of rat skeletal muscle to short-term disuse. Am. J. Physiol. Cell Physiol. 2017, 312, C627–C637. [Google Scholar] [CrossRef]
- Uchida, T.; Sakashita, Y.; Kitahata, K.; Yamashita, Y.; Tomida, C.; Kimori, Y.; Komatsu, A.; Hirasaka, K.; Ohno, A.; Nakao, R.; et al. Reactive oxygen species upregulate expression of muscle atrophy-associated ubiquitin ligase Cbl-b in rat L6 skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2018, 314, C721–C731. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Shimizu, M.; Mizunoya, W.; Wariishi, H.; Tatsumi, R.; Buchman, V.L.; Ikeuchi, Y. Differential expression of sarcoplasmic and myofibrillar proteins of rat soleus muscle during denervation atrophy. Biosci. Biotechnol. Biochem. 2009, 73, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Midrio, M.; Danieli-Betto, D.; Megighian, A.; Betto, R. Early effects of denervation on sarcoplasmic reticulum properties of slow-twitch rat muscle fibres. Pflug. Arch. Eur. J. Physiol. 1997, 434, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Castets, P.; Rion, N.; Theodore, M.; Falcetta, D.; Lin, S.; Reischl, M.; Wild, F.; Guerard, L.; Eickhorst, C.; Brockhoff, M.; et al. mTORC1 and PKB/Akt control the muscle response to denervation by regulating autophagy and HDAC4. Nat. Commun. 2019, 10, 3187. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Thompson, L.V. Skeletal muscle denervation investigations: Selecting an experimental control wisely. Am. J. Physiol. Cell Physiol. 2019, 316, C456–C461. [Google Scholar] [CrossRef]
- Sacheck, J.M.; Hyatt, J.P.; Raffaello, A.; Jagoe, R.T.; Roy, R.R.; Edgerton, V.R.; Lecker, S.H.; Goldberg, A.L. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 140–155. [Google Scholar] [CrossRef]
- Furlow, J.D.; Watson, M.L.; Waddell, D.S.; Neff, E.S.; Baehr, L.M.; Ross, A.P.; Bodine, S.C. Altered gene expression patterns in muscle ring finger 1 null mice during denervation- and dexamethasone-induced muscle atrophy. Physiol. Genom. 2013, 45, 1168–1185. [Google Scholar] [CrossRef]
- Moresi, V.; Williams, A.H.; Meadows, E.; Flynn, J.M.; Potthoff, M.J.; McAnally, J.; Shelton, J.M.; Backs, J.; Klein, W.H.; Richardson, J.A.; et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 2010, 143, 35–45. [Google Scholar] [CrossRef]
- Luo, L.; Martin, S.C.; Parkington, J.; Cadena, S.M.; Zhu, J.; Ibebunjo, C.; Summermatter, S.; Londraville, N.; Patora-Komisarska, K.; Widler, L.; et al. HDAC4 Controls Muscle Homeostasis through Deacetylation of Myosin Heavy Chain, PGC-1alpha, and Hsc70. Cell Rep. 2019, 29, 749–763. [Google Scholar] [CrossRef]
- Londhe, P.; Guttridge, D.C. Inflammation induced loss of skeletal muscle. Bone 2015, 80, 131–142. [Google Scholar] [CrossRef]
- Hsu, C.G.; Talukder, M.A.H.; Yue, L.; Turpin, L.C.; Noble, M.; Elfar, J.C. Human equivalent dose of oral 4-aminopyridine differentiates nerve crush injury from transection injury and improves post-injury function in mice. Neural Regen. Res. 2020, 15, 2098–2107. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, Q.; Huang, Z.; Zhu, J.; Qiu, J.; Ma, W.; Yang, X.; Ding, F.; Sun, H. Isoquercitrin Delays Denervated Soleus Muscle Atrophy by Inhibiting Oxidative Stress and Inflammation. Front. Physiol. 2020, 11, 988. [Google Scholar] [CrossRef]
- Scalabrin, M.; Pollock, N.; Staunton, C.A.; Brooks, S.V.; McArdle, A.; Jackson, M.J.; Vasilaki, A. Redox responses in skeletal muscle following denervation. Redox Biol. 2019, 26, 101294. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, L.; Michelucci, A.; Ambrogini, P.; Sartini, S.; Guarnier, F.A.; Fusella, A.; Zamparo, I.; Mammucari, C.; Protasi, F.; Boncompagni, S. Muscle activity prevents the uncoupling of mitochondria from Ca(2+) Release Units induced by ageing and disuse. Arch. Biochem. Biophys. 2019, 663, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sugiura, Y.; Chen, F.; Lee, K.F.; Ye, Q.; Lin, W. Blocking skeletal muscle DHPRs/Ryr1 prevents neuromuscular synapse loss in mutant mice deficient in type III Neuregulin 1 (CRD-Nrg1). PLoS Genet. 2019, 15, e1007857. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E.; Krebs, J. Why Calcium? How Calcium Became the Best Communicator. J. Biol. Chem. 2016, 291, 20849–20857. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Hamilton, R.; Jernigan, A.; Zhang, Y.; Sabia, M.; Rahman, M.M.; Li, Y.; Wei, R.; Chaudhuri, A.; Van Remmen, H. Genetic ablation of 12/15-lipoxygenase but not 5-lipoxygenase protects against denervation-induced muscle atrophy. Free Radic. Biol. Med. 2014, 67, 30–40. [Google Scholar] [CrossRef]
- Aweida, D.; Rudesky, I.; Volodin, A.; Shimko, E.; Cohen, S. GSK3-beta promotes calpain-1-mediated desmin filament depolymerization and myofibril loss in atrophy. J. Cell Biol. 2018, 217, 3698–3714. [Google Scholar] [CrossRef]
- Lin, Y.; Brady, M.J.; Wolanske, K.; Holbert, R.; Ruderman, N.B.; Yaney, G.C. Alterations of nPKC distribution, but normal Akt/PKB activation in denervated rat soleus muscle. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E318–E325. [Google Scholar] [CrossRef][Green Version]
- Nikawa, T.; Ishidoh, K.; Hirasaka, K.; Ishihara, I.; Ikemoto, M.; Kano, M.; Kominami, E.; Nonaka, I.; Ogawa, T.; Adams, G.R.; et al. Skeletal muscle gene expression in space-flown rats. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 522–524. [Google Scholar] [CrossRef]
- Gosztonyi, G.; Naschold, U.; Grozdanovic, Z.; Stoltenburg-Didinger, G.; Gossrau, R. Expression of Leu-19 (CD56, N-CAM) and nitric oxide synthase (NOS) I in denervated and reinnervated human skeletal muscle. Microsc. Res. Tech. 2001, 55, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Nakada, S.; Yamashita, Y.; Machida, S.; Miyagoe-Suzuki, Y.; Arikawa-Hirasawa, E. Perlecan Facilitates Neuronal Nitric Oxide Synthase Delocalization in Denervation-Induced Muscle Atrophy. Cells 2020, 9, 2524. [Google Scholar] [CrossRef] [PubMed]
- Turinsky, J.; Damrau-Abney, A. Akt1 kinase and dynamics of insulin resistance in denervated muscles in vivo. Am. J. Physiol. 1998, 275, R1425–R1430. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Vallega, G.; Kandror, K.V.; Pilch, P.F. Insulin-mediated translocation of GLUT-4-containing vesicles is preserved in denervated muscles. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E1019–E1026. [Google Scholar] [CrossRef] [PubMed]
- Willmann, R.; Pun, S.; Stallmach, L.; Sadasivam, G.; Santos, A.F.; Caroni, P.; Fuhrer, C. Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J. 2006, 25, 4050–4060. [Google Scholar] [CrossRef]
- Biswas, A.K.; Acharyya, S. Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer 2020, 20, 274–284. [Google Scholar] [CrossRef]
- Shukla, S.K.; Markov, S.D.; Attri, K.S.; Vernucci, E.; King, R.J.; Dasgupta, A.; Grandgenett, P.M.; Hollingsworth, M.A.; Singh, P.K.; Yu, F.; et al. Macrophages potentiate STAT3 signaling in skeletal muscles and regulate pancreatic cancer cachexia. Cancer Lett. 2020, 484, 29–39. [Google Scholar] [CrossRef]
- Quan-Jun, Y.; Yan, H.; Yong-Long, H.; Li-Li, W.; Jie, L.; Jin-Lu, H.; Jin, L.; Peng-Guo, C.; Run, G.; Cheng, G. Selumetinib Attenuates Skeletal Muscle Wasting in Murine Cachexia Model through ERK Inhibition and AKT Activation. Mol. Cancer Ther. 2017, 16, 334–343. [Google Scholar] [CrossRef]
- Li, Y.P.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M.B. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 362–370. [Google Scholar] [CrossRef]
- Mulder, S.E.; Dasgupta, A.; King, R.J.; Abrego, J.; Attri, K.S.; Murthy, D.; Shukla, S.K.; Singh, P.K. JNK signaling contributes to skeletal muscle wasting and protein turnover in pancreatic cancer cachexia. Cancer Lett. 2020, 491, 70–77. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Benny Klimek, M.E.; Aydogdu, T.; Link, M.J.; Pons, M.; Koniaris, L.G.; Zimmers, T.A. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem. Biophys. Res. Commun. 2010, 391, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Busquets, S.; Toledo, M.; Orpi, M.; Massa, D.; Porta, M.; Capdevila, E.; Padilla, N.; Frailis, V.; Lopez-Soriano, F.J.; Han, H.Q.; et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J. Cachexia Sarcopenia Muscle 2012, 3, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Winbanks, C.E.; Murphy, K.T.; Bernardo, B.C.; Qian, H.; Liu, Y.; Sepulveda, P.V.; Beyer, C.; Hagg, A.; Thomson, R.E.; Chen, J.L.; et al. Smad7 gene delivery prevents muscle wasting associated with cancer cachexia in mice. Sci. Transl. Med. 2016, 8, 348ra398. [Google Scholar] [CrossRef] [PubMed]
- Secli, L.; Sorge, M.; Morotti, A.; Brancaccio, M. Blocking Extracellular Chaperones to Improve Cardiac Regeneration. Front. Bioeng. Biotechnol. 2020, 8, 411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, Z.; Ding, H.; Zhou, Y.; Doan, H.A.; Sin, K.W.T.; Zhu, Z.J.; Flores, R.; Wen, Y.; Gong, X.; et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat. Commun. 2017, 8, 589. [Google Scholar] [CrossRef]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Sacheck, J.M.; Ohtsuka, A.; McLary, S.C.; Goldberg, A.L. IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E591–E601. [Google Scholar] [CrossRef]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef]
- Costelli, P.; Muscaritoli, M.; Bossola, M.; Penna, F.; Reffo, P.; Bonetto, A.; Busquets, S.; Bonelli, G.; Lopez-Soriano, F.J.; Doglietto, G.B.; et al. IGF-1 is downregulated in experimental cancer cachexia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R674–R683. [Google Scholar] [CrossRef]
- White, J.P.; Baynes, J.W.; Welle, S.L.; Kostek, M.C.; Matesic, L.E.; Sato, S.; Carson, J.A. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS ONE 2011, 6, e24650. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Penna, F.; Aversa, Z.; Mercantini, P.; Baccino, F.M.; Costelli, P.; Ziparo, V.; Lucia, S.; Rossi Fanelli, F.; Muscaritoli, M. Early changes of muscle insulin-like growth factor-1 and myostatin gene expression in gastric cancer patients. Muscle Nerve 2013, 48, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; von Haehling, S.; Doehner, W.; Palus, S.; Anker, S.D.; Springer, J. IGF-1 treatment reduces weight loss and improves outcome in a rat model of cancer cachexia. J. Cachexia Sarcopenia Muscle 2011, 2, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Lundholm, K.; Korner, U.; Gunnebo, L.; Sixt-Ammilon, P.; Fouladiun, M.; Daneryd, P.; Bosaeus, I. Insulin treatment in cancer cachexia: Effects on survival, metabolism, and physical functioning. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 2699–2706. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.G.; Gomes-Marcondes, M.C. Metformin treatment modulates the tumour-induced wasting effects in muscle protein metabolism minimising the cachexia in tumour-bearing rats. BMC Cancer 2016, 16, 418. [Google Scholar] [CrossRef] [PubMed]
- Honors, M.A.; Kinzig, K.P. The role of insulin resistance in the development of muscle wasting during cancer cachexia. J. Cachexia Sarcopenia Muscle 2012, 3, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Szentesi, P.; Csernoch, L.; Dux, L.; Keller-Pinter, A. Changes in Redox Signaling in the Skeletal Muscle with Aging. Oxidative Med. Cell. Longev. 2019, 2019, 4617801. [Google Scholar] [CrossRef]
- Blasco, A.; Gras, S.; Modol-Caballero, G.; Tarabal, O.; Casanovas, A.; Piedrafita, L.; Barranco, A.; Das, T.; Pereira, S.L.; Navarro, X.; et al. Motoneuron deafferentation and gliosis occur in association with neuromuscular regressive changes during ageing in mice. J. Cachexia Sarcopenia Muscle 2020. [Google Scholar] [CrossRef]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Carter, H.N.; Chen, C.C.; Hood, D.A. Mitochondria, muscle health, and exercise with advancing age. Physiology 2015, 30, 208–223. [Google Scholar] [CrossRef]
- Beltran Valls, M.R.; Wilkinson, D.J.; Narici, M.V.; Smith, K.; Phillips, B.E.; Caporossi, D.; Atherton, P.J. Protein carbonylation and heat shock proteins in human skeletal muscle: Relationships to age and sarcopenia. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Capanni, C.; Squarzoni, S.; Petrini, S.; Villanova, M.; Muscari, C.; Maraldi, N.M.; Guarnieri, C.; Caldarera, C.M. Increase of neuronal nitric oxide synthase in rat skeletal muscle during ageing. Biochem. Biophys. Res. Commun. 1998, 245, 216–219. [Google Scholar] [CrossRef]
- Leiter, J.R.; Upadhaya, R.; Anderson, J.E. Nitric oxide and voluntary exercise together promote quadriceps hypertrophy and increase vascular density in female 18-mo-old mice. Am. J. Physiol. Cell Physiol. 2012, 302, C1306–C1315. [Google Scholar] [CrossRef] [PubMed]
- Hord, J.M.; Botchlett, R.; Lawler, J.M. Age-related alterations in the sarcolemmal environment are attenuated by lifelong caloric restriction and voluntary exercise. Exp. Gerontol. 2016, 83, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Richmonds, C.R.; Boonyapisit, K.; Kusner, L.L.; Kaminski, H.J. Nitric oxide synthase in aging rat skeletal muscle. Mech. Ageing Dev. 1999, 109, 177–189. [Google Scholar] [CrossRef]
- Samengo, G.; Avik, A.; Fedor, B.; Whittaker, D.; Myung, K.H.; Wehling-Henricks, M.; Tidball, J.G. Age-related loss of nitric oxide synthase in skeletal muscle causes reductions in calpain S-nitrosylation that increase myofibril degradation and sarcopenia. Aging Cell 2012, 11, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.T.; Koopman, R.; Naim, T.; Leger, B.; Trieu, J.; Ibebunjo, C.; Lynch, G.S. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 4433–4442. [Google Scholar] [CrossRef]
- Hughes, D.C.; Marcotte, G.R.; Marshall, A.G.; West, D.W.D.; Baehr, L.M.; Wallace, M.A.; Saleh, P.M.; Bodine, S.C.; Baar, K. Age-related Differences in Dystrophin: Impact on Force Transfer Proteins, Membrane Integrity, and Neuromuscular Junction Stability. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 640–648. [Google Scholar] [CrossRef]
- Ramaswamy, K.S.; Palmer, M.L.; van der Meulen, J.H.; Renoux, A.; Kostrominova, T.Y.; Michele, D.E.; Faulkner, J.A. Lateral transmission of force is impaired in skeletal muscles of dystrophic mice and very old rats. J. Physiol. 2011, 589, 1195–1208. [Google Scholar] [CrossRef]
- Gannon, J.; Staunton, L.; O’Connell, K.; Doran, P.; Ohlendieck, K. Phosphoproteomic analysis of aged skeletal muscle. Int. J. Mol. Med. 2008, 22, 33–42. [Google Scholar] [CrossRef]
- Russ, D.W.; Grandy, J.S. Increased desmin expression in hindlimb muscles of aging rats. J. Cachexia Sarcopenia Muscle 2011, 2, 175–180. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meyer, G.A.; Kiss, B.; Ward, S.R.; Morgan, D.L.; Kellermayer, M.S.; Lieber, R.L. Theoretical predictions of the effects of force transmission by desmin on intersarcomere dynamics. Biophys. J. 2010, 98, 258–266. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ansved, T.; Edstrom, L. Effects of age on fibre structure, ultrastructure and expression of desmin and spectrin in fast- and slow-twitch rat muscles. J. Anat. 1991, 174, 61–79. [Google Scholar] [PubMed]
- Marcucci, L.; Reggiani, C. Increase of resting muscle stiffness, a less considered component of age-related skeletal muscle impairment. Eur. J. Transl. Myol. 2020, 30, 8982. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
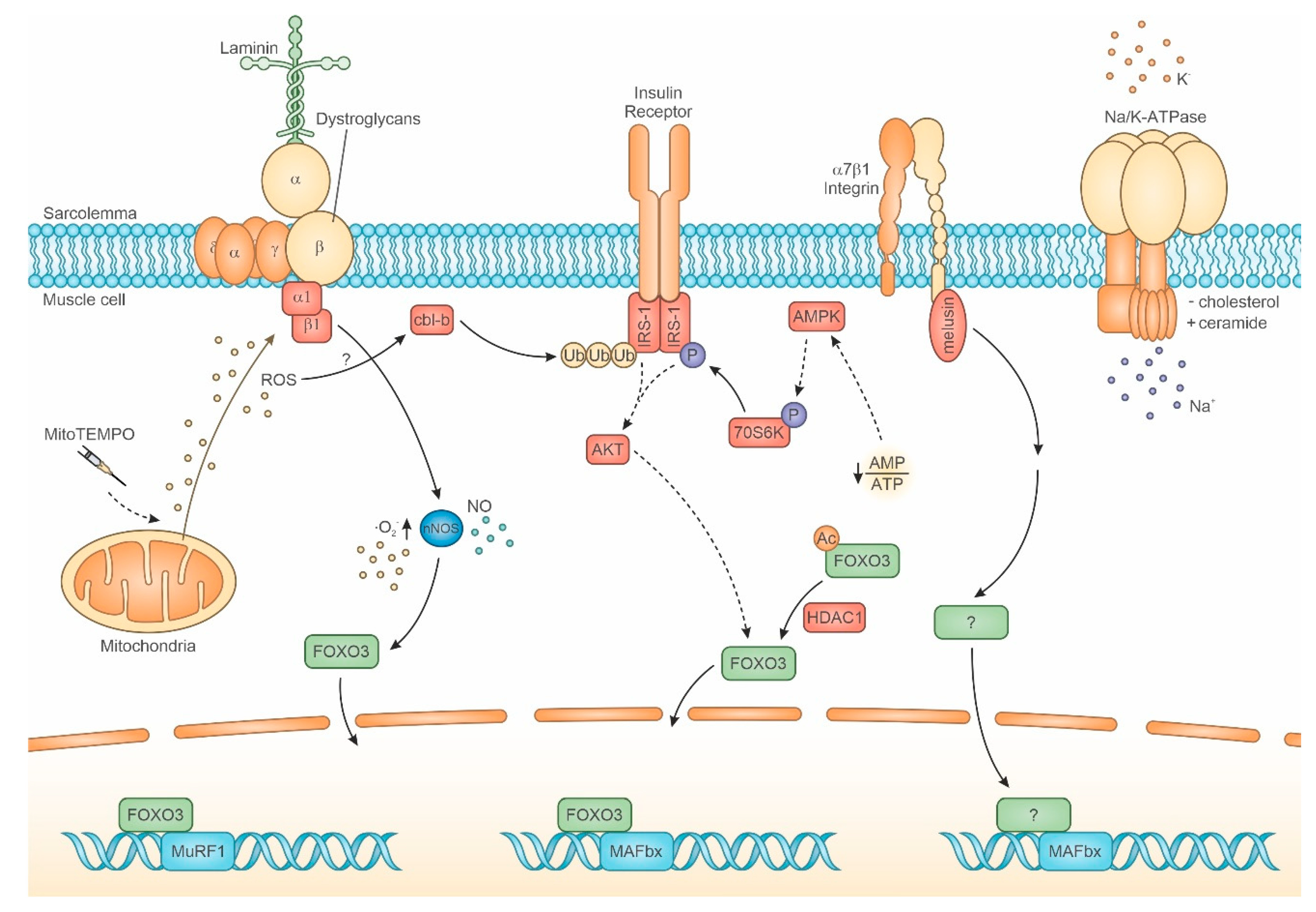{kind=link}
| Time | Localization | Event | Consequences | Reference |
|---|---|---|---|---|
| 6 h | costamere | loss of melusin | increased FoxO3-independent transcription of MAFbx-Atrogin-1 | [128] |
| redistribution of non-muscle α-actinin and β- and γ- actin | subsarcolemmal cytoskeleton reorganization | [229,230] | ||
| loss of active sarcolemmal nNOS | FoxO3 nuclear translocation | [30] | ||
| decreased sarcolemmal NO production | AMPK inactivity | [20] | ||
| sarcoplasm | increased accumulation of active/uncoupled nNOS | increased NO/ROS production | [30] | |
| mitochondria | ROS production | loss of active sarcolemmal nNOS | [30] | |
| ROS production | covalent S-S tropomyosin species | [30] | ||
| sarcolemma | loss of cholesterol | membrane instability loss of Na/K pump activity | [210,231] | |
| inhibition of 2subunit of Na/K pump | membrane instability | [231] | ||
| nucleus | increased FoxO3 localization | transcriptional activity | [30] | |
| decreased nNOS transcription | decreased nNOS protein | [30] | ||
| 12 h | sarcoplasm | AMPK inactivity | p70S6K activation loss of Na/K pump activity | [20,211] |
| ceramide accumulation | sarcolemma instability | [210] | ||
| increased phospo-p70S6K | pSer-IRS-1 | [20,31] | ||
| 24 h | sarcolemma/costamere | decreased levels of IRS-1 | impaired Akt activation | [31] |
| decreased integrin signaling activation | impaired Akt, ERK1/2 activation | [31,128] | ||
| nucleus | increased FoxO3 transcription | increased protein levels and atrogene upregulation | [31,68,128,227] | |
| myosin gene down-regulation | fiber type transition | [2] | ||
| multiple transcriptional changes | phenotype change | [68] | ||
| sarcoplasm/nucleus | increased p53 protein levels | MuRF-1 and p21 upregulation | [57] | |
| 48 h | SR/ER | decreased Grp94/gp96 levels | decreased nNOS delivery to sarcolemma | [30] |
| sarcoplasm | increased protein ubiquitination and deacetylation by HDAC1 | increased protein catabolism, FoxO3 activation | [32,227] | |
| increased Bax/Bcl2 ratio; mitochondrial release of AIF; caspase -3 and caspase-8 activation | increased myonuclei apoptosis | [73] | ||
| reduced myofiber cross-sectional area | earliest morphological evidence of muscle atrophy | [30] |
| Time | Localization | Event | Consequences | Reference |
|---|---|---|---|---|
| 0.5–6 h | nucleus | increased gene transcription for: calcium-release channels and calcium-binding proteins oxidative stress YAP | increased sarcoplasmic calcium binding; increased oxidative stress; increased YAP | [59,80,87] |
| decreased gene transcription for: focal adhesion and extracellular receptors | reduced mechanotransduction | [87] | ||
| 3 h | sarcolemma | neuromuscolar junction disruption | AChR clustering | [256] |
| 6 h | sarcoplasm | increased protein levels of Hippo kinase MST1 and YAP | reduced YAP signaling | [22] |
| nucleus | YAP localization | increased YAP signaling | [22] | |
| 24 h | sarcolemma/costamere | inhibition of IR signaling without Akt inhibition | decreased glucose uptake | [254] |
| sarcoplasm | increased Monoamine oxidase A | oxidative stress | [87,244] | |
| DAG-sensitive nPKC binding to intracellular membranes | impaired insulin-stimulated glycogen synthesis | [250] | ||
| nucleus | PCG- and - genes down-regulation | reduced mitochondriogenesis | [237] | |
| up-regulation of pro-inflammatory genes | activation of NF- B pathway | [59,87,213] | ||
| 48 h | SR | decreased Ca2+ uptake | reduced levels of stored Ca2+ | [234] |
| reduced levels of stored Ca2+ | ER stress response | [21] | ||
| 48–72 h | sarcoplasm | increased protein ubiquitination and deacetylation by HDAC4 | increased protein catabolism, FoxO3 activation | [32,227,240] |
| increased muscle lipo-hydroperoxides by phospholipase A2 | oxidative stress via NADPH-oxidase | [59,248] | ||
| reduced muscle mass | earliest evidence of muscle atrophy | [233,235,237] | ||
| nucleus | increased ATF4 expression | ER stress response | [59] | |
| 72 h | costamere | active GSK3- and Ca2+-calmodulin kinase | desmin phosphorylation/ubiquitination | [249] |
| reduced syndecan 4 | reduced integrin signalling | [147] | ||
| mitochondria | misplacement at A band | reduced connection with triads and Ca2+ uptake | [245] | |
| increased ROS production and expression of mitochondrial anti-oxidant enzymes | oxidative stress | [103,104,244] | ||
| nucleus | FoxO3, HDAC4 upregulation | atrogene up-regulation | [87,235,237,238,239] | |
| GLUT-4 down-regulation | reduced glucose import | [255] | ||
| sarcoplasm | increased peroxiredoxin 6 | increased phospholipase A2 activity | [244] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorza, L.; Sorge, M.; Seclì, L.; Brancaccio, M. Master Regulators of Muscle Atrophy: Role of Costamere Components. Cells 2021, 10, 61. https://doi.org/10.3390/cells10010061
Gorza L, Sorge M, Seclì L, Brancaccio M. Master Regulators of Muscle Atrophy: Role of Costamere Components. Cells. 2021; 10(1):61. https://doi.org/10.3390/cells10010061
Chicago/Turabian StyleGorza, Luisa, Matteo Sorge, Laura Seclì, and Mara Brancaccio. 2021. "Master Regulators of Muscle Atrophy: Role of Costamere Components" Cells 10, no. 1: 61. https://doi.org/10.3390/cells10010061
APA StyleGorza, L., Sorge, M., Seclì, L., & Brancaccio, M. (2021). Master Regulators of Muscle Atrophy: Role of Costamere Components. Cells, 10(1), 61. https://doi.org/10.3390/cells10010061