Context Matters: NOTCH Signatures and Pathway in Cancer Progression and Metastasis

 ,
,  , ,
, ,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Notch Signaling: Principles and Involvement in Physiological and Pathological Processes
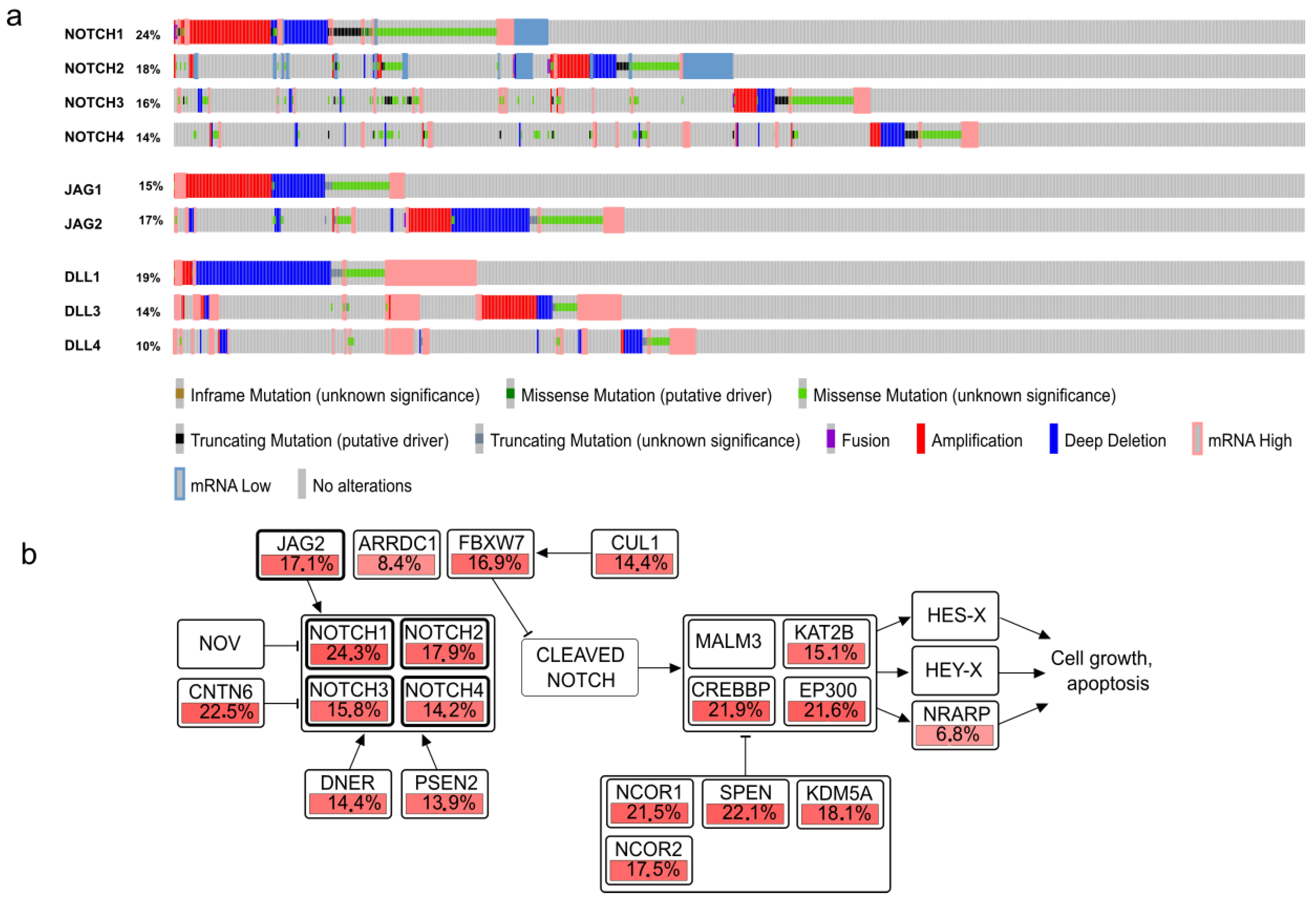
3. The Relevance of NOTCH Mutations, Amplification, Pathways, and Signatures
3.1. Gain of Function NOTCH Mutations and Their Consequences
3.2. Loss-of-Function NOTCH Mutations in Cancers and Its Consequences
3.3. Ambivalence of NOTCH Signaling in Cancer Initiation and Progression
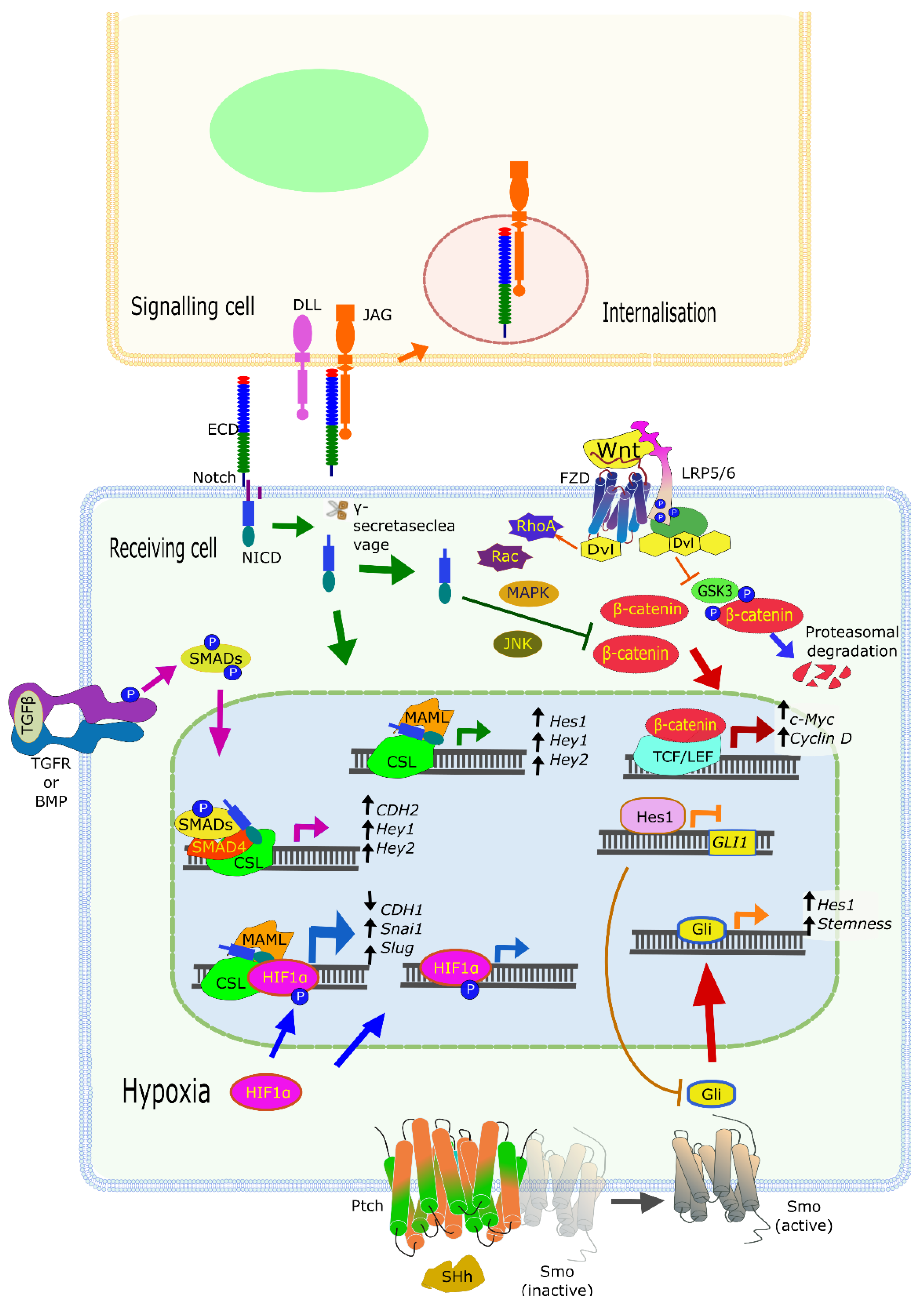
3.4. Context Truly Matters with Notch: CROSSTALK with Other Signaling Pathways
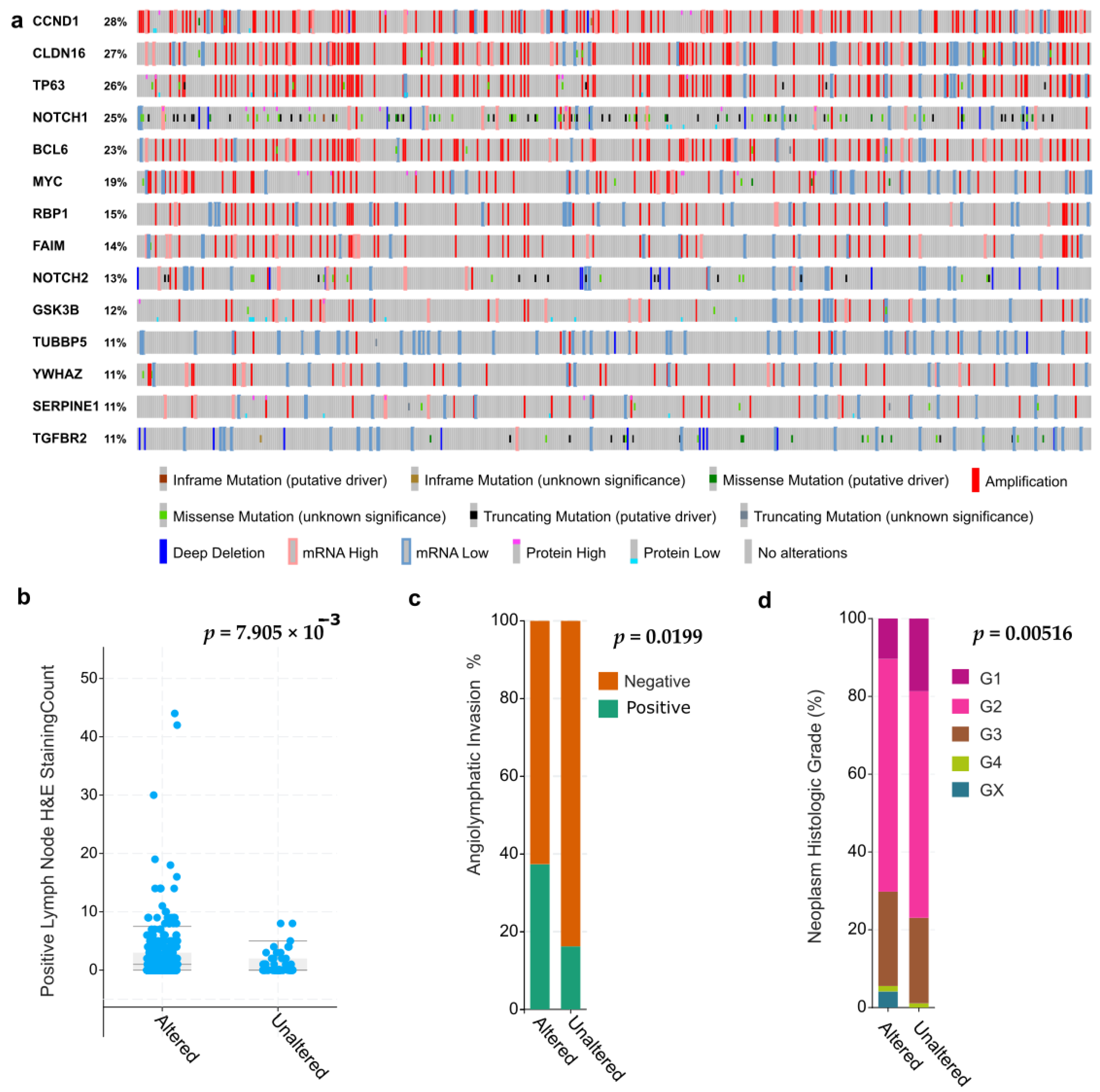
3.5. A “Holistic” Approach: NOTCH-Related Signatures, Gene Sets, and Pathways
3.6. Functional Validation of NOTCH Signaling and Personalized Cancer Medicine
4. NOTCH and EMT Signatures
5. Notch in Cell Migration/Invasion
6. Notch Signaling and Metastasis
7. Notch Signaling in (Neo-)Angiogenesis
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kopan, R.; Ilagan, M.X.G. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Rowe, R.G.; Lim, K.C.; Lin, Y.; Willis, A.; Tang, Y.; Li, X.Y.; Nor, J.E.; Maillard, I.; Weiss, S.J. A Snail1/Notch1 signalling axis controls embryonic vascular development. Nat. Commun. 2014, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Phng, L.K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef] [PubMed]
- High, F.A.; Min, M.L.; Pear, W.S.; Loomes, K.M.; Kaestner, K.H.; Epstein, J.A. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. USA 2008, 105, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Boareto, M.; Jolly, M.K.; Ben-Jacob, E.; Onuchic, J.N. Jagged mediates differences in normal and tumor angiogenesis by affecting tip-stalk fate decision. Proc. Natl. Acad. Sci. USA 2015, 112, E3836–E3844. [Google Scholar] [CrossRef] [PubMed]
- Van Engeland, N.C.A.; Suarez Rodriguez, F.; Rivero-Müller, A.; Ristori, T.; Duran, C.L.; Stassen, O.M.J.A.; Antfolk, D.; Driessen, R.C.H.; Ruohonen, S.; Ruohonen, S.T.; et al. Vimentin regulates Notch signaling strength and arterial remodeling in response to hemodynamic stress. Sci. Rep. 2019, 9, 12415. [Google Scholar] [CrossRef]
- Loerakker, S.; Stassen, O.M.J.A.; ter Huurne, F.M.; Boareto, M.; Bouten, C.V.C.; Sahlgren, C.M. Mechanosensitivity of Jagged–Notch signaling can induce a switch-type behavior in vascular homeostasis. Proc. Natl. Acad. Sci. USA 2018, 115, E3682–E3691. [Google Scholar] [CrossRef]
- Duarte, A.; Hirashima, M.; Benedito, R.; Trindade, A.; Diniz, P.; Bekman, E.; Costa, L.; Henrique, D.; Rossant, J. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004, 18, 2474–2478. [Google Scholar] [CrossRef]
- Politi, K.; Feirt, N.; Kitajewski, J. Notch in mammary gland development and breast cancer. Semin. Cancer Biol. 2004, 14, 341–347. [Google Scholar] [CrossRef]
- Stanger, B.Z.; Datar, R.; Murtaugh, L.C.; Melton, D.A. Direct regulation of intestinal fate by Notch. Proc. Natl. Acad. Sci. USA 2005, 102, 12443–12448. [Google Scholar] [CrossRef]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, R605–R615. [Google Scholar] [CrossRef] [PubMed]
- Fortini, M.E. Notch Signaling: The Core Pathway and Its Posttranslational Regulation. Dev. Cell 2009, 16, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Graziani, I.; Eliasz, S.; De Marco, M.A.; Chen, Y.; Pass, H.I.; De May, R.M.; Strack, P.R.; Miele, L.; Bocchetta, M. Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer Res. 2008, 68, 9678–9685. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Lemieux, M.E.; Thomas, T.; Rogers, J.M.; Lipper, C.H.; Lee, W.; Johnson, C.; Sholl, L.M.; South, A.P.; Marto, J.A.; et al. IER5, a dna damage response gene, is required for notch-mediated induction of squamous cell differentiation. Elife 2020, 9, e58081. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Weinmaster, G.; Roberts, V.J. A homolog of Drosophila Notch expressed during mammalian development. Development 1991, 113, 199–205. [Google Scholar]
- Milner, L.A.; Kopan, R.; Martin, D.I.K.; Bernstein, I.D. A human homologue of the Drosophila developmental gene, Notch, is expressed in CD34+ hematopoietic precursors. Blood 1994, 83, 2057–2062. [Google Scholar] [CrossRef]
- Larsson, C.; Lardelli, M.; White, I.; Lendahl, U. The human NOTCH1, 2, and 3 genes are located at chromosome positions 9q34, 1p13-p11, and 19p13.2-p13.1 in regions of neoplasia-associated translocation. Genomics 1994, 24, 253–258. [Google Scholar] [CrossRef]
- Thélu, J.; Rossio, P.; Favier, B. Notch signalling is linked to epidermal cell differentiation level in basal cell carcinoma, psoriasis and wound healing. BMC Dermatol. 2002, 2, 7. [Google Scholar] [CrossRef]
- Sriuranpong, V.; Borges, M.W.; Ravi, R.K.; Arnold, D.R.; Nelkin, B.D.; Baylin, S.B.; Ball, D.W. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001, 61, 3200–3205. [Google Scholar] [CrossRef]
- Jundt, F.; Anagnostopoulos, I.; Förster, R.; Mathas, S.; Stein, H.; Dörken, B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood 2002, 99, 3398–3403. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris IV, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.M.; Zhao, W.L.; Fu, J.F.; Shi, J.Y.; Pan, Q.; Hu, J.; Gao, X.D.; Chen, B.; Li, J.M.; Xiong, S.M.; et al. NOTCH1 mutations in T-cell acute lymphoblastic leukemia: Prognostic significance and implication in multifactorial leukemogenesis. Clin. Cancer Res. 2006, 12, 3043–3049. [Google Scholar] [CrossRef]
- Trabucco, S.E.; Gowen, K.; Maund, S.L.; Sanford, E.; Fabrizio, D.A.; Hall, M.J.; Yakirevich, E.; Gregg, J.P.; Stephens, P.J.; Frampton, G.M.; et al. A Novel Next-Generation Sequencing Approach to Detecting Microsatellite Instability and Pan-Tumor Characterization of 1000 Microsatellite Instability-High Cases in 67,000 Patient Samples. J. Mol. Diagn. 2019, 21, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Vettore, A.L.; Ramnarayanan, K.; Poore, G.; Lim, K.; Ong, C.K.; Huang, K.K.; Leong, H.S.; Chong, F.T.; Lim, T.K.-H.; Lim, W.K.; et al. Mutational landscapes of tongue carcinoma reveal recurrent mutations in genes of therapeutic and prognostic relevance. Genome Med. 2015, 7, 98–112. [Google Scholar] [CrossRef]
- Rampias, T.; Vgenopoulou, P.; Avgeris, M.; Polyzos, A.; Stravodimos, K.; Valavanis, C.; Scorilas, A.; Klinakis, A. A new tumor suppressor role for the Notch pathway in bladder cancer. Nat. Med. 2014, 20, 1199–1205. [Google Scholar] [CrossRef]
- Chen, C.; Wang, X.; Huang, S.; Wang, L.; Han, L.; Yu, S. Prognostic roles of Notch receptor mRNA expression in human ovarian cancer. Oncotarget 2017, 8, 32731–32740. [Google Scholar] [CrossRef][Green Version]
- Vujovic, F.; Hunter, N.; Farahani, R.M. Notch pathway: A bistable inducer of biological noise? Cell Commun. Signal. 2019, 17, 133. [Google Scholar] [CrossRef]
- Parmigiani, E.; Taylor, V.; Giachino, C. Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma. Cells 2020, 9, 2304. [Google Scholar] [CrossRef]
- Tao, J.; Jiang, M.-M.; Jiang, L.; Salvo, J.S.; Zeng, H.-C.; Dawson, B.; Bertin, T.K.; Rao, P.H.; Chen, R.; Donehower, L.A.; et al. Notch activation as a driver of osteogenic sarcoma. Cancer Cell 2014, 26, 390–401. [Google Scholar] [CrossRef]
- Augert, A.; Eastwood, E.; Ibrahim, A.H.; Wu, N.; Grunblatt, E.; Basom, R.; Liggitt, D.; Eaton, K.D.; Martins, R.; Poirier, J.T.; et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci. Signal. 2019, 12, eaau2922. [Google Scholar] [CrossRef] [PubMed]
- Chanrion, M.; Kuperstein, I.; Barrière, C.; El Marjou, F.; Cohen, D.; Vignjevic, D.; Stimmer, L.; Paul-Gilloteaux, P.; Bièche, I.; Tavares, S.D.R.; et al. Concomitant Notch activation and p53 deletion trigger epithelial-to-mesenchymal transition and metastasis in mouse gut. Nat. Commun. 2014, 5, 5005. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhang, L.; He, C.; Xu, F.; Liu, J.; Hu, Z.; Zhao, L.; Tian, Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell. Biochem. 2012, 113, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Gaykalova, D.A.; Ochs, M.F.; Mambo, E.; Arnaoutakis, D.; Liu, Y.; Loyo, M.; Agrawal, N.; Howard, J.; Li, R.; et al. Activation of the NOTCH pathway in head and neck cancer. Cancer Res. 2014, 74, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.; Bindea, G.; Albu, R.I.; Mlecnik, B.; Machiels, J.-P. Cetuximab promotes epithelial to mesenchymal transition and cancer associated fibroblasts in patients with head and neck cancer. Oncotarget 2015, 6, 34288–34299. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, P.; Nair, S.; Kaur, E.; Aich, J.; Dani, P.; Sethunath, V.; Gardi, N.; Chandrani, P.; Godbole, M.; Sonawane, K.; et al. Notch pathway activation is essential for maintenance of stem-like cells in early tongue cancer. Oncotarget 2016, 7, 50437–50449. [Google Scholar] [CrossRef]
- Das, T.; Zhong, R.; Spiotto, M.T. Notch Signaling and Human Papillomavirus-Associated Oral Tumorigenesis. Adv. Exp. Med. Biol. 2021, 1287, 105–122. [Google Scholar] [CrossRef]
- Gelsomino, L.; Panza, S.; Giordano, C.; Barone, I.; Gu, G.; Spina, E.; Catalano, S.; Fuqua, S.; Andò, S. Mutations in the estrogen receptor alpha hormone binding domain promote stem cell phenotype through notch activation in breast cancer cell lines. Cancer Lett. 2018, 428, 12–20. [Google Scholar] [CrossRef]
- Miao, K.; Lei, J.H.; Valecha, M.V.; Zhang, A.; Xu, J.; Wang, L.; Lyu, X.; Chen, S.; Miao, Z.; Zhang, X.; et al. NOTCH1 activation compensates BRCA1 deficiency and promotes triple-negative breast cancer formation. Nat. Commun. 2020, 11, 3265. [Google Scholar] [CrossRef]
- Yang, J.; Hou, Y.; Zhou, M.; Wen, S.; Zhou, J.; Xu, L.; Tang, X.; Du, Y.-E.; Hu, P.; Liu, M. Twist induces epithelial-mesenchymal transition and cell motility in breast cancer via ITGB1-FAK/ILK signaling axis and its associated downstream network. Int. J. Biochem. Cell Biol. 2016, 71, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.M.S.; Forghanifard, M.M.; Kordi-Tamandani, D.M.; Abbaszadegan, M.R. MAML1 regulates EMT markers expression through NOTCH-independent pathway in breast cancer cell line MCF7. Biochem. Biophys. Res. Commun. 2019, 510, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol. Cancer 2015, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.-K.; Kaushik, N.; Suh, Y.; Yoo, K.-C.; Cui, Y.-H.; Kim, M.-J.; Lee, H.-J.; Kim, I.-G.; Lee, S.-J. Radiation driven epithelial-mesenchymal transition is mediated by Notch signaling in breast cancer. Oncotarget 2016, 7, 53430–53442. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, Y.; Faronato, M.; Filipovic, A.; Vircillo, V.; Magnani, L.; Coombes, R.C. Nicastrin and Notch4 drive endocrine therapy resistance and epithelial to mesenchymal transition in MCF7 breast cancer cells. Breast Cancer Res. 2014, 16, R62. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Lee, H.-W.; Baek, J.-H.; Cho, Y.-H.; Kang, H.G.; Jeong, J.S.; Song, J.; Park, H.-S.; Chun, K.-H. Activation of nuclear PTEN by inhibition of Notch signaling induces G2/M cell cycle arrest in gastric cancer. Oncogene 2016, 35, 251–260. [Google Scholar] [CrossRef]
- Spitschak, A.; Meier, C.; Kowtharapu, B.; Engelmann, D.; Pützer, B.M. MiR-182 promotes cancer invasion by linking RET oncogene activated NF-κB to loss of the HES1/Notch1 regulatory circuit. Mol. Cancer 2017, 16, 24. [Google Scholar] [CrossRef]
- Niki, M.; Nakajima, K.; Ishikawa, D.; Nishida, J.; Ishifune, C.; Tsukumo, S.-I.; Shimada, M.; Nagahiro, S.; Mitamura, Y.; Yasutomo, K. MicroRNA-449a deficiency promotes colon carcinogenesis. Sci. Rep. 2017, 7, 10696. [Google Scholar] [CrossRef]
- Lin, X.; Wang, S.; Sun, M.; Zhang, C.; Wei, C.; Yang, C.; Dou, R.; Liu, Q.; Xiong, B. miR-195-5p/NOTCH2-mediated EMT modulates IL-4 secretion in colorectal cancer to affect M2-like TAM polarization. J. Hematol. Oncol. 2019, 12, 20. [Google Scholar] [CrossRef]
- Peng, J.-H.; Wang, X.-L.; Ran, L.; Song, J.-L.; Zhang, Z.-T.; Liu, X.; Li, H.-Y. Inhibition of Notch signaling pathway enhanced the radiosensitivity of breast cancer cells. J. Cell. Biochem. 2018, 119, 8398–8409. [Google Scholar] [CrossRef]
- Liang, X.; Vacher, S.; Boulai, A.; Bernard, V.; Baulande, S.; Bohec, M.; Bièche, I.; Lerebours, F.; Callens, C. Targeted next-generation sequencing identifies clinically relevant somatic mutations in a large cohort of inflammatory breast cancer. Breast Cancer Res. 2018, 20, 88. [Google Scholar] [CrossRef] [PubMed]
- Luiken, S.; Fraas, A.; Bieg, M.; Sugiyanto, R.; Goeppert, B.; Singer, S.; Ploeger, C.; Warsow, G.; Marquardt, J.U.; Sticht, C.; et al. NOTCH target gene HES5 mediates oncogenic and tumor suppressive functions in hepatocarcinogenesis. Oncogene 2020, 39, 3128–3144. [Google Scholar] [CrossRef] [PubMed]
- Demehri, S.; Turkoz, A.; Kopan, R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 2009, 16, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, L.; Avila, J.L.; Demarest, R.M.; Troutman, S.; Allen, M.; Ratti, F.; Rustgi, A.K.; Stanger, B.Z.; Radtke, F.; Adsay, V.; et al. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res. 2010, 70, 4280–4286. [Google Scholar] [CrossRef]
- Yeh, C.-H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef]
- Fukusumi, T.; Califano, J.A. The NOTCH Pathway in Head and Neck Squamous Cell Carcinoma. J. Dent. Res. 2018, 97, 645–653. [Google Scholar] [CrossRef]
- Fahim, Y.; Yousefi, M.; Izadpanah, M.H.; Forghanifard, M.M. TWIST1 correlates with Notch signaling pathway to develop esophageal squamous cell carcinoma. Mol. Cell. Biochem. 2020, 474, 181–188. [Google Scholar] [CrossRef]
- Gao, Y.-B.; Chen, Z.-L.; Li, J.-G.; Hu, X.-D.; Shi, X.-J.; Sun, Z.-M.; Zhang, F.; Zhao, Z.-R.; Li, Z.-T.; Liu, Z.-Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef]
- Schwaederle, M.; Elkin, S.K.; Tomson, B.N.; Carter, J.L.; Kurzrock, R. Squamousness: Next-generation sequencing reveals shared molecular features across squamous tumor types. Cell Cycle 2015, 14, 2355–2361. [Google Scholar] [CrossRef]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Invest. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef] [PubMed]
- Sawangarun, W.; Mandasari, M.; Aida, J.; Morita, K.-I.; Kayamori, K.; Ikeda, T.; Sakamoto, K. Loss of Notch1 predisposes oro-esophageal epithelium to tumorigenesis. Exp. Cell Res. 2018, 372, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Ribeiro, M.; Ben-Jacob, E.; Devi, G.R.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Jackstadt, R.; van Hooff, S.R.; Leach, J.D.; Cortes-Lavaud, X.; Lohuis, J.O.; Ridgway, R.A.; Wouters, V.M.; Roper, J.; Kendall, T.J.; Roxburgh, C.S.; et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell 2019, 36, 319–336.e7. [Google Scholar] [CrossRef] [PubMed]
- Capaccione, K.M.; Hong, X.; Morgan, K.M.; Liu, W.; Bishop, J.M.; Liu, L.; Markert, E.; Deen, M.; Minerowicz, C.; Bertino, J.R.; et al. Sox9 mediates Notch1-induced mesenchymal features in lung adenocarcinoma. Oncotarget 2014, 5, 3636–3650. [Google Scholar] [CrossRef]
- Lindsey, S.; Langhans, S.A. Crosstalk of Oncogenic Signaling Pathways during Epithelial-Mesenchymal Transition. Front. Oncol. 2014, 4, 358. [Google Scholar] [CrossRef]
- Giachino, C.; Boulay, J.L.; Ivanek, R.; Alvarado, A.; Tostado, C.; Lugert, S.; Tchorz, J.; Coban, M.; Mariani, L.; Bettler, B.; et al. A Tumor Suppressor Function for Notch Signaling in Forebrain Tumor Subtypes. Cancer Cell 2015, 28, 730–742. [Google Scholar] [CrossRef]
- Brat, D.J.; Verhaak, R.G.W.; Al-dape, K.D.; Yung, W.K.A.; Salama, S.R.; Cooper, L.A.D.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Zhao, D.; Mo, Y.; Li, M.T.; Zou, S.W.; Cheng, Z.L.; Sun, Y.P.; Xiong, Y.; Guan, K.L.; Lei, Q.Y. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J. Clin. Invest. 2014, 124, 5453–5465. [Google Scholar] [CrossRef]
- Brzozowa-Zasada, M.; Piecuch, A.; Michalski, M.; Segiet, O.; Kurek, J.; Harabin-Słowińska, M.; Wojnicz, R. Notch and its oncogenic activity in human malignancies. Eur. Surg. 2017, 49, 199–209. [Google Scholar] [CrossRef]
- Petrovic, J.; Zhou, Y.; Fasolino, M.; Goldman, N.; Schwartz, G.W.; Mumbach, M.R.; Nguyen, S.C.; Rome, K.S.; Sela, Y.; Zapataro, Z.; et al. Oncogenic Notch Promotes Long-Range Regulatory Interactions within Hyperconnected 3D Cliques. Mol. Cell 2019, 73, 1174–1190.e12. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Ibaseta, A.; Fischer, M.M.; Cancilla, B.; O’Young, G.; Cristea, S.; Luca, V.C.; Yang, D.; Jahchan, N.S.; Hamard, C.; et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017, 545, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Mitani, Y.; Rao, X.; Zafereo, M.; Zhang, J.; Zhang, J.; Futreal, P.A.; Lozano, G.; El-Naggar, A.K. Spatio-Temporal Genomic Heterogeneity, Phylogeny, and Metastatic Evolution in Salivary Adenoid Cystic Carcinoma. J. Natl. Cancer Inst. 2017, 109, djx033. [Google Scholar] [CrossRef] [PubMed]
- Bolós, V.; Blanco, M.; Medina, V.; Aparicio, G.; Díaz-Prado, S.; Grande, E. Notch signalling in cancer stem cells. Clin. Transl. Oncol. Off. Publ. Fed. Spanish Oncol. Soc. Natl. Cancer Inst. Mex. 2009, 11, 11–19. [Google Scholar] [CrossRef]
- Ali, S.A.; Justilien, V.; Jamieson, L.; Murray, N.R.; Fields, A.P. Protein Kinase Cι Drives a NOTCH3-dependent Stem-like Phenotype in Mutant KRAS Lung Adenocarcinoma. Cancer Cell 2016, 29, 367–378. [Google Scholar] [CrossRef]
- Moore, G.; Annett, S.; McClements, L.; Robson, T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells 2020, 9, 1503. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Tetsu, O.; McCormick, F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398, 422–426. [Google Scholar] [CrossRef]
- Niehrs, C.; Acebron, S.P. Mitotic and mitogenic Wnt signalling. EMBO J. 2012, 31, 2705–2713. [Google Scholar] [CrossRef]
- Hadjihannas, M.V.; Brückner, M.; Behrens, J. Conductin/axin2 and Wnt signalling regulates centrosome cohesion. EMBO Rep. 2010, 11, 317–324. [Google Scholar] [CrossRef]
- Koni, M.; Pinnarò, V.; Brizzi, M.F. The Wnt Signalling Pathway: A Tailored Target in Cancer. Int. J. Mol. Sci. 2020, 21, 7697. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.; Cheng, P.; King, I.N.; Andersen, P.; Shenje, L.; Nigam, V.; Srivastava, D. Notch post-translationally regulates β-catenin protein in stem and progenitor cells. Nat. Cell Biol. 2011, 13, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Hattori, M.; Hirai, N.; Shinozuka, Y.; Hirata, H.; Kageyama, R.; Sakai, T.; Minato, N. Hes1 Directly Controls Cell Proliferation through the Transcriptional Repression of p27Kip1. Mol. Cell. Biol. 2005, 25, 4262–4271. [Google Scholar] [CrossRef] [PubMed]
- Carlson, P.; Dasgupta, A.; Grzelak, C.A.; Kim, J.; Barrett, A.; Coleman, I.M.; Shor, R.E.; Goddard, E.T.; Dai, J.; Schweitzer, E.M.; et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. 2019, 21, 238–250. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, C.; Zhang, Z.; Wang, Q.; Wei, H.; Shi, W.; Li, J.; Wang, Z.; Ou, Y.; Wang, W.; et al. Jagged1-Notch1-deployed tumor perivascular niche promotes breast cancer stem cell phenotype through Zeb1. Nat. Commun. 2020, 11, 5129. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Celià-Terrassa, T.; Kumar, S.; Hang, X.; Wei, Y.; Choudhury, A.; Hwang, J.; Peng, J.; Nixon, B.; Grady, J.J.; et al. Notch ligand Dll1 mediates cross-talk between mammary stem cells and the macrophageal niche. Science 2018, 360, eaan4153. [Google Scholar] [CrossRef]
- Xiong, S.; Wang, R.; Chen, Q.; Luo, J.; Wang, J.; Zhao, Z.; Li, Y.; Wang, Y.; Wang, X.; Cheng, B. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am. J. Cancer Res. 2018, 8, 302–316. [Google Scholar]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T.C.L.D.S.E. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Schreck, K.C.; Taylor, P.; Marchionni, L.; Gopalakrishnan, V.; Bar, E.E.; Gaiano, N.; Eberhart, C.G. The Notch target Hes1 directly modulates Gli1 expression and hedgehog signaling: A potential mechanism of therapeutic resistance. Clin. Cancer Res. 2010, 16, 6060–6070. [Google Scholar] [CrossRef]
- Ingram, W.J.; McCue, K.I.; Tran, T.H.; Hallahan, A.R.; Wainwright, B.J. Sonic Hedgehog regulates Hes1 through a novel mechanism that is independent of canonical Notch pathway signalling. Oncogene 2008, 27, 1489–1500. [Google Scholar] [CrossRef]
- Chang, W.H.; Lai, A.G. Aberrations in Notch-Hedgehog signalling reveal cancer stem cells harbouring conserved oncogenic properties associated with hypoxia and immunoevasion. Br. J. Cancer 2019, 121, 666–678. [Google Scholar] [CrossRef]
- Loganathan, S.K.; Schleicher, K.; Malik, A.; Quevedo, R.; Langille, E.; Teng, K.; Oh, R.H.; Rathod, B.; Tsai, R.; Samavarchi-Tehrani, P.; et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 2020, 367, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, S.K.; Schramek, D. In vivo CRISPR screens reveal potent driver mutations in head and neck cancers. Mol. Cell. Oncol. 2020, 7, 1758541. [Google Scholar] [CrossRef] [PubMed]
- Reyna, M.A.; Haan, D.; Paczkowska, M.; Verbeke, L.P.C.; Vazquez, M.; Kahraman, A.; Pulido-Tamayo, S.; Barenboim, J.; Wadi, L.; Dhingra, P.; et al. Pathway and network analysis of more than 2500 whole cancer genomes. Nat. Commun. 2020, 11, 729. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.P.A.; Martignetti, L.; Robine, S.; Barillot, E.; Zinovyev, A.; Calzone, L. Mathematical Modelling of Molecular Pathways Enabling Tumour Cell Invasion and Migration. PLoS Comput. Biol. 2015, 11, e1004571. [Google Scholar] [CrossRef] [PubMed]
- Fre, S.; Pallavi, S.K.; Huyghe, M.; Laé, M.; Janssen, K.-P.; Robine, S.; Artavanis-Tsakonas, S.; Louvard, D. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 6309–6314. [Google Scholar] [CrossRef]
- Xue, A.G.; Chan, M.; Gujral, T.S. Pan-cancer analysis of the developmental pathways reveals non-canonical wnt signaling as a driver of mesenchymal-type tumors. Transl. Res. 2020, 224, 1–15. [Google Scholar] [CrossRef]
- Kim, W.; Khan, S.K.; Gvozdenovic-Jeremic, J.; Kim, Y.; Dahlman, J.; Kim, H.; Park, O.; Ishitani, T.; Jho, E.-H.; Gao, B.; et al. Hippo signaling interactions with Wnt/β-catenin and Notch signaling repress liver tumorigenesis. J. Clin. Investig. 2017, 127, 137–152. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, Z.; Xiong, X.; Zhong, Y.; Zhang, W.; Dong, Y.; Li, J.; Zhu, Z.; Zhang, W.; Wu, H.; et al. Membrane-tethered Notch1 exhibits oncogenic property via activation of EGFR-PI3K-AKT pathway in oral squamous cell carcinoma. J. Cell. Physiol. 2019, 234, 5940–5952. [Google Scholar] [CrossRef]
- Brijwani, N.; Jain, M.; Dhandapani, M.; Zahed, F.; Mukhopadhyay, P.; Biswas, M.; Khatri, D.; Radhakrishna, V.D.; Majumder, B.; Radhakrishnan, P.; et al. Rationally co-targeting divergent pathways in KRAS wild-type colorectal cancers by CANscript technology reveals tumor dependence on Notch and Erbb2. Sci. Rep. 2017, 7, 1502. [Google Scholar] [CrossRef]
- Mangolini, M.; Götte, F.; Moore, A.; Ammon, T.; Oelsner, M.; Lutzny-Geier, G.; Klein-Hitpass, L.; Williamson, J.C.; Lehner, P.J.; Dürig, J.; et al. Notch2 controls non-autonomous Wnt-signalling in chronic lymphocytic leukaemia. Nat. Commun. 2018, 9, 3839. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, H.; Chen, Y.; Qiao, G.; Jiang, W.; Ni, P.; Liu, X.; Ma, L. miR-598 inhibits metastasis in colorectal cancer by suppressing JAG1/Notch2 pathway stimulating EMT. Exp. Cell Res. 2017, 352, 104–112. [Google Scholar] [CrossRef]
- Tveriakhina, L.; Schuster-Gossler, K.; Jarrett, S.M.; Andrawes, M.B.; Rohrbach, M.; Blacklow, S.C.; Gossler, A. The ectodomains determine ligand function in vivo and selectivity of DLL1 and DLL4 toward NOTCH1 and NOTCH2 in vitro. Elife 2018, 7, e40045. [Google Scholar] [CrossRef] [PubMed]
- Nolin, E.; Gans, S.; Llamas, L.; Bandyopadhyay, S.; Brittain, S.M.; Bernasconi-Elias, P.; Carter, K.P.; Loureiro, J.J.; Thomas, J.R.; Schirle, M.; et al. Discovery of a ZIP7 inhibitor from a Notch pathway screen. Nat. Chem. Biol. 2019, 15, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Philipovskiy, A.; Dwivedi, A.K.; Gamez, R.; McCallum, R.; Mukherjee, D.; Nahleh, Z.; Aguilera, R.J.; Gaur, S. Association between tumor mutation profile and clinical outcomes among Hispanic Latina women with triple-negative breast cancer. PLoS ONE 2020, 15, e0238262. [Google Scholar] [CrossRef]
- KleinJan, A.; Tindemans, I.; Montgomery, J.E.; Lukkes, M.; de Bruijn, M.J.W.; van Nimwegen, M.; Bergen, I.; Moellering, R.E.; Hoogsteden, H.C.; Boon, L.; et al. The Notch pathway inhibitor stapled α-helical peptide derived from mastermind-like 1 (SAHM1) abrogates the hallmarks of allergic asthma. J. Allergy Clin. Immunol. 2018, 142, 76–85.e8. [Google Scholar] [CrossRef]
- Krämer, A.; Mentrup, T.; Kleizen, B.; Rivera-Milla, E.; Reichenbach, D.; Enzensperger, C.; Nohl, R.; Täuscher, E.; Görls, H.; Ploubidou, A.; et al. Small molecules intercept Notch signaling and the early secretory pathway. Nat. Chem. Biol. 2013, 9, 731–738. [Google Scholar] [CrossRef]
- Lehal, R.; Zaric, J.; Vigolo, M.; Urech, C.; Frismantas, V.; Zangger, N.; Cao, L.; Berger, A.; Chicote, I.; Loubéry, S.; et al. Pharmacological disruption of the Notch transcription factor complex. Proc. Natl. Acad. Sci. USA 2020, 117, 16292–16301. [Google Scholar] [CrossRef]
- Fabbro, D.; Bauer, M.; Murone, M.; Lehal, R. Notch Inhibition in Cancer: Challenges and Opportunities. Chimia (Aarau) 2020, 74, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhang, J.; Yoo, S.Y.; Bengtsson, L.; Moorthy, S.; Neskey, D.M.; Zhao, M.; Ortega Alves, M.V.; Chang, K.; Drummond, J.; et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013, 3, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Rettig, E.M.; Bishop, J.A.; Agrawal, N.; Chung, C.H.; Sharma, R.; Zamuner, F.; Li, R.J.; Koch, W.M.; Califano, J.A.; Guo, T.; et al. HEY1 is expressed independent of NOTCH1 and is associated with poor prognosis in head and neck squamous cell carcinoma. Oral Oncol. 2018, 82, 168–175. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Long, J.; Li, L.; Zhao, Z.-B.; Wei, F.; Yao, Y.; Qiu, W.-J.; Wu, Z.-X.; Luo, Q.-Q.; Liu, W.; et al. Mutations in the notch signalling pathway are associated with enhanced anti-tumour immunity in colorectal cancer. J. Cell. Mol. Med. 2020, 24, 12176–12187. [Google Scholar] [CrossRef]
- Fender, A.W.; Nutter, J.M.; Fitzgerald, T.L.; Bertrand, F.E.; Sigounas, G. Notch-1 Promotes Stemness and Epithelial to Mesenchymal Transition in Colorectal Cancer. J. Cell. Biochem. 2015, 116, 2517–2527. [Google Scholar] [CrossRef]
- Fischer, M.; Yen, W.-C.; Kapoun, A.M.; Wang, M.; O’Young, G.; Lewicki, J.; Gurney, A.; Hoey, T. Anti-DLL4 inhibits growth and reduces tumor-initiating cell frequency in colorectal tumors with oncogenic KRAS mutations. Cancer Res. 2011, 71, 1520–1525. [Google Scholar] [CrossRef]
- Steinway, S.N.; Zañudo, J.G.T.; Michel, P.J.; Feith, D.J.; Loughran, T.P.; Albert, R. Combinatorial interventions inhibit TGFβ-driven epithelial-to-mesenchymal transition and support hybrid cellular phenotypes. npj Syst. Biol. Appl. 2015, 1, 15014. [Google Scholar] [CrossRef]
- Karve, K.; Netherton, S.; Deng, L.; Bonni, A.; Bonni, S. Regulation of epithelial-mesenchymal transition and organoid morphogenesis by a novel TGFβ-TCF7L2 isoform-specific signaling pathway. Cell Death Dis. 2020, 11, 704. [Google Scholar] [CrossRef]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef]
- Fujiki, K.; Inamurai, H.; Miyayamai, T.; Matsuokai, M. Involvement of Notch1 signaling in malignant progression of A549 cells subjected to prolonged cadmium exposure. J. Biol. Chem. 2017, 292, 7942–7953. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Imanaka, N.; Chen, J.; Griffin, J.D. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br. J. Cancer 2010, 102, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Fu, Y.X.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Caiado, F.; Carvalho, T.; Rosa, I.; Remédio, L.; Costa, A.; Matos, J.; Heissig, B.; Yagita, H.; Hattori, K.; Da Silva, J.P.; et al. Bone marrow-derived CD11b+Jagged2+ cells promote epithelial-to-mesenchymal transition and metastasization in colorectal cancer. Cancer Res. 2013, 73, 4233–4246. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Ying, X.; Lin, P.C.; Zhou, B.P. Twist-mediated Epithelial-mesenchymal Transition Promotes Breast Tumor Cell Invasion via Inhibition of Hippo Pathway. Sci. Rep. 2016, 6, 24606. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lee, D.K.; Feng, Z.; Xu, Y.; Bu, W.; Li, Y.; Liao, L.; Xu, J. Breast tumor cell-specific knockout of Twist1 inhibits cancer cell plasticity, dissemination, and lung metastasis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 11494–11499. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qin, L.; Sun, T.; Wu, H.; He, T.; Yang, Z.; Mo, Q.; Liao, L.; Xu, J. Twist1 promotes breast cancer invasion and metastasis by silencing Foxa1 expression. Oncogene 2017, 36, 1157–1166. [Google Scholar] [CrossRef]
- Qiao, W.; Jia, Z.; Liu, H.; Liu, Q.; Zhang, T.; Guo, W.; Li, P.; Deng, M.; Li, S. Prognostic and clinicopathological value of Twist expression in breast cancer: A meta-analysis. PLoS ONE 2017, 12, e0186191. [Google Scholar] [CrossRef]
- Wang, Y.; Song, W.; Kan, P.; Huang, C.; Ma, Z.; Wu, Q.; Yao, X.; Zhang, B. Overexpression of epsin 3 enhances migration and invasion of glioma cells by inducing epithelial-mesenchymal transition. Oncol. Rep. 2018, 40, 3049–3059. [Google Scholar] [CrossRef]
- Li, J.; Li, Q.; Lin, L.; Wang, R.; Chen, L.; Du, W.; Jiang, C.; Li, R. Targeting the Notch1 oncogene by miR-139-5p inhibits glioma metastasis and epithelial-mesenchymal transition (EMT). BMC Neurol. 2018, 18, 133. [Google Scholar] [CrossRef]
- Chen, D.W.; Wang, H.; Bao, Y.F.; Xie, K. Notch signaling molecule is involved in the invasion of MiaPaCa2 cells induced by CoCl2 via regulating epithelial-mesenchymal transition. Mol. Med. Rep. 2018, 17, 4965–4972. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Piao, M.H.; Feng, C.S.; Yuan, Y. Isoflurane promotes epithelial-to-mesenchymal transition and metastasis of bladder cancer cells through HIF-1α-β-catenin/Notch1 pathways. Life Sci. 2020, 258, 118154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, G.; Zhou, L.; Li, P.; Yun, M.; Shi, Q.; Wang, T.; Wu, X. Notch signalling induces epithelial-mesenchymal transition to promote metastasis in oral squamous cell carcinoma. Int. J. Mol. Med. 2018, 42, 1909–1916. [Google Scholar] [CrossRef]
- Mutvei, A.P.; Landor, S.K.J.; Fox, R.; Braune, E.B.; Tsoi, Y.L.; Phoon, Y.P.; Sahlgren, C.; Hartman, J.; Bergh, J.; Jin, S.; et al. Notch signaling promotes a HIF2α-driven hypoxic response in multiple tumor cell types. Oncogene 2018, 37, 6083–6095. [Google Scholar] [CrossRef]
- Xing, F.; Okuda, H.; Watabe, M.; Kobayashi, A.; Pai, S.K.; Liu, W.; Pandey, P.R.; Fukuda, K.; Hirota, S.; Sugai, T.; et al. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene 2011, 30, 4075–4086. [Google Scholar] [CrossRef]
- Ciria, M.; García, N.A.; Ontoria-Oviedo, I.; González-King, H.; Carrero, R.; De La Pompa, J.L.; Montero, J.A.; Sepúlveda, P. Mesenchymal Stem Cell Migration and Proliferation Are Mediated by Hypoxia-Inducible Factor-1α Upstream of Notch and SUMO Pathways. Stem Cells Dev. 2017, 26, 973–985. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, H.; Jiang, X.; Qian, C.; Liu, Z.; Luo, D. Factors involved in cancer metastasis: A better understanding to “seed and soil” hypothesis. Mol. Cancer 2017, 16, 176. [Google Scholar] [CrossRef]
- Natsuizaka, M.; Whelan, K.A.; Kagawa, S.; Tanaka, K.; Giroux, V.; Chandramouleeswaran, P.M.; Long, A.; Sahu, V.; Darling, D.S.; Que, J.; et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat. Commun. 2017, 8, 1758. [Google Scholar] [CrossRef]
- Boareto, M.; Jolly, M.K.; Goldman, A.; Pietilä, M.; Mani, S.A.; Sengupta, S.; Ben-Jacob, E.; Levine, H.; Onuchic, J.N. Notch-Jagged signalling can give rise to clusters of cells exhibiting a hybrid epithelial/mesenchymal phenotype. J. R. Soc. Interface 2016, 13, 20151106. [Google Scholar] [CrossRef]
- Luo, H.; Liu, W.H.; Liang, H.Y.; Yan, H.T.; Lin, N.; Li, D.Y.; Wang, T.; Tang, L.J. Differentiation-inducing therapeutic effect of Notch inhibition in reversing malignant transformation of liver normal stem cells via MET. Oncotarget 2018, 9, 18885–18895. [Google Scholar] [CrossRef]
- Pelullo, M.; Nardozza, F.; Zema, S.; Quaranta, R.; Nicoletti, C.; Besharat, Z.M.; Felli, M.P.; Cerbelli, B.; d’Amati, G.; Palermo, R.; et al. Kras/ADAM17-Dependent Jag1-ICD Reverse Signaling Sustains Colorectal Cancer Progression and Chemoresistance. Cancer Res. 2019, 79, 5575–5586. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Cao, J.; Liang, Z.; Lin, Q.; Wang, J.; Yang, X.; Zhang, R.; Zong, J.; Du, X.; Peng, Y.; et al. Estrogen receptor α-NOTCH1 axis enhances basal stem-like cells and epithelial-mesenchymal transition phenotypes in prostate cancer. Cell Commun. Signal. 2019, 17, 50. [Google Scholar] [CrossRef] [PubMed]
- Bocci, F.; Jolly, M.K.; Tripathi, S.C.; Aguilar, M.; Hanash, S.M.; Levine, H.; Onuchic, J.N. Numb prevents a complete epithelial-mesenchymal transition by modulating Notch signaling. J. R. Soc. Interface 2017, 14, 20170512. [Google Scholar] [CrossRef] [PubMed]
- Irshad, S.; Bansal, M.; Guarnieri, P.; Davis, H.; Al Haj Zen, A.; Baran, B.; Pinna, C.M.A.; Rahman, H.; Biswas, S.; Bardella, C.; et al. Bone morphogenetic protein and Notch signalling crosstalk in poor-prognosis, mesenchymal-subtype colorectal cancer. J. Pathol. 2017, 242, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Liu, P.; Zheng, M.; Wang, Z.; Gao, Y.; Liang, K.; Wang, H.; Tan, X. The MiR-1224-5p/elf3 axis regulates malignant behaviors of pancreatic cancer via pi3k/akt/notch signaling pathways. Onco. Targets. Ther. 2020, 13, 3449–3466. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xu, J.; Liu, B.; He, X.; Zhou, L.; Hu, X.; Qiao, F.; Zhang, A.; Xu, X.; Zhang, H.; et al. IL6 blockade potentiates the anti-tumor effects of γ-secretase inhibitors in Notch3-expressing breast cancer. Cell Death Differ. 2018, 25, 330–339. [Google Scholar] [CrossRef]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, L.; Zhao, S.; Zhao, X.Y.; Min, P.X.; Ma, Y.D.; Wang, Y.Y.; Chen, Y.; Tang, S.J.; Zhang, Y.J.; et al. Non-canonical notch signaling regulates actin remodeling in cell migration by activating PI3K/AKT/Cdc42 pathway. Front. Pharmacol. 2019, 10, 370. [Google Scholar] [CrossRef]
- LaFoya, B.; Munroe, J.A.; Mia, M.M.; Detweiler, M.A.; Crow, J.J.; Wood, T.; Roth, S.; Sharma, B.; Albig, A.R. Notch: A multi-functional integrating system of microenvironmental signals. Dev. Biol. 2016, 418, 227–241. [Google Scholar] [CrossRef]
- Giovannini, C.; Minguzzi, M.; Genovese, F.; Baglioni, M.; Gualandi, A.; Ravaioli, M.; Milazzo, M.; Tavolari, S.; Bolondi, L.; Gramantieri, L. Molecular and proteomic insight into Notch1 characterization in hepatocellular carcinoma. Oncotarget 2016, 7, 39609–39626. [Google Scholar] [CrossRef]
- Sun, T.; Patil, R.; Galstyan, A.; Klymyshyn, D.; Ding, H.; Chesnokova, A.; Cavenee, W.K.; Furnari, F.B.; Ljubimov, V.A.; Shatalova, E.S.; et al. Blockade of a Laminin-411–Notch Axis with CRISPR/Cas9 or a Nanobioconjugate Inhibits Glioblastoma Growth through Tumor-Microenvironment Cross-talk. Cancer Res. 2019, 79, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Zhou, X.; Li, T.; Liu, P.; Hai, L.; Tong, L.; Ma, H.; Tao, Z.; Xie, Y.; Zhang, C.; et al. Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J. Exp. Clin. Cancer Res. 2019, 38, 339. [Google Scholar] [CrossRef] [PubMed]
- Nandagopal, N.; Santat, L.A.; LeBon, L.; Sprinzak, D.; Bronner, M.E.; Elowitz, M.B. Dynamic Ligand Discrimination in the Notch Signaling Pathway. Cell 2018, 172, 869–880.e19. [Google Scholar] [CrossRef] [PubMed]
- Luca, V.C.; Kim, B.C.; Ge, C.; Kakuda, S.; Wu, D.; Roein-Peikar, M.; Haltiwanger, R.S.; Zhu, C.; Ha, T.; Garcia, K.C. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 2017, 355, 1320–1324. [Google Scholar] [CrossRef]
- Antfolk, D.; Sjöqvist, M.; Cheng, F.; Isoniemi, K.; Duran, C.L.; Rivero-Muller, A.; Antila, C.; Niemi, R.; Landor, S.; Bouten, C.V.C.; et al. Selective regulation of Notch ligands during angiogenesis is mediated by vimentin. Proc. Natl. Acad. Sci. USA 2017, 114, E4574–E4581. [Google Scholar] [CrossRef]
- Li, L.; Zhao, F.; Lu, J.; Li, T.; Yang, H.; Wu, C.; Liu, Y. Notch-1 signaling promotes the malignant features of human breast cancer through NF-κB activation. PLoS ONE 2014, 9, e95912. [Google Scholar] [CrossRef]
- Hafeez, B.B.; Adhami, V.M.; Asim, M.; Siddiqui, I.A.; Bhat, K.M.; Zhong, W.; Saleem, M.; Din, M.; Setaluri, V.; Mukhtar, H. Targeted knockdown of notchl inhibits invasion of human prostate cancer cells concomitant with inhibition of matrix metalloproteinase-9 and urokinase plasminogen activator. Clin. Cancer Res. 2009, 15, 452–459. [Google Scholar] [CrossRef]
- Lai, X.X.; Li, G.; Lin, B.; Yang, H. Interference of Notch 1 inhibits the proliferation and invasion of breast cancer cells: Involvement of the β-catenin signaling pathway. Mol. Med. Rep. 2018, 17, 2472–2478. [Google Scholar] [CrossRef]
- Díaz, B.; Yuen, A.; Iizuka, S.; Higashiyama, S.; Courtneidge, S.A. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J. Cell Biol. 2013, 201, 279–292. [Google Scholar] [CrossRef]
- Pignatelli, J.; Bravo-Cordero, J.J.; Roh-Johnson, M.; Gandhi, S.J.; Wang, Y.; Chen, X.; Eddy, R.J.; Xue, A.; Singer, R.H.; Hodgson, L.; et al. Macrophage-dependent tumor cell transendothelial migration is mediated by Notch1/Mena INV-initiated invadopodium formation. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 2010, 12, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Wieland, E.; Rodriguez-Vita, J.; Liebler, S.S.; Mogler, C.; Moll, I.; Herberich, S.E.; Espinet, E.; Herpel, E.; Menuchin, A.; Chang-Claude, J.; et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017, 31, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Sonoshita, M.; Aoki, M.; Fuwa, H.; Aoki, K.; Hosogi, H.; Sakai, Y.; Hashida, H.; Takabayashi, A.; Sasaki, M.; Robine, S.; et al. Suppression of colon cancer metastasis by aes through inhibition of notch signaling. Cancer Cell 2011, 19, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Sonoshita, M.; Kakizaki, F.; Aoyama, N.; Itatani, Y.; Uegaki, M.; Sakamoto, H.; Kobayashi, T.; Inoue, T.; Kamba, T.; et al. Amino-terminal enhancer of split gene AES encodes a tumor and metastasis suppressor of prostate cancer. Cancer Sci. 2017, 108, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Hultgren, N.W.; Fang, J.S.; Ziegler, M.E.; Ramirez, R.N.; Phan, D.T.T.; Hatch, M.M.S.; Welch-Reardon, K.M.; Paniagua, A.E.; Kim, L.S.; Shon, N.N.; et al. Slug regulates the Dll4-Notch-VEGFR2 axis to control endothelial cell activation and angiogenesis. Nat. Commun. 2020, 11, 5400. [Google Scholar] [CrossRef]
- Mendonça, L.; Trindade, A.; Carvalho, C.; Correia, J.; Badenes, M.; Gigante, J.; Duarte, A. Metastasis is impaired by endothelial-specific Dll4 loss-of-function through inhibition of epithelial-to-mesenchymal transition and reduction of cancer stem cells and circulating tumor cells. Clin. Exp. Metastasis 2019, 36, 365–380. [Google Scholar] [CrossRef]
- Mizukoshi, K.; Okazawa, Y.; Haeno, H.; Koyama, Y.; Sulidan, K.; Komiyama, H.; Saeki, H.; Ohtsuji, N.; Ito, Y.; Kojima, Y.; et al. Metastatic seeding of human colon cancer cell clusters expressing the hybrid epithelial/mesenchymal state. Int. J. Cancer 2020, 146, 2547–2562. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Onuchic, J.N.; Levine, H.; Ben-Jacob, E. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; Lebleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Jordan, N.V.; Bardia, A.; Wittner, B.S.; Benes, C.; Ligorio, M.; Zheng, Y.; Yu, M.; Sundaresan, T.K.; Licausi, J.A.; Desai, R.; et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 2016, 537, 102–106. [Google Scholar] [CrossRef]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Piva, F.; Marinelli, O.; Maggi, F.; Bianchi, F.; Bittoni, A.; Berardi, R.; Giampieri, R.; et al. Expression Profiling of Circulating Tumor Cells in Pancreatic Ductal Adenocarcinoma Patients: Biomarkers Predicting Overall Survival. Front. Oncol. 2019, 9, 874. [Google Scholar] [CrossRef] [PubMed]
- Sprouse, M.L.; Welte, T.; Boral, D.; Liu, H.N.; Yin, W.; Vishnoi, M.; Goswami-Sewell, D.; Li, L.; Pei, G.; Jia, P.; et al. PMN-MDSCs Enhance CTC Metastatic Properties through Reciprocal Interactions via ROS/Notch/Nodal Signaling. Int. J. Mol. Sci 2019, 20, 1916. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Debeb, B.G.; Aceto, N.; Farach-Carson, M.C.; Woodward, W.A.; Levine, H. Inflammatory breast cancer: A model for investigating cluster-based dissemination. npj Breast Cancer 2017, 3, 21. [Google Scholar] [CrossRef]
- Pisani, P.; Airoldi, M.; Allais, A.; Valletti, P.A.; Battista, M.; Benazzo, M.; Briatore, R.; Cacciola, S.; Cocuzza, S.; Colombo, A.; et al. Metastatic disease in head & neck oncology. Acta Otorhinolaryngol. Ital. 2020, 40, S1–S86. [Google Scholar] [CrossRef]
- Donato, C.; Kunz, L.; Giner, F.C.; Paasinen-Sohns, A.; Strittmatter, K.; Szczerba, B.M.; Scherrer, R.; Di Maggio, N.; Heusermann, W.; Biehlmaier, O.; et al. Hypoxia Triggers the Intravasation of Clustered Circulating Tumor Cells. Cell Rep. 2020, 32, 108105. [Google Scholar] [CrossRef]
- Oshi, M.; Newman, S.; Tokumaru, Y.; Yan, L.; Matsuyama, R.; Endo, I.; Nagahashi, M.; Takabe, K. Intra-tumoral angiogenesis is associated with inflammation, immune reaction and metastatic recurrence in breast cancer. Int. J. Mol. Sci. 2020, 21, 6708. [Google Scholar] [CrossRef]
- Zeng, Q.; Li, S.; Chepeha, D.B.; Giordano, T.J.; Li, J.; Zhang, H.; Polverini, P.J.; Nor, J.; Kitajewski, J.; Wang, C.Y. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell 2005, 8, 13–23. [Google Scholar] [CrossRef]
- Jia, X.; Wang, W.; Xu, Z.; Wang, S.; Wang, T.; Wang, M.; Wu, M. A humanized anti-DLL4 antibody promotes dysfunctional angiogenesis and inhibits breast tumor growth. Sci. Rep. 2016, 6, 27985. [Google Scholar] [CrossRef]
- Qiu, X.X.; Chen, L.; Wang, C.H.; Lin, Z.X.; Chen, B.J.; You, N.; Chen, Y.; Wang, X.F. The Vascular Notch Ligands Delta-Like Ligand 4 (DLL4) and Jagged1 (JAG1) Have Opposing Correlations with Microvascularization but a Uniform Prognostic Effect in Primary Glioblastoma: A Preliminary Study. World Neurosurg. 2016, 88, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Oon, C.E.; Bridges, E.; Sheldon, H.; Sainson, R.C.A.; Jubb, A.; Turley, H.; Leek, R.; Buffa, F.; Harris, A.L.; Li, J.L. Role of Delta-like 4 in Jagged1-induced tumour angiogenesis and tumour growth. Oncotarget 2017, 8, 40115–40131. [Google Scholar] [CrossRef] [PubMed][Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Misiorek, J.O.; Przybyszewska-Podstawka, A.; Kałafut, J.; Paziewska, B.; Rolle, K.; Rivero-Müller, A.; Nees, M. Context Matters: NOTCH Signatures and Pathway in Cancer Progression and Metastasis. Cells 2021, 10, 94. https://doi.org/10.3390/cells10010094
Misiorek JO, Przybyszewska-Podstawka A, Kałafut J, Paziewska B, Rolle K, Rivero-Müller A, Nees M. Context Matters: NOTCH Signatures and Pathway in Cancer Progression and Metastasis. Cells. 2021; 10(1):94. https://doi.org/10.3390/cells10010094
Chicago/Turabian StyleMisiorek, Julia O., Alicja Przybyszewska-Podstawka, Joanna Kałafut, Beata Paziewska, Katarzyna Rolle, Adolfo Rivero-Müller, and Matthias Nees. 2021. "Context Matters: NOTCH Signatures and Pathway in Cancer Progression and Metastasis" Cells 10, no. 1: 94. https://doi.org/10.3390/cells10010094
APA StyleMisiorek, J. O., Przybyszewska-Podstawka, A., Kałafut, J., Paziewska, B., Rolle, K., Rivero-Müller, A., & Nees, M. (2021). Context Matters: NOTCH Signatures and Pathway in Cancer Progression and Metastasis. Cells, 10(1), 94. https://doi.org/10.3390/cells10010094