Abstract
The RAF/MEK/ERK signaling pathway regulates diverse cellular processes as exemplified by cell proliferation, differentiation, motility, and survival. Activation of ERK1/2 generally promotes cell proliferation, and its deregulated activity is a hallmark of many cancers. Therefore, components and regulators of the ERK pathway are considered potential therapeutic targets for cancer, and inhibitors of this pathway, including some MEK and BRAF inhibitors, are already being used in the clinic. Notably, ERK1/2 kinases also have pro-apoptotic functions under certain conditions and enhanced ERK1/2 signaling can cause tumor cell death. Although the repertoire of the compounds which mediate ERK activation and apoptosis is expanding, and various anti-cancer compounds induce ERK activation while exerting their anti-proliferative effects, the mechanisms underlying ERK1/2-mediated cell death are still vague. Recent studies highlight the importance of dual-specificity phosphatases (DUSPs) in determining the pro- versus anti-apoptotic function of ERK in cancer. In this review, we will summarize the recent major findings in understanding the role of ERK in apoptosis, focusing on the major compounds mediating ERK-dependent apoptosis. Studies that further define the molecular targets of these compounds relevant to cell death will be essential to harnessing these compounds for developing effective cancer treatments.
1. Introduction
The MAPK signaling pathway, comprised of RAS, RAF, MEK, and ERK is an evolutionarily conserved signaling cascade that plays a fundamental role in the control of key cellular processes, including cell survival and proliferation. Aberrant activation of RAS/RAF/MEK/ERK signaling is frequently found in various cancers, due to genetic and epigenetic alterations, rendering them attractive therapeutic targets (Figure 1) [1,2,3]. Indeed, the majority of solid tumors are explicitly characterized by their mutations in the RAS/RAF/MEK/ERK genes, wherein highly selective small molecule inhibitors of BRAF and MEK1/2 are currently used for cancer therapy (Figure 1) [4,5,6,7,8]. RAS is one of the most well-known proto-oncogenes and its gain-of-function mutations occur in approximately 30% of all human cancers [9]. RAF mutations are also frequent, particularly in melanoma [10]. Intriguingly, mutations in MEK are less identified (melanoma 3–8%, colorectal 3%) and ERK mutations have rarely been reported as drivers in human cancers despite the well-recognized importance of ERK activation in cancer malignancy [11].
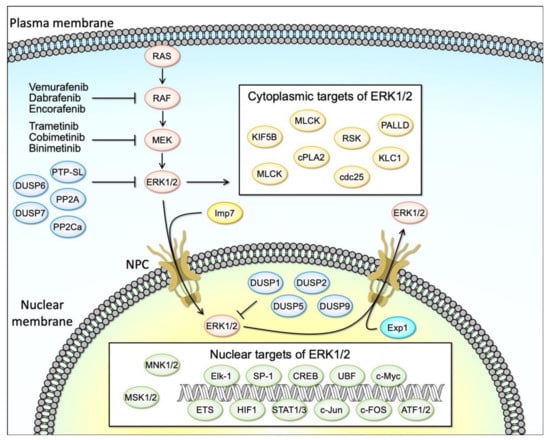
Figure 1.
The regulation of RAF/MEK/ERK signaling pathway. The RAF/MEK/ERK signaling pathway regulates diverse cellular processes through the phosphorylation of cytoplasmic and nuclear proteins. Activated ERK1/2 translocates to the nucleus by Imp7 (Importin 7) and inactive ERK1/2 is nuclear exported by Exp1 (Exportin 1). Serine/threonine phosphatases (PP2A and PP2Ca), tyrosine phosphatases (PTP-SL), and dual-specificity phosphatases (DUSP6, 7) directly dephosphorylate ERK1/2 in the cytoplasm. Dual-specificity phosphatases (DUSP1, 2, 5, and 9) dephosphorylate ERK1/2 in the nucleus. Abnormal activation of RAF/MEK/ERK1/2 signaling is known in various cancer cells. BRAF inhibitors (vemurafenib, dabrafenib, and encorafenib), and MEK inhibitors (trametinib, cobimetinib, and binimetinib) prevent ERK1/2 activation and are currently used for cancer therapy.
Contrary to the well-established role of ERK MAPK signaling in promoting cell proliferation, survival, and tumorigenesis, growing evidence suggests that the RAF/MEK/ERK signaling can mediate pro-apoptotic signaling in vitro and in vivo [12]. In addition, various compounds with anti-cancer properties were shown to induce apoptosis in an ERK activation-dependent manner [13]. These include betulinic acid, quercetin, piperlongumine, kaempferol, and phorbol esters (DAG mimetics), and an increasing number of the compounds have been reported to exert apoptosis-inducing effects through ERK activation [14,15,16,17,18]. Furthermore, ERK activity is implicated in the mechanisms of apoptosis induced by several DNA-damaging chemotherapeutic reagents for cancer treatment as exemplified by cisplatin or etoposide (or doxorubicin) [19,20,21]. In many cases, sustained and marked ERK activation seems to be implicated in mediating apoptosis [22,23]. Impressive data have particularly highlighted the importance of the cellular threshold for active ERK1/2 levels in determining the growth arrest versus death responses mediated by ERK1/2 signaling [24]. However, despite the growing evidence of the pro-apoptotic role of ERK and the expanding repertoire of drugs capable of inducing ERK activation and apoptosis, the mechanisms underlying this Janus-faced behavior of the ERK1/2 pathway in terms of cell survival regulation remain to be fully elucidated. Recently, a chemical genetic approach using yeast genetics has identified ACA-28 and its lead compounds as ERK signaling modulators, and these compounds turned out to stimulate ERK signaling thereby inducing apoptosis specifically in ERK-active cancerous situations [25,26]. Interestingly, ACA-28 was shown to induce ERK activation and apoptosis, at least in part, via downregulation of the dual-specificity phosphatase DUSP6, a negative feedback regulator for the RAS–ERK pathway, which plays a critical role in determining the threshold of ERK MAPK activation [27].
Regarding the detailed information/literature on the anti-apoptotic role of the ERK signaling pathway in proliferation and tumorigenesis, the molecular and structural regulation of this pathway, or the possibilities of targeting this pathway for cancer therapeutics, readers are directed to excellent reviews elsewhere [7,28,29,30,31,32,33]. In this review, we will summarize recent advances regarding understanding the role of ERK in apoptosis with a special focus on the role of DUSPs in this aspect.
2. Overview of the RAS/RAF/MEK/ERK1/2 Signaling Pathway
The RAS/RAF/MEK/ERK pathway (hereinafter referred to as the ERK cascade) is a highly conserved signaling mechanism, which is used to connect extracellular signals from cell surface receptors to machinery that regulates multiple critical physiological processes, including growth, proliferation, differentiation, migration, and apoptosis (Figure 1) [28]. The unique feature of MAPK signaling regulation is its sequential phosphorylation cascade which, in combination with feedback loops at multiple levels, allows tight regulation of ERK signaling activity in physiological conditions.
The non-oncogenic MAPK cascade during normal growth conditions is finely regulated to ensure that ERK activation and de-activation meet the requirements of transcriptional and translational events for cell growth. ERK is activated by various extracellular stimuli such as growth factors, cytokines, hormones, and heat or oxidative stresses through receptor tyrosine kinases (RTKs) or EGFRs [34,35,36,37,38]. These receptors then transmit activating signals to the small GTPase RAS, which acts as a binary switch in growth signaling. Upon stimulation by upstream receptors, RAS switches from the GDP-bound inactive form to the GTP-bound active form. The activated RAS then goes through a conformational change in its downstream effector-binding region, leading to the binding and regulation of downstream effectors, including RAF. Activated RAF phosphorylates and activates serine/threonine-protein kinases MEK1/2, which in turn phosphorylate and activate ERK1/2 kinases [39].
It should be mentioned that there are several MAP3K isoforms (Raf-A,B,C, Mos, and MEKK1/3), and some of the alternative spliced isoforms of RAF proteins, as exemplified by a dominant-negative antagonist of A-Raf (DA Raf), antagonize their full-length counterparts and the downstream ERK pathway [40]. Interestingly, a splice isoform of human Raf1 that causes protein truncation and loss of the C-terminal kinase domain (Raf1-tr) exhibited increased nuclear localization compared with full-length Raf1. Raf1-tr was shown to have increased binding to DNA-dependent protein kinase (DNA-PK), which inhibits DNA-PK function and causes amplification of irradiation- and bleomycin-induced DNA damage. Importantly, the expression levels of Raf1-tr impacts cancer cells’ resistance/sensitivity to bleomycin-induced apoptosis. Thus, these various isoforms and spliced variants exert various cellular and pathophysiological functions, which may be mediated by antagonizing their full-length counterparts [41]. Alternative spliced isoforms of MEK (ERK1b) and ERK (ERK1c) exist as well [42], extending the signaling specificity of the cascade even further.
Activated ERK1/2 kinases phosphorylate numerous substrates which regulate cell cycle progression, differentiation, and survival (Figure 1) [30]. These substrates include cytosolic signaling proteins, p90 ribosomal S6 kinase (RSK) and MAPK-interacting serine/threonine kinase (MNK), as well as transcription factors, such as ETS domain-containing protein Elk-1, proto-oncogenes c-FOS, c-Jun and c-Myc, and cyclic AMP-dependent transcription factor ATF2, located in the nucleus [29]. RSK (also known as MAPKAP-K1) and MSK (for mitogen- and stress-activated protein kinase) constitute a family of protein kinases that mediate signal transduction downstream of MAP kinase cascades [43]. RSK is activated by the ERK family in response to various stimuli as exemplified by growth factors. In contrast, MSK is also activated by ERK in response to such stimuli, but in addition, it can be activated by p38 family MAP kinases in response to various cellular stress stimuli and pro-inflammatory cytokines/factors. In mammals, four RSK genes (RSK1-RSK4) and two MSK genes (MSK1 and MSK2) have been identified. These enzymes were among the first substrates of ERK to be discovered and have proven to be multifunctional mediators of ERK signal transduction. While the RSK isoforms promote cell survival through the inactivation of several apoptotic effectors, RSK1 and RSK2 were also shown to phosphorylate and inhibit upstream signaling components, including SOS1 and GAB2 [28]. Transcription factors targeted by the RAS–ERK pathway regulate gene expression involved in cellular proliferation, differentiation, and cell cycle progression (e.g., Cyclin D) [28,44,45]. Multiple distinct substrates of ERK1/2 have been identified to date in different subcellular compartments including the cytosol, the nucleus, or the endomembranes [46].
Positive and negative feedback plays a critical role in the maintenance of ERK activation balance and cellular homeostasis. The activity of the ERK MAPK pathway is tightly regulated by the competing actions of upstream kinases and inhibitory phosphatases as well as by the scaffolding proteins [30]. In mammals, scaffold proteins identified thus far include KSR1/2, MORG1, IQGAP1/3, β-arrestins, and LAMTOR3 [28]. Scaffold proteins fine-tune ERK signals and provide signal fidelity by isolating the ERK signaling complex from external interferences. Especially, scaffold proteins play a central role as spatial regulators of ERKs signals [47], by dictating ERK localization in the cell membrane and endomembranes via interactions with ERK. Notably, in addition to scaffold proteins, ERK signaling regulators also impact the spatio-temporal dynamics of the ERK signaling activity. For example, MEK not only activates ERK but also anchors ERK in the cytoplasm. When the signaling is in an inactive state, ERK is localized to the cytoplasm. In unstimulated cells, MEK1 binds to ERK1/2. MEK1 contains a nuclear export signal (NES) that directs its location and that of the MEK–ERK complex to the cytoplasm. The binding of MEK1 to ERK may represent a mechanism for preventing the accumulation of ERK in the nucleus where it could phosphorylate its nuclear substrates. The phosphorylation and activation of MEK1 in response to mitogenic stimuli result in dissociation of the MEK–ERK complex and the phosphorylation and activation of ERK by MEK1 [48]. The ERK cascade is inactivated by the dephosphorylation of its components by several phosphatases. These include serine/threonine phosphatases, such as PP2A and PP2Ca, tyrosine phosphatases, such as PTP-SL, as well as dual-specificity phosphatases (DUSPs), such as DUSP6, and 7 (cytosol), and DUSP1, 2, 5, and 9 (in the nucleus). DUSP5 can bind and sequester inactive ERK2 at the nucleus [49], whereas DUSP6 can retain inactive ERK2 in the cytoplasm [50], thus affecting the spatio-temporal dynamics of ERK signaling. Reciprocally, ERK activation affects DUSPs at multiple levels, through transcriptional and post-transcriptional mechanisms [51,52,53,54]. For example, DUSP6 protein expression correlates with ERK signaling activation in lung cancer cell lines, and regulation of DUSP6 is mediated at the promoter level by the ETS1 transcription factor, a well-known nuclear target of activated ERK. Another example of the negative feedback regulation of ERK signaling includes direct negative regulation of upstream kinases of the ERK signaling cascade by ERK phosphorylation. Activated cytoplasmic ERK1/2 can directly phosphorylate EGFR, RAF, and MEK, which impairs signaling as a rapid feedback response [55].
3. Targeting the Oncogenic Activation of the RAS/RAF/MEK/ERK Signaling for Cancer Therapy
3.1. Roles of ERK Signaling in Proliferation and Survival
Unlimited cell proliferation and a lack of apoptosis are important biological characteristics of tumors [56]. The ERK MAPK signaling cascade promotes proliferation and has an anti-apoptotic effect. The ERK cascade activates transcription of several proliferative signaling networks driven by FOS, Elk-1, Jun, and ETS family transcription factors, as well as Myc (Figure 1) [29]. These factors support cancer cell proliferation by promoting cell cycle entry, angiogenesis, and survival.
Apoptosis, or programmed cell death, is an essential process leading to the removal of damaged cells without affecting normal cells, following DNA damage or during development [57]. Apoptotic cell death is associated with several conserved features and culminates in the activation of cysteine–aspartic proteases (caspases), which degrade cellular components to prepare dying cells for clearance by phagocytes with minimal stress to surrounding cells and tissues [58]. Dysregulation of apoptosis has a major role in the cause of various diseases, including neurodegenerative diseases, autoimmune disorders, and cancer [59,60,61]. There are at least two broad signaling pathways that lead to apoptosis. Namely, apoptosis is initiated by either internal or external stimuli and mediated via two distinct pathways: the intrinsic pathway (mitochondria-mediated) and the extrinsic pathway [62]. The intrinsic apoptosis pathway begins when an injury occurs within the cell, which is mainly regulated by the B-cell lymphoma 2 (Bcl-2) family of intracellular proteins. Intrinsic stresses such as oncogenes, direct DNA damage, hypoxia, and survival factor deprivation, can activate the intrinsic apoptotic pathway. p53 is a sensor of cellular stress and is a critical activator of the intrinsic pathway. The extrinsic pathway begins outside a cell when conditions in the extracellular environment determine that a cell must die. Apoptotic signaling through the extrinsic pathway engaged when extracellular ligands such as TNF (tumor necrosis factor), Fas-L (Fas ligand), and TRAIL (TNF-related apoptosis-inducing ligand) are attached to the extracellular domain of the DR (transmembrane receptors), i.e., the type 1 TNF receptor (TNFR1), Fas (also called CD95/Apo-1), and TRAIL receptors.
Genetic and pharmacological evidence suggests that ERK signaling mostly provides prosurvival cues in response to serum and receptor tyrosine kinase (RTK) activation. The Bcl-2 family controls apoptosis at the mitochondria by maintaining a balance between pro-and anti-apoptotic members. The Bcl-2 family comprises anti-apoptotic (pro-survival) and pro-apoptotic members. Pro-survival members, such as Bcl-xL, are structurally similar to Bcl-2, whereas pro-apoptotic proteins Bax and Bak (Bcl-2 Antagonist Killer-1) are structurally similar to Bcl-2 and Bcl-xL and antagonize their pro-survival functions. ERK1/2 signaling regulates the activities and levels of Bcl-2 family proteins such as the pro-apoptotic protein BIM and the anti-apoptotic protein MCL-1, thereby promoting the survival of cancer cells. ERK1/2 activation can also inhibit apoptosis induced by the death receptors Fas, or TNF (extrinsic death pathway). In contrast to ERK activation, manipulation to dampen ERK1/2 signaling in response to these stimuli promotes apoptosis.
3.2. Roles of ERK Signaling in Tumor Development
In addition to the regulation of cellular physiological functions, such as cell proliferation, differentiation, and cell cycle, the ERK MAPK signaling is also involved in tumor formation. Elevated activation of the ERK MAPK signaling pathway has been detected in various human tumors, such as ovarian, colon, breast, thyroid, pancreatic, brain, and lung cancers [63,64,65]. Continuous activation of the ERK MAPK signaling pathway can promote the transformation of normal cells into tumors, while inhibition of the ERK MAPK signaling pathway can inhibit tumor growth in vivo [66]. Furthermore, the ERK signaling pathway plays a role in several steps of tumor development, including tumor invasion and metastasis, degradation of the tumor extracellular matrix, angiogenesis, and tumor cell migration. The phosphorylation by ERK of proteins such as myosin light chain kinase, calpain, focal adhesion kinase, and paxillin promotes cancer cell migration. Therefore, aberrant activation of ERK MAPK signaling causes de novo cell transformation and promotes tumor growth and progression.
3.3. Oncogenic Activation of ERK1/2 in Human Cancers
The MAP kinase cascade is probably the most important oncogenic driver of human cancers. Aberrant activation of any signaling molecule that functions upstream of ERK1/2 would culminate in the constitutive activation of these kinases, leading to tumorigenesis. Therefore, components of the ERK signaling cascade are considered attractive therapeutic targets for cancer treatment and the blockade of this signaling module by targeted inhibitors is an important anti-tumor strategy [1,5,7,33]. The ERK cascade is one of the most commonly dysregulated pathways in human cancers. KRAS is the most commonly mutated isoform in various tumor types, followed by NRAS and HRAS [67,68]. In particular, KRAS is mutated exclusively in pancreatic ductal adenocarcinoma. Oncogenic RAS mutations exhibit stable GTP-binding and constitutive activation of RAS and downstream RAF/MEK/ERK and PI3K/AKT signaling. Similarly, mutations involving BRAF, which are prevalent in malignant melanoma, exert an oncogenic effect by activating the downstream MEK/ERK MAPK pathway, resulting in uncontrolled cellular proliferation. Unlike RAS and BRAF, activating mutations in MEK are rarely found in human tumors. Intriguingly, ERK mutations are very rare in the cancer genome, although a recent paper identified ERK2 mutants as rare cancer-associated gain- and loss-of-function gene products [69].
3.4. Anticancer Agents Targeting the ERK Cascade
These findings propelled the development of highly selective and potent small-molecule inhibitors of kinases of the RAS-effector signaling cascades, including RAF, MEK, or ERK inhibitors for cancer therapeutics [70]. RAF monomer (BRAF (V600)) inhibitors (such as vemurafenib, dabrafenib, and encorafenib) are clinically approved for the treatment of BRAFV600 mutant melanoma (Figure 1) [33]. Given the critical role of MEK in ERK phosphorylation/activation, acting downstream of RAS and RAF, it has become an attractive drug target providing the promise of a novel therapy for RAS- and RAF-driven tumors. Indeed, two MEK inhibitors (trametinib (GSK) and cobimetinib), either as single agents or together with the RAF inhibitors (vemurafenib (Zelboraf; Genentech/Plexxikon) and dabrafenib (Tafinlar; GlaxoSmithKline)), have been approved for the treatment of patients with metastatic melanoma expressing the mutated RAF paralogue BRAF(V600E) (Figure 1). Despite intensive efforts by the investigators to search for an effective RAS inhibitor for more than three decades, oncogenic RAS mutants have been considered “undruggable” until recently due to their high affinity with GTP and the lack of a proper binding pocket for small molecule inhibitor binding [71]. It has been demonstrated that KRAS G12C could be targeted by using a covalent small molecule that docks in the switch II pocket and cross-links with Cys12 [72]. Currently, several potent covalent inhibitors against mutant-specific RAS (KRAS-G12C) are being developed and some of these molecules are in clinical trials. In contrast to the advanced development and clinical evaluation of the above-mentioned RAF and MEK inhibitors, only limited progress has been made in the ERK inhibitor development and they are still undergoing clinical trials. In contrast to RAF inhibitors, these inhibitors have a lower therapeutic index since they strongly inhibit this signaling pathway in normal cells [72,73].
Therefore, the close association of the RAS–ERK signaling activation with various clinical tumors together with the findings that the blockade of ERK signaling by specific inhibitors of RAF or MEK suppress tumor proliferation at the clinical level are consistent with the well-recognized concept that RAS–ERK pathway activation exerts anti-apoptotic functions.
4. Role for the ERK Cascade in Apoptosis
Notably, contrary to the well-recognized anti-apoptotic role of ERK1/2, accumulating evidence showed that ERK1/2 can be also pro-apoptotic (Figure 2). This anti-survival aspect of ERK1/2 has been observed in several physiological contexts, including neuronal cell death [74], brain injury following ischemia [75,76], development, tumor-suppressive mechanisms, and/or tumor responses to chemotherapy [77,78,79,80,81].
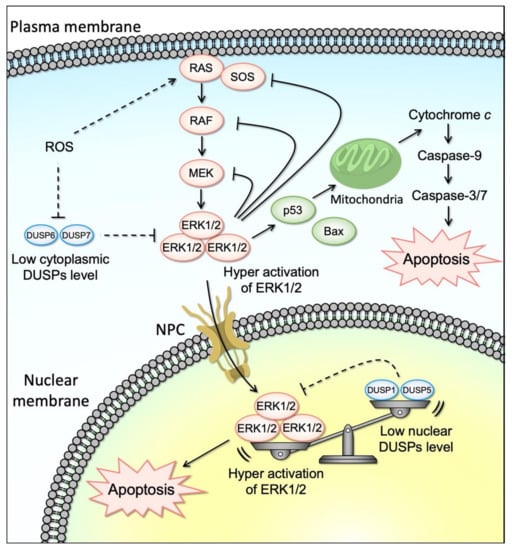
Figure 2.
Activation of ERK1/2 upstream factors or down-regulation of DUSP6/7 by various stimuli (such as ROS) induces the activation of ERK1/2 in the cytoplasm. Activated cytoplasmic ERK1/2 induces the mitochondrial apoptosis or is translocated to the nucleus through the NPC (nuclear pore complex). Down-regulation of DUSP1/5 by the stimuli (such as ROS) also induces the activation of ERK1/2 in the nucleus. Consequently, either down-regulation of cytoplasmic or nuclear DUSPs leads to the accumulation of activated ERK1/2 in the nucleus. Sustained ERK1/2 activation above a certain threshold activates specific pro-apoptotic targets and induces apoptosis.
Here, we will focus on the possible mechanisms of the pro-apoptotic role of the ERK pathway in cancers by listing and analyzing the major compounds/stimuli that induce apoptosis by stimulating ERK1/2. We will also provide future perspectives on the potential and limitations of these agents as a novel cancer therapeutic approach.
4.1. Compounds/Stimuli That Induce Apoptosis through ERK Activation
Various agents and stimuli, which have been reported to induce apoptosis through activation of the ERK pathway are summarized (Table 1). The repertoire of the stimuli/compounds contains a broad spectrum ranging from H2O2, nitric oxide (donors), UV, γ-irradiation, phorbol esters, glutamate, cadmium, zinc, bufalin, indomethacin, to antitumor agents, including cisplatin, doxorubicin, etoposide, carboplatin, and taxol. The list of the compounds or stimuli that can induce cancer cell death by activating ERK also includes various natural compounds including resveratrol, chelerythrine, shikonin, betulinic acid, baicalein, icaritin, quercetin, piperlongumine, kaempferol, and recently identified ACA-28 and its lead derivatives.

Table 1.
List of the compounds that induce ERK activation-dependent apoptosis.
In most cases, the demonstration of whether the apoptosis is induced through ERK activation is experimentally investigated either by the MEK inhibitor (PD98059, U0126), ERK silencing, or the expression of the dominant-negative MEK. A paper used both MEK inhibitor and DUSP 5/6 overexpression for ERK inhibition and apoptosis cancellation [82]. Special caution should be taken when considering the results in Table 1 that use old generation inhibitors of MEK that are not as specific as the recently developed drugs were used. For example, it was shown that PD98059 can directly affect mitochondria and induce the production of reactive oxygen species [83]. Moreover, U0126 was shown to act as a direct ROS scavenger [84], implicating potential side-effects of these inhibitors on ROS signaling. In contrast, Wan et al., confirmed that U0126 did not affect ROS increase induced by 2’-dihydroxy-4,4’-dimethoxydihydrochalcone. Thus, the effect of MEK inhibitors on ROS production remains controversial. Furthermore, future work will be needed if SiRNA was used to reduce ERK activity without experiments to rule out the possible off-target effects. Some recent papers used PD184352, which is a novel, potent, and highly selective ATP non-competitive MEK1/2 inhibitor to confirm that the apoptosis is induced through ERK activation [85,86]. Future work with rigorous confirmation will further validate the properties of these compounds to induce ERK activation-dependent apoptosis.
4.2. Mechanisms of ERK-Induced Apoptosis
Depending on the cell types and the nature of the stimuli, ERK activation is associated with the intrinsic apoptotic pathway characterized by the release of cytochrome c from the mitochondria and activation of initiator caspase-9, or with the extrinsic apoptotic pathway, which relies on the activation of an initiator caspase-8.
In vivo studies showed that ERK1/2 kinases act upstream of caspase-3 in cisplatin-induced cell death in renal cells [87,182]. Furthermore, cisplatin-induced apoptosis required ERK activation to induce mitochondria membrane depolarization and cytochrome c release, and caspase-3 activation. Several studies suggested that ERK may act on mitochondria to induce cytochrome c release through a pro-apoptotic molecule Bax and/or p53. Cisplatin or H2O2-induced expression of Bax and p53 were decreased by the ERK pathway inhibition [183,184,185].
ERK also regulates the extrinsic pathway and mediates apoptosis induction. Inhibition of ERK reduced TNF-α expression, caspase-3 activation, and apoptosis in renal tissues [186]. Furthermore, inhibition of ERK activity by a MEK inhibitor U0126 abolished the increase in IL-1β mRNA induced by brain ischemia [187]. Thus, ERK may promote the expression of death ligand thereby mediating apoptosis via the extrinsic pathway.
In addition, ERK can promote cell death by suppressing the survival signaling pathways, including PI3K/AKT signaling [188,189]. Shinha et al. reported that ERK activation in response to survival factor deprivation may lead to cell death via the suppression of the survival mediated by the Akt signaling pathway [188]. Similar cross-talk between ERK and Akt signaling pathways, the two major signaling pathways relevant for survival as well as tumorigenesis was reported by various reports [190].
4.3. DNA Damage and p53 in the ERK Activation-Induced Apoptosis
DNA-damaging agents/stimuli are one of the largest groups that induce apoptosis via ERK activation. Chemotherapeutic agents, such as etoposide, adriamycin, platinum compounds (cisplatin), or ionizing and ultraviolet irradiation activate ERK1/2 and induced apoptosis in primary cells and various cancer cell lines [191,192]. Inhibition of ERK1/2 activity by the MEK inhibitor PD98059 attenuates apoptosis induced by DNA-damaging agents, while forced activation of ERK by the constitutively active MEK1 mutant sensitizes cells to DNA damage-induced apoptosis [191]. DNA damage is well known to activate p53 via ATM and ATR, two members of the PI3K family. Tumor protein p53 is a nuclear transcription factor that regulates the expression of a wide variety of genes involved in apoptosis, growth arrest, or senescence in response to genotoxic or cellular stresses. DNA damage induces a rapid increase in p53 protein levels and a subsequent increase in its transcriptional activity [193]. This then leads to the transcriptional activation of a number of genes whose products trigger cell-cycle arrest, DNA repair, or apoptosis, including pro-apoptotic Bcl-2 family members. The p53-mediated apoptosis pathway is one of the major apoptosis signaling pathways involving the stimulation of both the extrinsic and intrinsic pathways. p53 transcriptionally upregulates pro-apoptotic genes, Bax, Noxa, PUMA (p53-Upregulated Modulator of Apoptosis), and BID [194]. p53 has also been shown to transcriptionally repress the anti-apoptotic genes Bcl2, Bcl-xL, and Survivin [195,196]. Furthermore, p53 stimulates apoptosis via extrinsic pathway by upregulating mRNA encoding Fas and DR5 in response to DNA damage, thereby promoting cell death through caspase-3 [194].
Then, what is the functional connection between ERK activation and p53 in mediating DNA damage-induced apoptosis? Some studies showed that apoptosis induced by various DNA damaging agents correlates with p53 upregulation and modulation of Bcl-2 family protein in a MEK-dependent manner, suggesting that ERK activation stimulates p53 transactivation [107,114,142]. Consistently, the upregulation/accumulation of p53 by ERK activation is associated with p53 phosphorylation by ERK at Ser15 (although this Serine residue is not followed by a proline and thus does not represent a typical consensus phosphorylation site for ERK), which stabilizes p53 by inhibiting the association with Mdm2, a ubiquitin ligase for p53 [197]. In addition, phosphorylation of the Thr55 residue of p53 by ERK2 is implicated in the doxorubicin-induced p53 activation and cell death in MCF-7 breast cancer cells [198]. Furthermore, manipulating p53 activity or expression by various approaches, including p53 siRNA, a dominant-negative p53 mutant, p53 inhibitor Pifithrin-alpha, p53-deficient cells, also showed that ERK-activated apoptosis requires p53 expression [107,114,142,152,154,199]. These reports support the role of ERK in p53-mediated apoptosis in cancer cells.
However, Tang et al., reported that etoposide-induced ERK activation takes place downstream of ATM and is independent of p53 [191], based on the findings that the MEK inhibitor failed to abolish p53 stabilization upon DNA damage and that the ERK activation in response to etoposide requires ATM but not p53. Similarly, RAS-mediated activation of ERK by cisplatin induces cell death independently of p53 in osteosarcoma and neuroblastoma cell lines, and activated ERK has been implicated in doxorubicin-mediated cell death, independent of p53 status [20,21].
Conversely, p53 may act as one of the upstream regulators of ERK activation for the induction of apoptosis in carboplatin-treated cervical cancer cells [200]. The authors reported that a requirement of ERK activation in carboplatin-induced apoptosis in SiHa and CaSki cells and that abrogation of p53 transactivation activity by a p53 inhibitor (Pifithrin-α) or dominant-negative mutant of p53 resulted in a decrease in activation of ERK in carboplatin-treated cells [200]. Another example of p53-mediated ERK activation includes ERK1/2 activation mediated by the Nutlin-3-induced mitochondrial translocation of p53. Nutlin-3, an MDM2 antagonist, induced the phosphorylation of EGFR, (MEK)1/2, and ERK1/2 in U2OS human osteosarcoma cells, which was canceled by p53 silencing [201].
Moreover, Heo et al. reported that DNA damage induced both the phosphorylation of p53 at Ser 15 and ERK [202]. Inhibition of p53 by a dominant-negative mutant or in p53−/− fibroblast cells abolished ERK phosphorylation and ERK inhibitor prevented p53 phosphorylation, indicating that phosphorylations of p53 and phospho-ERK are interdependent with each other. These results indicate that ATM mediates interdependent activation of p53 and ERK through the formation of a ternary complex between phopsho-p53 and phospho-ERK in response to DNA damage to cause growth arrest. Thus, ERK’s role in phosphorylating p53 in response to DNA-damaging drugs seems to be dependent on the cell line being treated.
Collectively, ERK activation plays an important role in DNA damage-induced apoptosis dependently or independently of p53.
4.4. ROS and ERK Activation-Induced Cell Death
Another important signature shared by many stimuli/compounds capable of inducing ERK-mediated apoptosis is their property of simulating ROS. These include chemical oxidants such as H2O2, and nitric oxide donors, or heat, cisplatin, etoposide, doxorubicin, indomethacin, cearoin, icaritin, triptolide, and piperlongumine (Table 1). Especially, a close association between DNA damage-inducing stimuli/compounds and ROS production was reported [203]. In most cases, the demonstration of the implication of ROS in the mechanism of ERK-induced apoptosis is experimentally performed with the use of different ROS inhibitors [17,188,204,205,206,207,208,209,210].
For example, triptolide-treatment induced ERK activation in a dose- and time-dependent manner in MDA-MB-231 breast cancer cells [37]. ERK activation was crucial in mediating triptolide-induced caspase-dependent apoptosis. Triptolide-induced ERK activation modulated the expression of the Bcl-2 protein family member Bax but was not involved in the downregulation of Bcl-xL expression. ROS act upstream of ERK activation, as the MEK inhibitor U0126 did not inhibit the generation of ROS induced by triptolide treatment. Sensitization of anti-tumor agents by a compound associated with ROS-mediated ERK activation was also reported [211]. Curcumin potentiates antitumor activity of cisplatin in bladder cancer cell lines via ROS-mediated activation of ERK1/2 [114]. Importantly, NAC (ROS scavenger) and U0126 (MEK inhibitor) inhibited upregulation of p53 and apoptosis as well as downregulation of survival proteins induced by co-treatment with curcumin plus cisplatin. Thus, co-treatment with curcumin and cisplatin synergistically induced apoptosis through ROS-mediated activation of ERK1/2 in bladder cancer.
It has long been recognized that an increase of ROS can induce or mediate the activation of the MAPK pathways, including ERK MAPK [212], and various cellular stimuli that induce ROS production can activate MAPK pathways in multiple cell types [213]. Although the precise mechanism(s) for the ROS-mediated ERK activation remains unknown, ROS can activate ERK signaling at least in two ways. Namely, activation of EGFRs, and deactivation of MAPK phosphatases (MKPs or DUSPs). ROS can specifically and reversibly react with proteins, altering their activity, localization, and half-life [214]. It is most likely that the signaling molecule action of ROS may depend upon their ability to react with the cysteine residues of a certain group of target proteins. ROS can rapidly oxidize the highly reactive thiol groups to form a disulfide bond.
Indeed, many growth factors such as EGFRs or PDGFRs, as well as MKPs/DUSPs, have cysteine-rich motifs, and they can be targets of ROS. EGFRs lie upstream of ERK and are most commonly activated through ligand-induced dimerization or oligomerization and are generally involved in the regulation of cell proliferation, survival, migration, and differentiation [215]. Interestingly, ROS have been shown to activate EGFRs even in the absence of its ligand [216], which is referred to as “receptor transactivation”. Moreover, it has been reported that oxidation of the upstream activators of ERK MAPK, including RAS, RAF, or PKC can be activated by ROS [207,208,217,218,219,220,221]. Thus, ROS can activate ERK signaling via oxidation/activation of the upstream regulators of ERK MAPK.
By contrast, the function of MKPs/DUSPs is to dephosphorylate and, therefore, inactivate ERKs, p38 MAPKs, and JNKs. Hydrogen peroxide activates MAPK pathways, which leads to the induction of MKP-1 expression, and MKP-1 expression correlates with the inactivation of MAPK pathways [222]. Moreover, overexpression of MKP-1 renders MCF-7 cells resistant to hydrogen peroxide-induced cell death by inhibiting MAPK activation, while loss of MKP-1 by downregulation via small interfering RNA (siRNA) or deletion of the MKP-1 gene sensitizes cells to hydrogen peroxide-induced cell death through MAPK activation. This suggests that the expression levels of the MKP-1 affect MAPK activation and the sensitivity of the cells to apoptosis induced by hydrogen peroxide.
Importantly, oxidation of catalytic cysteine within the active site of DUSPs inactivates phosphatase activities of DUSPs as well as various tyrosine phosphatases [223,224]. In addition, intracellular ROS accumulation activates ERK, which triggers proteasomal degradation of DUSPs. Indeed, ROS have been shown to inhibit phosphatase activities of ERK-directed phosphatases, DUSP1 and DUSP6, by oxidation of their catalytic Cys residues. Furthermore, intracellular ROS accumulation such as hydrogen peroxide causes DUSP6 phosphorylation on Ser159 and Ser197 residues, leading to ubiquitination and degradation of DUSP6 in ovarian cancer cells. This oxidation and/or degradation-mediated inactivation/downregulation of DUSPs might contribute to the sustained ERK signaling activity, a hallmark of ERK-dependent apoptosis, which will be described later.
4.5. DUSP and Negative Regulators of the ERK Cascade in the ERK Activation-Induced Cell Death
The importance of DUSP regulation in the ERK activation-mediated cell death was reported by Sugiura’s lab following their discovery of a novel ERK signaling modulator ACA-28 and its lead compound ACAGT-007. These compounds exhibited the property of inducing ERK-dependent apoptosis in human melanoma cells [25,26]. ACA-28 was isolated through a chemical genetic approach to search for compounds that can modulate ERK MAPK signaling using the fission yeast model system based on the counteractive genetic interaction between calcineurin and MAPK Pmk1 in S. pombe [26,225]. The unique feature of ACA-28 was to induce ERK-dependent apoptosis specifically in ERK-active human melanoma cell lines, but not in normal human melanocyte (NHEM). Similarly, using HER2-overexpressing A4-15 cells as a model system to recapitulate the cancerous situation with aberrantly active ERK, ACA-28 was shown to induce ERK-dependent apoptosis specifically in HER2-overexpressing cells, but not in the parental NIH/3T3 cells. Moreover, the IC50 of ACA28 against melanoma cell lines (SK-MEL-28) was much superior to that against normal cells (10.4 μM vs 5.2 μM, respectively). These results suggest that ACA-28 preferentially kills melanoma cells, and the mechanisms of the selective toxicity depend on the ERK activation status of each cell line. The analysis of DUSP6, a major ERK phosphatase in the cytosol, based on the assumption that DUSP6 regulation may be a key event to determine the ERK activation status, revealed that DUSP6 expression levels correlate with ERK activation status [27]. Namely, DUSP6 was overexpressed in HER2-overexpressing ERK-active cells as compared with NIH/3T3 cells. Furthermore, the MEK inhibitor U0126 abolished ERK activation, which led to DUSP6 downregulation. Notably, ACA-28 was shown to downregulate DUSP6 protein, at least in part, via the proteasome, and induced apoptosis in A4-15 cells by stimulating ERK phosphorylation. Consistently, DUSP6 silencing specifically induced apoptosis in ERK-active HER2 overexpressing cells, but not in NIH/3T3 cells. Thus, ACA-28 induced DUSP6 downregulation in cancer cells with highly active ERK, thereby inducing ERK hyperphosphorylation and cell death. Our study, together with the following papers showed that despite their predictive role as tumor suppressors, the ERK-inhibitory MKPs/DUSPs can be linked to tumor progression, in other words, ERK-active cancer cells are addicted to DUSPs for growth. Thus, controlling the DUSP expression can be a novel measure for cancer therapy. Consistent with the concept that DUSPs can be tumor-promoting, the MKP/DUSP inhibitor NSC 95397 reduced cell viability and anchorage-independent growth of colon cancer cell lines through inhibition of proliferation and apoptosis induction by regulating cell-cycle-related proteins and caspases [103]. Further, by using the MEK inhibitor U0126, the authors provided mechanistic evidence that the antineoplastic effects of NSC 95397 were achieved by inhibiting MKP/DUSP activity followed by ERK1/2 phosphorylation. Thus, MKP/DUSP inhibition by NSC 95397 might serve as an effective therapeutic intervention for colon cancer through regulating MKP/DUSP and ERK1/2 pathways.
Intriguingly, Unni et al. showed that synthetic lethality induced by co-expression of mutant KRAS and EGFR is mediated through hyperactivation of ERK in lung adenocarcinoma cells, implying that tumors with mutant oncogenes in the RAS pathway must retain the ERK activity to avoid toxicities that hamper tumor growth [226,227]. DUSP6 was upregulated in EGFR- or KRAS-mutant lung adenocarcinoma cells with high ERK phosphorylation levels, which enables tumor cell growth by suppressing hyperactive ERK. Consistently, knockdown or inhibition of DUSP6 elevated phosphorylated ERK and reduced the viability of lung adenocarcinoma cells with either KRAS or EGFR oncogenic mutations, indicating that cancer cells with ERK hyperactivation with oncogene activation are addicted to DUSP high-expression for their viability.
An additional example of the negative regulators of the ERK cascade in the ERK-dependent apoptosis includes Spry proteins, a family of endogenous proteins that negatively regulate the ERK signaling pathway. Intriguingly, similar to DUSP proteins, some cancer cells, including glioblastoma, are dependent on Spry proteins for their growth by adaptation, which makes Spry proteins play an onco-promoting role contrary to their original role as a negative regulator of the EGFR-activated ERK signaling pathway. The authors concluded that an antitumoral effect of SPRY2 inhibition is based on excessive activation of ERK signaling and DNA damage response, resulting in reduced cell proliferation and increased cytotoxicity, proposing SPRY2 as a promising pharmacological target in glioblastoma patients [228]. Altogether, compounds targeting negative regulators of the ERK cascade, such as DUSP6 and SPRY proteins, might offer a treatment strategy for certain cancers by inducing the toxic effects of RAS-mediated hyperactive ERK signaling.
4.6. Kinetics and Distribution of Phosphorylated ERK and Cell Death
In general, direct activation of the ERK cascade is always transient, peaking at 5–10 min after stimulation and reducing back to basal level 30–90 min thereafter. The second and third waves of activation may appear at later time points, due to autocrine loops or other ERK-required processes, such as mitosis [229]. Several compounds listed in Table 1, including H. pylori secreted protein HP1286, Etoposide, Qizhen capsule, Perfluorooctane sulfonate, Perfluorohexanesulfonate, and Cypermethrin induced ERK activation at an early stage, such as 5–15 min upon compound treatment, and U0126 abolished ERK activation and canceled apoptosis. However, various agents implicated in ERK-dependent apoptosis have also been reported to induce sustained ERK activation (Table 1). These include kaempferol, Cadmium, and Pemetrexed [17,117,123,135]. For example, Tong et al. reported that Icaritin causes sustained ERK1/2 activation and induces apoptosis in human endometrial cancer cells [23]. Icaritin treatment induced the expression of pro-apoptotic protein Bax with a concomitant decrease of Bcl-2 expression. Furthermore, icaritin induced sustained phosphorylation of ERK1/2 in Hec1A cells, and a MEK inhibitor U0126 blocked the ERK1/2 activation by icaritin and abolished the icaritin-induced growth inhibition and apoptosis. This type of long-term effects can arise from the indirect effect of other signaling pathways on the ERK activation, including the ROS-mediated oxidation and inactivation of DUSPs and other tyrosine phosphatases PP1/2A as described above (ROS and ERK Activation-induced Cell Death). It should be noted that many papers listed in Table 1 lack the information on the short-term effects of the compounds on ERK activation as the authors investigated ERK activation at time points wherein apoptosis was investigated (ex, 24 h). So, future work will be needed to acquire precise information in this regard.
Interestingly, transient versus sustained phosphorylation of ERK has been involved in the underlying mechanism to determine the anti-versus pro-apoptotic effects of the sex steroid estradiol. 17β-estradiol transiently induced ERK phosphorylation in osteocytic cells, whereas the estradiol-induced ERK phosphorylation in osteoclasts was sustained for at least 24 h [230]. Conversion of sustained ERK phosphorylation to a transient one abrogated the pro-apoptotic effect, whereas prolongation of ERK activation converted the anti-apoptotic effect of 17β-estradiol to a pro-apoptotic one. The authors also analyzed the effect of the nuclear export system using leptomycin (the exportin inhibitor) and further suggested that the length of time that phospho-ERKs are retained in the nucleus is responsible for the pro-versus anti-apoptotic effects of estrogen. This study suggested the intriguing possibility that not only the kinetics but also the subcellular distribution of phosphorylated ERK and its substrates in each compartment may be critical in determining the fate of the cells for anti- versus pro-apoptosis. This concept is consistent with the finding that sustained ERK activation alone, such as in models expressing constitutively active forms of upstream kinases, is not sufficient to promote cell death [231,232,233,234,235,236,237,238]. Similar sustained nuclear ERK activity was reported with apoptosis induced by tamoxifen, zinc, and doxorubicin [204,239,240].
In this regard, DUSPs again play key roles in determining both the kinetics and the distribution of phosphorylated ERK1/2. For example, DUSP6 not only serves as inactivating enzyme for ERK1/2, but it also serves as an anchor for inactive ERK in the cytosol and even participates in the transport of dephosphorylated ERKs from the nucleus to the cytosol [241]. In line with this, Kim et al. found that ROS produced during senescence of human primary fibroblasts inactivate the cytosolic ERK phosphatase DUSP6, resulting in cytoplasmic sequestration of active ERK.
Downregulation of DUSPs similar to what was observed with ACA-28 in cancer cell lines has been reported with camptothecin (CPT) in colon cancer [242]. Lee et al. reported that MKP1/DUSP1 inhibition and sustained ERK1/2 activation were implicated in CPT-induced human colon cancer cell death [242]. The authors found that CPT promoted nuclear accumulation of active ERK and prolonged ERK activity through inhibition of MKP1, implicating the function of the RAS/RAF/ERK pathway activation in cancer cell death. Among several colon cancer cell lines, CPT was shown to selectively increase the activation of ERK1/2 in HCT116 cells by downregulating MKP1 protein levels posttranscriptionally. Importantly, CPT-induced repression of MKP1 alters the nuclear-cytoplasmic location of activated (phosphorylated) ERK1/2 as evidenced by the nuclear accumulation of phospho-ERK1/2 upon CPT treatment. Thus, enhanced nuclear phospho-ERK1/2 levels were associated with MKP downregulation. These data imply that CPT-induced MKP1 protein degradation prevents the inactivation of phospho-ERK1/2 by nuclear MKP1, allowing ERK1/2 activity to be sustained in the nucleus (Figure 2).
An additional example of the role of DUSPs in the spatio-temporal ERK signaling activation and their implication in tumorigenesis was demonstrated by Kidger et al. [243]. DUSP5 both inactivates and anchors ERK in the nucleus. The authors reported that the MKP DUSP5 terminates ERK signaling in the nucleus but, surprisingly, promotes ERK activation in the cytoplasm by relieving feedback inhibition of upstream RAF and MEK kinase activation mediated by ERK phosphorylation of each kinase (Figure 2). Importantly, DUSP5 nuclear localization is required for its effect to control cytoplasmic ERK Phosphorylation. Paradoxically, DUSP5 facilitates oncogenic mutant BRAFV600E-driven cell proliferation and transformation, suggesting that DUSP5 plays an oncogenic role in BRAFV600E-driven cancers. Consistently, BRAFV600E expression combined with Dusp5 deletion causes ERK hyperactivation and proliferative arrest, thus indicating that DUSP5 can be a novel therapeutic for cancer therapy. Notably, DUSP5 deletion does not influence normal cell proliferation in primary MEFs. Furthermore, mice entirely lacking DUSP5 develop normally and display no obvious adult phenotype, suggesting that the physiological role of DUSP5 may only be fully apparent in the event of stress or pathological challenge.
In normal cells, the subcellular localization of ERK is also tightly regulated by scaffold proteins [244,245]. Consistently, in addition to DUSPs, several cytoplasmic ERK anchoring proteins, such as DAPK, PEA-15, and Bik, have been implicated in the mechanisms of ERK1/2-mediated cell death [238,246,247]. Activated ERK interacts with these cytoplasmic anchor proteins, which sequester active ERK1/2 in the cytoplasm. Inhibition of ERK1/2 nuclear localization impairs ERK1/2-mediated survival signals and augments the pro-apoptotic signals of DAPK by ERK phosphorylation of the cytoplasmic DAPK. Consistently, mouse testes deficient in PEA-15 exhibited nuclear ERK activation [248]. Cytoplasmic sequestration of active ERK by binding to PEA-15 has been reported to induce autophagy, whereas sustained cytoplasmic ERK activity induces senescence in human primary fibroblasts. Thus, anchoring proteins for ERK, including docking phosphatases can be involved in the mechanisms of ERK-dependent cell death by affecting the distribution of phosphorylated ERK1/2 either in the cytosol or the nucleus, thereby determining the cell fate for various modes of cell death.
4.7. The Cellular Threshold for ERK-Dependent Cell Death
In addition to the kinetics of ERK activation, Park’s lab provided evidence regarding the threshold of active ERK that determines the ERK-dependent apoptosis versus growth arrest [24]. The authors reported that ERK1/2 overexpression switches Raf-induced growth arrest responses to caspase-dependent apoptotic death responses. Upon titration of active ERK levels by the MEK1/2 inhibitor AZD6244 reverts the death responses to growth arrest responses, suggesting that a cellular threshold for active ERK1/2 levels exists and affects the cell fate between death and growth arrest. Interestingly, kinase activity is necessary for ERK1/2 to mediate death signaling. Therefore, ERK1/2 mediates death signaling depending on kinase activity and specific physical interactions.
Consistent with these findings, our lab reported the unique feature of ACA-28 to induce apoptosis preferentially in ERK-active cancer cells, including melanoma cell lines (SK-MEL-28, SK-MEL-2, and MeWO) or HER2-overexpressing cells, but not in normal melanocyte or NIH/3T3 cells, respectively. These results suggest that cancerous situations with higher ERK phosphorylation levels were favorable for ACA-28 to induce apoptosis, consistent with a cellular threshold hypothesis. Thus, the high-ERK activation status in each cell may be a critical determinant/prerequisite for ERK-dependent cell death.
4.8. ERK Activation and Other Types of Cell Death
In this review, we focused on the pro-apoptotic function of the ERK signaling pathway, especially in the context of cancer cells. Recent studies highlighted the importance of the ERK cascade in various types of cell death other than apoptosis, including autophagy-dependent cell death, necroptosis, ferroptosis, pyroptosis, and parthanatos [249]. Importantly, ERK activation can also mediate other types of cell death or growth inhibitory mechanisms, including autophagy or oncogene-induced senescence [250,251,252,253]. Autophagy is an evolutionarily conserved catabolic process where cytoplasmic components and intracellular organelles are sequestered into autophagosomes and transferred to the lysosome for degradation [254]. Autophagy was initially identified as a cell survival mechanism as it constitutes an adaptive response to different kinds of stress by which the cells avoid cell death. However, in some settings, it can also contribute to a form of cell death, described as type II programmed cell death. In cancer settings, autophagy can also perform a dual role, protecting cell survival or contributing to cell death. In some tumor cells, autophagy can be a protective response enabling survival against anticancer treatments through blockade of the apoptotic pathway. Meanwhile, other tumor cells undergo autophagic cell death in response to cancer therapies [255].
Interestingly, some of the compounds executing ERK-dependent cell death (Table 1) simultaneously induce apoptosis and autophagy. These include cisplatin, ginsenoside Rg5 (Rg5), triptolide, morusine, scutellarin, and derrone (Table 1). For example, morusin induced a pro-apoptotic effect through the intrinsic mitochondrial apoptotic pathway and pro-autophagic effect as evidenced by the cytochrome c release and caspase-3 cleavage, as well as the increased level of LC3-II and decreased level of SQSTM1/p62, respectively [95]. ROS-mediated ERK signaling activation may constitute an underlying mechanism functionally connecting apoptosis and autophagy since the NAC treatment canceled both types of cell death associated with several compounds such as Rg5, moursine, physalin B, and derrone. For example, physalin B, a naturally occurring secosteroid, induces mitochondria-mediated ROS generation, which leads to the ubiquitin-proteasome pathway inhibition and incomplete non-canonical autophagy response [118]. Autophagosome/lysosome fusion is blocked due to the inhibition of functions of microtubules and F-actin microfilaments, leading to autophagy substrates accumulation which accelerates apoptosis. Besides, MAPK pathways are sustainedly activated and partially mediate autophagy response and apoptosis in physalin B-treated cells. Similarly, derrone, one of the major compounds of unripe fruits, induces autophagic cell death through induction of ROS and sustained ERK activation in the non-small cell lung cancer cell line [97]. In addition, activation of ERK-p53 and ERK-mediated phosphorylation of Bcl-2 is involved in autophagic cell death in several cancer cell lines, thus indicating that both ROS and p53 can also contribute to simultaneous induction of ERK-induced apoptosis and autophagic cell death.
What is the connection between autophagy and apoptosis? In the case of Rg5, suppression of apoptosis weakens Rg5-induced autophagy, while inhibition of autophagy attenuates Rg5-induced apoptosis in gastric cancer, indicating that Rg5 could simultaneously induce autophagic and apoptotic cell death in gastric cancer and that the two processes stimulate each other via activating ROS-mediated MAPK pathways [96]. Meanwhile, curcumin treatment induced autophagy via the ERK1/2 signaling pathway activation to protect chondrocytes from apoptosis [256], thus indicating the complicated interaction between apoptosis and autophagy, which can either collaborate or compete with each other.
It should be mentioned that cytosolic sequestration of phosphorylated ERK by PEA-15 can promote autophagy [257], again suggesting the importance of controlling spatial dynamics of ERK in the cell death induced by ERK activation.
5. Conclusions and Perspectives: Promises and Challenges for ERK-Induced Apoptosis in Cancer
ERK has largely been considered a survival signaling pathway. However, growing literature, as shown in this review, indicates that the ERK signaling pathway mediates apoptosis induced by various stimuli in different settings. Although the molecular mechanisms by which ERK mediates apoptosis remain largely elusive, several characteristics mediators of ERK activation-induced apoptosis, such as ROS and DNA damage as well as p53, are becoming clear. In addition, the importance of the spatio-temporal dynamics of ERK activation and the emerging role of the anchor proteins for ERK1/2 were highlighted. Especially, DUSPs can play critical roles in determining both the kinetics and the subcellular distribution of the ERK activation. For example, ROS can stimulate ERK signaling via activation of upstream activators and inactivation of catalytic activity of DUSPs, which leads to sustained ERK activation due to the lack of negative feedback responses elicited by DUSPs. Furthermore, compounds like ACA-28 can promote downregulation of DUSPs, which is responsible for the selective apoptosis induction in cancer cells harboring the oncogenic mutation(s) of the ERK signaling pathway. In addition to the role of ERK inactivating enzymes, DUSPs as anchor proteins for ERK can also impact the subcellular distribution of phosphorylated ERK. Thus, DUSPs can play key roles in determining ERK functions for survival or apoptosis by affecting both the kinetics/threshold (duration and magnitude) as well as the compartments of ERK activation. Sustained ERK activity in the cytoplasm might promote senescence or autophagy, whereas the sustained nuclear activity of ERK may lead to apoptosis. These distinct physiological ERK functions can be defined by ERK substrates in each compartment and the kinetics of ERK activation. For example, inhibition of ERK translocation to the nucleus hampers its access to the transcription factor substrates thereby blocking the proliferative response. Cytosolic ERK1/2, besides inhibiting the survival and proliferative signals in the nucleus, may further potentiate the catalytic activities of some pro-apoptotic proteins in the cytoplasm. Moreover, strong ERK activation above a certain threshold might activate specific pro-apoptotic targets. Similarly, sustained activation of ERK could activate additional targets that are not usually activated by transient ERK signaling. Elucidating the downstream targets of ERK1/2 responsible for the execution of distinct modes of cell death as well as the molecular mechanisms of the cellular threshold for ERK to exert apoptosis or survival will be explored in future studies.
Inhibition of the ERK cascade (in other words, targeting the pro-survival function of ERK signaling) is the mainstream strategy for the EGFR/RAS/RAF-driven tumors. Unfortunately, however, clinical responses to the ERK cascade inhibitors are variable across patients and the efficacy of these drugs is limited and temporary. For example, despite the outstanding responses obtained with BRAF inhibitors vemurafenib and dabrafenib, the majority of patients progress within a year with a median progression-free survival of fewer than 10 months. This insufficient clinical efficacy represents tumor adaptation, largely due to the tumors’ innate or adaptive resistance to the therapies aimed at blocking the pathway activity. The most frequent response to clinical therapies is acquired resistance to drugs due to the reactivation of the ERK cascade, through mechanisms including RAF dimerization and resultant upregulation of ERK signaling. To conquer the resistance associated with ERK signaling inhibitors, several combination therapies with BRAF and MEK inhibitors, or with ERK signaling inhibitors and the PI3-kinase signaling inhibitors (PI3-kinase, mTOR, AKT) have been described. However, whether the use of such inhibitors might develop the risk of increased systemic toxicity will have to be carefully assessed.
In this regard, an emerging view that ERK1/2 can promote cell death and the strategy/compound of stimulating pro-apoptotic ERK function may reveal a new window of therapeutic opportunity for targeting cancers harboring oncogenic activations of the ERK signaling pathway. For example, sustained ERK activation status is favorable to promote cell death in several cancer cell lines [23,258,259]. Furthermore, modulating ERK activation in a given subcellular compartment might facilitate cancer cell death. It should be noted that ACA-28 preferentially kills cancer cells with high ERK activity and its lead compound ACAGT-007 showed a superior activity both in terms of efficacy and selectivity against high-ERK melanoma cells in vitro. DUSPs are highly expressed in some cancer cells with high ERK activity and this differential DUSP expression may underlie the selective apoptosis induction by ACA-28 and lead derivatives based on the cancer cells’ addiction to DUSPs.
For the strategy to utilize ERK-induced apoptosis as an anti-cancer target, an important hurdle to overcome includes finding a means to achieve selective apoptosis induction in cancer cells with aberrant ERK activity while sparing the viability of normal cells. One way to address this issue would be a drug delivery system targeting cells expressing high-ERK phosphorylation levels by using high DUSP6 or SPRY proteins as a marker. This idea is based on the reports cited in this review, showing that inhibition/silencing of DUSP6 protein or SPRY2 protein is toxic to the oncogenic mutant lung cancer cells or glioblastoma cells, respectively, while it does not affect the viability of normal cells [227,228]. Furthermore, these negative regulators are highly expressed in cancer cells compared with normal cells. Thus, how to achieve drug delivery targeting cells expressing high DUSP6 or high SPRY proteins? For example, Ota et al. reported selective targeting of cancer cells by utilizing lysine-specific demethylase 1 (LSD1) to trigger the controlled release of anticancer drug tamoxifen in cancer cells, wherein LSD1 is highly expressed [260]. Conjugates of the LSD1 inhibitor PCPA were used as prodrugs to selectively release tamoxifen by LSD1 inhibition. Importantly, the pro-drug inhibited the growth of breast cancer cells by the simultaneous inhibition of LSD1 enzymatic activity and the estrogen receptor by tamoxifen without hampering growth in normal cells, wherein only a low amount of LSD1 is expressed with the almost undetectable release of tamoxifen. Thus, the selective release of tamoxifen in cancer cells utilizing the high expression of LSD1 in cancer cells as compared with the normal cells can be an excellent prototype for future study to achieve cancer-cell selective delivery of compounds such as ACA-28 derivatives to induce ERK-dependent apoptosis in cancer cells, but not in normal cells.
Author Contributions
Conceptualization, R.S. (Reiko Sugiura); methodology, R.S. (Reiko Sugiura); software, R.S. (Ryosuke Satoh) and T.T.; validation, R.S. (Reiko Sugiura), R.S. (Ryosuke Satoh) and T.T.; formal analysis, R.S. (Ryosuke Satoh) and T.T.; investigation, R.S. (Reiko Sugiura), R.S. (Ryosuke Satoh) and T.T.; resources, (R.S.) Ryosuke Sugiura; data curation, R.S. (Reiko Sugiura), R.S. (Ryosuke Satoh) and T.T.; writing—original draft preparation, R.S. (Reiko Sugiura); writing—review and editing, R.S. (Reiko Sugiura), R.S. (Ryosuke Satoh) and T.T.; visualization, R.S. (Ryosuke Satoh) and T.T.; supervision, R.S. (Reiko Sugiura); project administration, R.S. (Reiko Sugiura); funding acquisition, R.S. (Reiko Sugiura). All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Education, Culture, Sports, Science and Technology (MEXT)-Supported Program for the Strategic Research Foundation at Private Universities, 2014–2018 Grant Number JPS1411037 (R. Sugiura), JSPS KAKENHI Grant Number JP19H03376 (R. Sugiura), and a grant by the Antiaging Project for Private Universities (R. Sugiura).
Acknowledgments
We are grateful to the members of the Laboratory of Molecular Phar-macogenomics for their support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK–RAS–RAF Signaling Pathway in Cancer Therapy. Expert Opin. Ther. Target 2012, 16, 103–119. [Google Scholar] [CrossRef]
- Beeram, M.; Patnaik, A.; Rowinsky, E.K. Raf: A Strategic Target for Therapeutic Development Against Cancer. J. Clin. Oncol. 2005, 23, 6771–6790. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef]
- Stern, D.F. Keeping Tumors Out of the MAPK Fitness Zone. Cancer Discov. 2018, 8, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF–MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Kidger, A.M.; Sipthorp, J.; Cook, S.J. ERK1/2 Inhibitors: New Weapons to Inhibit the RAS-Regulated RAF-MEK1/2-ERK1/2 Pathway. Pharmacol. Therapeut. 2018, 187, 45–60. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS–ERK Signalling in Cancer: Promises and Challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Wu, P.-K.; Park, J.-I. MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Burotto, M.; Chiou, V.L.; Lee, J.; Kohn, E.C. The MAPK Pathway across Different Malignancies: A New Perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef]
- Zhuang, S.; Schnellmann, R.G. A Death-Promoting Role for Extracellular Signal-Regulated Kinase. J. Pharmacol. Exp. Ther. 2006, 319, 991–997. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J. ERK and Cell Death: Mechanisms of ERK-induced Cell Death—Apoptosis, Autophagy and Senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Rieber, M.; Rieber, M.S. Signalling Responses Linked to Betulinic Acid-Induced Apoptosis Are Antagonized by MEK Inhibitor U0126 in Adherent or 3D Spheroid Melanoma Irrespective of P53 Status. Int. J. Cancer 2006, 118, 1135–1143. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Lee, D.-H.; Jeong, J.-H.; Guo, Z.S.; Lee, Y.J. Quercetin Augments TRAIL-Induced Apoptotic Death: Involvement of the ERK Signal Transduction Pathway. Biochem. Pharmacol. 2008, 75, 1946–1958. [Google Scholar] [CrossRef]
- Randhawa, H.; Kibble, K.; Zeng, H.; Moyer, M.P.; Reindl, K.M. Activation of ERK Signaling and Induction of Colon Cancer Cell Death by Piperlongumine. Toxicol. In Vitro 2013, 27, 1626–1633. [Google Scholar] [CrossRef]
- Lee, E.-R.; Kang, Y.-J.; Kim, J.-H.; Lee, H.T.; Cho, S.-G. Modulation of Apoptosis in HaCaT Keratinocytes via Differential Regulation of ERK Signaling Pathway by Flavonoids. J. Biological. Chem. 2005, 280, 31498–31507. [Google Scholar] [CrossRef][Green Version]
- Radaszkiewicz, K.A.; Beckerová, D.; Woloszczuková, L.; Radaszkiewicz, T.W.; Lesáková, P.; Blanářová, O.V.; Kubala, L.; Humpolíček, P.; Pachernik, J. 12-O-Tetradecanoylphorbol-13-Acetate Increases Cardiomyogenesis through PKC/ERK Signaling. Sci. Rep.-UK 2020, 10, 15922. [Google Scholar] [CrossRef]
- Lee, K.-I.; Su, C.-C.; Yang, C.-Y.; Hung, D.-Z.; Lin, C.-T.; Lu, T.-H.; Liu, S.-H.; Huang, C.-F. Etoposide Induces Pancreatic β-Cells Cytotoxicity via the JNK/ERK/GSK-3 Signaling-Mediated Mitochondria-Dependent Apoptosis Pathway. Toxicol. In Vitro 2016, 36, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Chouhan, S.; Singh, S.; Chhipa, R.R.; Ajay, A.K.; Bhat, M.K. Constitutively Activated ERK Sensitizes Cancer Cells to Doxorubicin: Involvement of P53-EGFR-ERK Pathway. J. Biosci. 2017, 42, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Woessmann, W.; Chen, X.; Borkhardt, A. Ras-Mediated Activation of ERK by Cisplatin Induces Cell Death Independently of P53 in Osteosarcoma and Neuroblastoma Cell Lines. Cancer Chemoth. Pharm. 2002, 50, 397–404. [Google Scholar] [CrossRef]
- Martin, P.; Pognonec, P. ERK and Cell Death: Cadmium Toxicity, Sustained ERK Activation and Cell Death. FEBS J. 2010, 277, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.-S.; Zhang, Q.-H.; Huang, X.; Fu, X.-Q.; Qi, S.-T.; Wang, Y.-P.; Hou, Y.; Sheng, J.; Sun, Q.-Y. Icaritin Causes Sustained ERK1/2 Activation and Induces Apoptosis in Human Endometrial Cancer Cells. PLoS ONE 2011, 6, e16781. [Google Scholar] [CrossRef]
- Hong, S.-K.; Wu, P.-K.; Park, J.-I. A Cellular Threshold for Active ERK1/2 Levels Determines Raf/MEK/ERK-Mediated Growth Arrest versus Death Responses. Cell Signal. 2018, 42, 11–20. [Google Scholar] [CrossRef]
- Satoh, R.; Hamada, N.; Yamada, A.; Kanda, Y.; Ishikawa, F.; Takasaki, T.; Tanabe, G.; Sugiura, R. Discovery of New Benzhydrol Biscarbonate Esters as Potent and Selective Apoptosis Inducers of Human Melanomas Bearing the Activated ERK Pathway: SAR Studies on an ERK MAPK Signaling Modulator, ACA-28. Bioorg. Chem. 2020, 103, 104137. [Google Scholar] [CrossRef] [PubMed]
- Satoh, R.; Hagihara, K.; Matsuura, K.; Manse, Y.; Kita, A.; Kunoh, T.; Masuko, T.; Moriyama, M.; Moriyama, H.; Tanabe, G.; et al. Identification of ACA-28, a 1’-acetoxychavicol Acetate Analogue Compound, as a Novel Modulator of ERK MAPK Signaling, Which Preferentially Kills Human Melanoma Cells. Genes Cells 2017, 22, 608–618. [Google Scholar] [CrossRef]
- Kanda, Y.; Mizuno, A.; Takasaki, T.; Satoh, R.; Hagihara, K.; Masuko, T.; Endo, Y.; Tanabe, G.; Sugiura, R. Downregulation of DUSP6, a Negative Regulator of Oncogenic ERK Signaling, by ACA-28 Induces Apoptosis in NIH/3T3 Cells Overexpressing HER2/ErbB2. Genes Cells Devoted Mol. Cell Mech. 2020, 26, 109–116. [Google Scholar] [CrossRef]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK Signalling: A Master Regulator of Cell Behaviour, Life and Fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Guo, Y.-J.; Pan, W.-W.; Liu, S.-B.; Shen, Z.-F.; Xu, Y.; Hu, L.-L. ERK/MAPK Signalling Pathway and Tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Roskoski, R. ERK1/2 MAP Kinases: Structure, Function, and Regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Balmanno, K.; Cook, S.J. Tumour Cell Survival Signalling by the ERK1/2 Pathway. Cell Death Differ. 2009, 16, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Pouyssegur, J. Targeting the ERK Signaling Pathway in Cancer Therapy. Ann. Med. 2006, 38, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Ng, D.C.H.; Bogoyevitch, M.A. The Mechanism of Heat Shock Activation of ERK Mitogen-Activated Protein Kinases in the Interleukin 3-Dependent ProB Cell Line BaF3. J. Biol. Chem. 2000, 275, 40856–40866. [Google Scholar] [CrossRef]
- Harris, D.; Bonfil, D.; CHuderland, D.; Kraus, S.; Seger, R.; Naor, Z. Activation of MAPK Cascades by GnRH: ERK and Jun N-Terminal Kinase Are Involved in Basal and GnRH-Stimulated Activity of the Glycoprotein Hormone LHβ-Subunit Promoter. Endocrinology 2002, 143, 1018–1025. [Google Scholar] [CrossRef]
- Tan, B.-J.; Chiu, G.N.C. Role of Oxidative Stress, Endoplasmic Reticulum Stress and ERK Activation in Triptolide-Induced Apoptosis. Int. J. Oncol. 2013, 42, 1605–1612. [Google Scholar] [CrossRef]
- Saud, K.; Herrera-Molina, R.; Bernhardi, R.V. Pro- and Anti-Inflammatory Cytokines Regulate the ERK Pathway: Implication of the Timing for the Activation of Microglial Cells. Neurotox. Res. 2005, 8, 277–287. [Google Scholar] [CrossRef]
- Rubinfeld, H.; Seger, R. The ERK Cascade: A Prototype of MAPK Signaling. Mol. Biotechnol. 2005, 31, 151–174. [Google Scholar] [CrossRef]
- Endo, T. Dominant-Negative Antagonists of the Ras–ERK Pathway: DA-Raf and Its Related Proteins Generated by Alternative Splicing of Raf. Exp. Cell Res. 2020, 387, 111775. [Google Scholar] [CrossRef]
- Wang, C.; Fung, G.; Deng, H.; Jagdeo, J.; Mohamud, Y.; Xue, Y.C.; Jan, E.; Hirota, J.A.; Luo, H. NLRP3 Deficiency Exacerbates Enterovirus Infection in Mice. FASEB J. 2019, 33, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Maik-Rachline, G.; Yadav, S.S.; Hanoch, T.; Seger, R. Mitotic HOOK3 Phosphorylation by ERK1c Drives Microtubule-Dependent Golgi Destabilization and Fragmentation. Iscience 2021, 24, 102670. [Google Scholar] [CrossRef]
- Hauge, C.; Frödin, M. RSK and MSK in MAP Kinase Signalling. J. Cell Sci. 2006, 119, 3021–3023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK Signal Pathways in the Regulation of Cell Proliferation in Mammalian Cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Chambard, J.-C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK Implication in Cell Cycle Regulation. Biochim. Et Biophys. Acta Bba-Mol. Cell Res. 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Yang, L.; Zheng, L.; Chng, W.J.; Ding, J.L. Comprehensive Analysis of ERK1/2 Substrates for Potential Combination Immunotherapies. Trends Pharmacol. Sci. 2019, 40, 897–910. [Google Scholar] [CrossRef]
- Casar, B.; Crespo, P. ERK Signals: Scaffolding Scaffolds? Front. Cell Dev. Biol. 2016, 4, 49. [Google Scholar] [CrossRef]
- Whitmarsh, A.J.; Davis, R.J. Signal Transduction by MAP Kinases: Regulation by Phosphorylation-Dependent Switches. Sci. Signal. 1999, 1999, pe1. [Google Scholar] [CrossRef]
- Mandl, M.; Slack, D.N.; Keyse, S.M. Specific Inactivation and Nuclear Anchoring of Extracellular Signal-Regulated Kinase 2 by the Inducible Dual-Specificity Protein Phosphatase DUSP5. Mol. Cell Biol. 2005, 25, 1830–1845. [Google Scholar] [CrossRef]
- Karlsson, M.; Mathers, J.; Dickinson, R.J.; Mandl, M.; Keyse, S.M. Both Nuclear-Cytoplasmic Shuttling of the Dual Specificity Phosphatase MKP-3 and Its Ability to Anchor MAP Kinase in the Cytoplasm Are Mediated by a Conserved Nuclear Export Signal. J. Biol. Chem. 2004, 279, 41882–41891. [Google Scholar] [CrossRef]
- Ekerot, M.; Stavridis, M.P.; Delavaine, L.; Mitchell, M.P.; Staples, C.; Owens, D.M.; Keenan, I.D.; Dickinson, R.J.; Storey, K.G.; Keyse, S.M. Negative-Feedback Regulation of FGF Signalling by DUSP6/MKP-3 Is Driven by ERK1/2 and Mediated by Ets Factor Binding to a Conserved Site within the DUSP6/MKP-3 Gene Promoter. Biochem. J. 2008, 412, 287–298. [Google Scholar] [CrossRef]
- Furukawa, T.; Tanji, E.; Xu, S.; Horii, A. Feedback Regulation of DUSP6 Transcription Responding to MAPK1 via ETS2 in Human Cells. Biochem. Biophys. Res. Commun. 2008, 377, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Brondello, J.-M.; Pouysségur, J.; McKenzie, F.R. Reduced MAP Kinase Phosphatase-1 Degradation After P42/P44MAPK-Dependent Phosphorylation. Science 1999, 286, 2514–2517. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-W.; Yang, J.-L. Cooperation of ERK and SCFSkp2 for MKP-1 Destruction Provides a Positive Feedback Regulation of Proliferating Signaling. J. Biol. Chem. 2006, 281, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Lake, D.; Corrêa, S.A.L.; Müller, J. Negative Feedback Regulation of the ERK1/2 MAPK Pathway. Cell Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled Demolition at the Cellular Level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Sankari, S.L.; Babu, N.A.; Rajesh, E.; Kasthuri, M. Apoptosis in Immune-Mediated Diseases. J. Pharm. Bioallied Sci. 2015, 7, S200–S202. [Google Scholar] [CrossRef]
- Mattson, M.P. Apoptosis in Neurodegenerative Disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–130. [Google Scholar] [CrossRef]
- Reed, J.C. Dysregulation of Apoptosis in Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1999, 17, 2941–2953. [Google Scholar] [CrossRef]
- Hongmei, Z.; Tobias, M. Extrinsic and Intrinsic Apoptosis Signal Pathway Review. In Apoptosis and Medicine; Tobias, M.N., Ed.; IntechOpen: London, UK, 2012; Available online: https://www.intechopen.com/chapters/38236 (accessed on 1 September 2021). [CrossRef]
- Bhartiya, D.; Singh, J. FSH–FSHR3–Stem Cells in Ovary Surface Epithelium: Basis for Adult Ovarian Biology, Failure, Aging, and Cancer. Reproduction 2015, 149, R35–R48. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Herr, D.R. G Protein-Coupled Receptor GPR19 Regulates E-Cadherin Expression and Invasion of Breast Cancer Cells. Biochim. Et Biophys. Acta Bba-Mol. Cell Res. 2017, 1864, 1318–1327. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Kwon, J.H.; Kang, S.H.; Kim, J.W.; Yang, Y.C. Increased MAPK Activity and MKP-1 Overexpression in Human Gastric Adenocarcinoma. Biochem. Biophys. Res. Commun. 1998, 250, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Sebolt-Leopold, J.S.; Dudley, D.T.; Herrera, R.; Becelaere, K.V.; Wiland, A.; Gowan, R.C.; Tecle, H.; Barrett, S.D.; Bridges, A.; Przybranowski, S.; et al. Blockade of the MAP Kinase Pathway Suppresses Growth of Colon Tumors in Vivo. Nat. Med. 1999, 5, 810–816. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS Oncogenes: Weaving a Tumorigenic Web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Baines, A.T.; Xu, D.; Der, C.J. Inhibition of Ras for Cancer Treatment: The Search Continues. Future Med. Chem. 2011, 3, 1787–1808. [Google Scholar] [CrossRef]
- Brenan, L.; Andreev, A.; Cohen, O.; Pantel, S.; Kamburov, A.; Cacchiarelli, D.; Persky, N.S.; Zhu, C.; Bagul, M.; Goetz, E.M.; et al. Phenotypic Characterization of a Comprehensive Set of MAPK1 /ERK2 Missense Mutants. Cell Rep. 2016, 17, 1171–1183. [Google Scholar] [CrossRef]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK Pathway, in Cancer Therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic Targeting of RAS: New Hope for Drugging the “Undruggable”. Biochim. Et Biophys. Acta Bba-Mol. Cell Res. 2020, 1867, 118570. [Google Scholar] [CrossRef]
- Nissan, M.H.; Rosen, N.; Solit, D.B. ERK Pathway Inhibitors: How Low Should We Go? Cancer Discov. 2013, 3, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Smalley, I.; Smalley, K.S.M. ERK Inhibition: A New Front in the War against MAPK Pathway-Driven Cancers? Cancer Discov. 2018, 8, 140–142. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.C.; Slack, R.S. Emerging Role for ERK as a Key Regulator of Neuronal Apoptosis. Sci. Stke Signal. Transduct. Knowl. Environ. 2004, 2004, PE45. [Google Scholar] [CrossRef]
- Subramaniam, S.; Unsicker, K. ERK and Cell Death: ERK1/2 in Neuronal Death: ERK1/2 in Neuronal Death. FEBS J. 2009, 277, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Levinthal, D.J.; Kulich, S.M.; Chalovich, E.M.; DeFranco, D.B. Oxidative Neuronal Injury. The Dark Side of ERK1/2. Eur. J. Biochem. FEBS 2004, 271, 2060–2066. [Google Scholar] [CrossRef]
- Kulich, S.M.; Chu, C.T. Sustained Extracellular Signal-Regulated Kinase Activation by 6-Hydroxydopamine: Implications for Parkinson’s Disease: Sustained ERK Activation by 6-Hydroxydopamine. J. Neurochem. 2001, 77, 1058–1066. [Google Scholar] [CrossRef]
- Stanciu, M.; Wang, Y.; Kentor, R.; Burke, N.; Watkins, S.; Kress, G.; Reynolds, I.; Klann, E.; Angiolieri, M.R.; Johnson, J.W.; et al. Persistent Activation of ERK Contributes to Glutamate-Induced Oxidative Toxicity in a Neuronal Cell Line and Primary Cortical Neuron Cultures. J. Biol. Chem. 2000, 275, 12200–12206. [Google Scholar] [CrossRef]
- Chambon, J.-P.; Soule, J.; Pomies, P.; Fort, P.; Sahuquet, A.; Alexandre, D.; Mangeat, P.-H.; Baghdiguian, S. Tail Regression in Ciona Intestinalis (Prochordate) Involves a Caspase-Dependent Apoptosis Event Associated with ERK Activation. Dev. Camb. Engl. 2002, 129, 3105–3114. [Google Scholar]
- Kawakami, Y.; Rodríguez-León, J.; Koth, C.M.; Büscher, D.; Itoh, T.; Raya, Á.; Ng, J.K.; Esteban, C.R.; Takahashi, S.; Henrique, D.; et al. MKP3 Mediates the Cellular Response to FGF8 Signalling in the Vertebrate Limb. Nat. Cell Biol. 2003, 5, 513–519. [Google Scholar] [CrossRef]
- Deschênes-Simard, X.; Kottakis, F.; Meloche, S.; Ferbeyre, G. ERKs in Cancer: Friends or Foes? Cancer Res. 2014, 74, 412–419. [Google Scholar] [CrossRef]
- Szydlowska, K.; Gozdz, A.; Dabrowski, M.; Zawadzka, M.; Kaminska, B. Prolonged Activation of ERK Triggers Glutamate-Induced Apoptosis of Astrocytes: Neuroprotective Effect of FK506. J. Neurochem. 2010, 113, 904–918. [Google Scholar] [CrossRef] [PubMed]
- Cerioni, L.; Palomba, L.; Cantoni, O. The Raf/MEK Inhibitor PD98059 Enhances ERK1/2 Phosphorylation Mediated by Peroxynitrite via Enforced Mitochondrial Formation of Reactive Oxygen Species. FEBS Lett. 2003, 547, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Ong, Q.; Guo, S.; Zhang, K.; Cui, B. U0126 Protects Cells against Oxidative Stress Independent of Its Function as a MEK Inhibitor. ACS Chem. Neurosci. 2015, 6, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.-M.; Jung, J.H.; Jeong, S.-J.; Sohn, E.J.; Kim, B.; Kim, S.-H. Tanshinone IIA Induces Autophagic Cell Death via Activation of AMPK and ERK and Inhibition of MTOR and P70 S6K in KBM-5 Leukemia Cells. Phytother. Res. 2013, 28, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Sun, R.; Xu, K.; Man, Z.; Ji, J.; Pu, Y.; Yin, L.; Zhang, J.; Pu, Y. Prodigiosin Induces Apoptosis and Inhibits Autophagy via the Extracellular Signal-Regulated Kinase Pathway in K562 Cells. Toxicol. In Vitro Int. J. Publ. Assoc. BiBRA 2019, 60, 107–115. [Google Scholar] [CrossRef]
- Thongnuanjan, P.; Soodvilai, S.; Fongsupa, S.; Chabang, N.; Vivithanaporn, P.; Tuchinda, P.; Soodvilai, S. Protective Effect of Panduratin A on Cisplatin-Induced Apoptosis of Human Renal Proximal Tubular Cells and Acute Kidney Injury in Mice. Biol. Pharm. Bull. 2021, 44, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.-C.; Lo, Y.-S.; Chuang, Y.-C.; Lin, C.-C.; Ho, H.-Y.; Hsieh, M.-J.; Lin, J.-T. Dehydrocrenatidine Extracted from Picrasma Quassioides Induces the Apoptosis of Nasopharyngeal Carcinoma Cells through the JNK and ERK Signaling Pathways. Oncol. Rep. 2021, 46, 166. [Google Scholar] [CrossRef]
- Deng, J.; Zhang, X.; Wu, H.; Gan, Y.; Ye, L.; Zheng, H.; Zhu, Z.; Liu, W.J.; Liu, H. ROS-ERK Pathway as Dual Mediators of Cellular Injury and Autophagy-Associated Adaptive Response in Urinary Protein-Irritated Renal Tubular Epithelial Cells. J. Diabetes Res. 2021, 2021, 1–8. [Google Scholar] [CrossRef]
- Wang, D.; Shi, S.; Ren, T.; Zhang, Y.; Guo, P.; Wang, J.; Wang, W. U0126 Pretreatment Inhibits Cisplatin-Induced Apoptosis and Autophagy in HEI-OC1 Cells and Cochlear Hair Cells. Toxicol. Appl. Pharm. 2021, 415, 115447. [Google Scholar] [CrossRef]
- An, J.; Li, L.; Zhang, X. Curcusone C Induces Apoptosis in Endometrial Cancer Cells via Mitochondria-Dependent Apoptotic and ERK Pathway. Biotechnol. Lett. 2021, 43, 329–338. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Huang, Z.-Y.; Tang, H.-H.; Kuo, W.-T.; Wu, S.-Y.; Lan, S.-H.; Chang, K.-H.; Lin, P.-L.; Lee, M.-F.; Cheng, H.-C.; et al. Pterostilbene Sensitizes Cisplatin-Resistant Human Bladder Cancer Cells with Oncogenic HRAS. Cancers 2020, 12, 2869. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Zheng, H.; Liu, Q.; Zhang, H.; Wang, X.; Shen, T.; Wang, S.; Ren, D. Morusin Induces Apoptosis and Autophagy via JNK, ERK and PI3K/Akt Signaling in Human Lung Carcinoma Cells. Chem.-Biol. Interact. 2020, 331, 109279. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, M.; Qin, L.; Song, Y.; Peng, A. DAXX Mediates High Phosphate-Induced Endothelial Cell Apoptosis in Vitro through Activating ERK Signaling. PEERJ 2020, 8, e9203. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.; Palau, V.E.; Mahboob, R.; Lightner, J.; Stone, W.; Krishnan, K. Upregulation of PERK and C-JUN by γ-Tocotrienol and Not α-Tocopherol Are Essential to the Differential Effect on Apoptosis in Prostate Cancer Cells. BMC Cancer 2020, 20, 428. [Google Scholar] [CrossRef]
- Liu, Y.; Fan, D. Ginsenoside Rg5 Induces G2/M Phase Arrest, Apoptosis and Autophagy via Regulating ROS-Mediated MAPK Pathways against Human Gastric Cancer. Biochem. Pharmacol. 2019, 168, 285–304. [Google Scholar] [CrossRef]
- Kang, M.-J.; Kim, S.-Y.; Kwon, E.-B.; Jo, Y.H.; Lee, M.K.; Lee, H.-S.; Moon, D.-O.; Kim, M.-O. Derrone Induces Autophagic Cell Death through Induction of ROS and ERK in A549 Cells. PLoS ONE 2019, 14, e0218659. [Google Scholar] [CrossRef]
- Yano, S.; Wu, S.; Sakao, K.; Hou, D.-X. Involvement of ERK1/2-Mediated ELK1/CHOP/DR5 Pathway in 6-(Methylsulfinyl)Hexyl Isothiocyanate-Induced Apoptosis of Colorectal Cancer Cells. Biosci. Biotechnol. Biochem. 2019, 83, 960–969. [Google Scholar] [CrossRef]
- Wang, T.; Yu, N.; Qian, M.; Feng, J.; Cao, S.; Yin, J.; Zhang, Q. ERK-Mediated Autophagy Promotes Inactivated Sendai Virus (HVJ-E)-Induced Apoptosis in HeLa Cells in an Atg3-Dependent Manner. Cancer Cell Int. 2018, 18, 200. [Google Scholar] [CrossRef]
- Zhou, D.; Zhai, W.; Zhang, M. Mesenchymal Stem Cell-Derived Extracellular Vesicles Promote Apoptosis in RSC96 Schwann Cells through the Activation of the ERK Pathway. Int. J. Clin. Exp. Pathol. 2018, 11, 5157–5170. [Google Scholar]
- Oliveira, M.D.; Barbosa, M.I.; de Souza, T.B.; Moreira, D.R.; Martins, F.T.; Villarreal, W.; Machado, R.P.; Doriguetto, A.C.; Soares, M.B.; Bezerra, D.P. A Novel Platinum Complex Containing a Piplartine Derivative Exhibits Enhanced Cytotoxicity, Causes Oxidative Stress and Triggers Apoptotic Cell Death by ERK/P38 Pathway in Human Acute Promyelocytic Leukemia HL-60 Cells. Redox Biol. 2019, 20, 182–194. [Google Scholar] [CrossRef]
- Jiao, Y.-N.; Wu, L.-N.; Xue, D.; Liu, X.-J.; Tian, Z.-H.; Jiang, S.-T.; Han, S.-Y.; Li, P.-P. Marsdenia Tenacissima Extract Induces Apoptosis and Suppresses Autophagy through ERK Activation in Lung Cancer Cells. Cancer Cell Int. 2018, 18, 149. [Google Scholar] [CrossRef]
- Sun, C.; Li, C.; Li, X.; Zhu, Y.; Su, Z.; Wang, X.; He, Q.; Zheng, G.; Feng, B. Scutellarin Induces Apoptosis and Autophagy in NSCLC Cells through ERK1/2 and AKT Signaling Pathways in Vitro and in Vivo. J. Cancer 2018, 9, 3247–3256. [Google Scholar] [CrossRef]
- Dubey, N.; Peng, B.-Y.; Lin, C.-M.; Wang, P.; Wang, J.; Chan, C.-H.; Wei, H.-J.; Deng, W.-P. NSC 95397 Suppresses Proliferation and Induces Apoptosis in Colon Cancer Cells through MKP-1 and the ERK1/2 Pathway. Int. J. Mol. Sci. 2018, 19, 1625. [Google Scholar] [CrossRef]
- Gao, H.; Zhang, Y.; Dong, L.; Qu, X.-Y.; Tao, L.-N.; Zhang, Y.-M.; Zhai, J.-H.; Song, Y.-Q. Triptolide Induces Autophagy and Apoptosis through ERK Activation in Human Breast Cancer MCF-7 Cells. Exp. Ther. Med. 2018, 15, 3413–3419. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; Zhu, J.; Chang, Q.; Zhou, H.; Shi, Z.; Min, L.; Cai, Y.; Guan, H. Alpha, 2′-Dihydroxy-4,4′-Dimethoxydihydrochalcone Inhibits Cell Proliferation, Invasion, and Migration in Gastric Cancer in Part via Autophagy. Biomed. Pharmacother. 2018, 98, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-H.; Liu, J.-F.; Chiang, Y.-C.; Hu, S.C.-S.; Hsu, L.-F.; Lin, Y.-C.; Lin, Z.-C.; Lee, H.-C.; Chen, M.-C.; Huang, C.-L.; et al. Artocarpin, an Isoprenyl Flavonoid, Induces P53-Dependent or Independent Apoptosis via ROS-Mediated MAPKs and Akt Activation in Non-Small Cell Lung Cancer Cells. Oncotarget 2017, 8, 28342–28358. [Google Scholar] [CrossRef] [PubMed]
- Tavares, R.; Pathak, S.K. Helicobacter Pylori Secreted Protein HP1286 Triggers Apoptosis in Macrophages via TNF-Independent and ERK MAPK-Dependent Pathways. Front. Cell Infect. Microbiol. 2017, 7, 58. [Google Scholar] [CrossRef]
- Ogura, K.; Terasaki, Y.; Miyoshi-Akiyama, T.; Terasaki, M.; Moss, J.; Noda, M.; Yahiro, K. Vibrio Cholerae Cholix Toxin-Induced HepG2 Cell Death Is Enhanced by Tumor Necrosis Factor-Alpha through ROS and Intracellular Signal-Regulated Kinases. Toxicol. Sci. 2017, 156, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Xu, Y.-L.; Tang, Z.-H.; Li, T.; Zhang, L.-L.; Chen, X.; Lu, J.-H.; Leung, C.-H.; Ma, D.-L.; Qiang, W.-A.; et al. Baicalein Induces Beclin 1- and Extracellular Signal-Regulated Kinase-Dependent Autophagy in Ovarian Cancer Cells. Am. J. Chin. Med. 2017, 45, 123–136. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, Y.; Yao, Y.; Li, J.; Wang, W.; Wu, X. Equol Induces Mitochondria-Dependent Apoptosis in Human Gastric Cancer Cells via the Sustained Activation of ERK1/2 Pathway. Mol. Cells 2016, 39, 742–749. [Google Scholar] [CrossRef]
- Kuo, H.-H.; Kakadiya, R.; Wu, Y.-C.; Su, T.-L.; Lee, T.-C.; Lin, Y.-W.; Yih, L.-H. Derivatives of 6-Cinnamamido-Quinoline-4-Carboxamide Impair Lysosome Function and Induce Apoptosis. Oncotarget 2015, 7, 38078–38090. [Google Scholar] [CrossRef]
- Chen, J.-C.; Hsieh, M.-J.; Chen, C.-J.; Lin, J.-T.; Lo, Y.-S.; Chuang, Y.-C.; Chien, S.-Y.; Chen, M.-K. Polyphyllin G Induce Apoptosis and Autophagy in Human Nasopharyngeal Cancer Cells by Modulation of AKT and Mitogen-Activated Protein Kinase Pathways in Vitro and in Vivo. Oncotarget 2016, 7, 70276–70289. [Google Scholar] [CrossRef] [PubMed]
- Park, B.H.; Lim, J.E.; Jeon, H.G.; Seo, S.I.; Lee, H.M.; Choi, H.Y.; Jeon, S.S.; Jeong, B.C. Curcumin Potentiates Antitumor Activity of Cisplatin in Bladder Cancer Cell Lines via ROS-Mediated Activation of ERK1/2. Oncotarget 2016, 7, 63870–63886. [Google Scholar] [CrossRef] [PubMed]
- Hasan, I.; Sugawara, S.; Fujii, Y.; Koide, Y.; Terada, D.; Iimura, N.; Fujiwara, T.; Takahashi, K.G.; Kojima, N.; Rajia, S.; et al. MytiLec, a Mussel R-Type Lectin, Interacts with Surface Glycan Gb3 on Burkitt’s Lymphoma Cells to Trigger Apoptosis through Multiple Pathways. Mar. Drugs 2015, 13, 7377–7389. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Li, J.; Shi, S.; Wang, X.; Liang, T.; Wu, B.; Li, Q. Sustained ERK Activation-Mediated Proliferation Inhibition of Farrerol on Human Gastric Carcinoma Cell Line by G0/G1-Phase Cell-Cycle Arrest. Eur. J. Cancer Prev. 2016, 25, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, W.; Wang, Y.; Dai, N.; Gu, J.; Yuan, Y.; Liu, X.; Bian, J.; Liu, Z.-P. Cadmium Induces Apoptosis in Primary Rat Osteoblasts through Caspase and Mitogen-Activated Protein Kinase Pathways. J. Vet. Sci. 2015, 16, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Han, W.; Li, J.; Hu, L.; Zhou, Y. Physalin B Not Only Inhibits the Ubiquitin-Proteasome Pathway but Also Induces Incomplete Autophagic Response in Human Colon Cancer Cells in Vitro. Acta Pharmacol. Sin. 2015, 36, 517–527. [Google Scholar] [CrossRef]
- Seo, H.J.; Choi, S.J.; Lee, J.-H. Paraquat Induces Apoptosis through Cytochrome C Release and ERK Activation. Biomol. Ther. 2014, 22, 503–509. [Google Scholar] [CrossRef]
- Cheng, L.; Xia, T.-S.; Wang, Y.-F.; Zhou, W.; Liang, X.-Q.; Xue, J.-Q.; Shi, L.; Wang, Y.; Ding, Q. The Apoptotic Effect of D Rhamnose β-Hederin, a Novel Oleanane-Type Triterpenoid Saponin on Breast Cancer Cells. PLoS ONE 2014, 9, e90848. [Google Scholar] [CrossRef]
- Forbes, A.; Davey, A.K.; Perkins, A.V.; Grant, G.D.; McFarland, A.J.; McDermott, C.M.; Anoopkumar-Dukie, S. ERK1/2 Activation Modulates Pyocyanin-Induced Toxicity in A549 Respiratory Epithelial Cells. Chem.-Biol. Interact. 2014, 208, 58–63. [Google Scholar] [CrossRef]
- Choi, M.; Park, H.; Oh, J.; Lee, E.; Park, S.; Yoon, S. Nonylphenol-induced Apoptotic Cell Death in Mouse TM4 Sertoli Cells via the Generation of Reactive Oxygen Species and Activation of the ERK Signaling Pathway. J. Appl. Toxicol. 2014, 34, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Yiran, Z.; Chenyang, J.; Jiajing, W.; Yan, Y.; Jianhong, G.; Jianchun, B.; Xuezhong, L.; Zongping, L. Oxidative Stress and Mitogen-Activated Protein Kinase Pathways Involved in Cadmium-Induced BRL 3A Cell Apoptosis. Oxid. Med. Cell. Longev. 2013, 2013, 1–12. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, M.-Y.; Hao, K.; Chen, X.-Q.; Du, J.-Z. CRHR1 Mediates P53 Transcription Induced by High Altitude Hypoxia through ERK 1/2 Signaling in Rat Hepatic Cells. Peptides 2013, 44, 8–14. [Google Scholar] [CrossRef]
- Tsai, S.-C.; Huang, W.-W.; Huang, W.-C.; Lu, C.-C.; Chiang, J.-H.; Peng, S.-F.; Chung, J.-G.; Lin, Y.-H.; Hsu, Y.-M.; Amagaya, S.; et al. ERK-Modulated Intrinsic Signaling and G2/M Phase Arrest Contribute to the Induction of Apoptotic Death by Allyl Isothiocyanate in MDA-MB-468 Human Breast Adenocarcinoma Cells. Int. J. Oncol. 2012, 41, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Lee, S.-W.; Choi, S.-H.; Kim, S.-H.; Kim, W.-J.; Jung, J.-Y. P38 MAP Kinase and ERK Play an Important Role in Nitric Oxide-Induced Apoptosis of the Mouse Embryonic Stem Cells. Toxicol. In Vitro 2013, 27, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-J.; Wang, H.-B.; Ma, X.-Q.; Chen, J.-H. β,β-Dimethylacrylshikonin Induces Mitochondria Dependent Apoptosis through ERK Pathway in Human Gastric Cancer SGC-7901 Cells. PLoS ONE 2012, 7, e41773. [Google Scholar] [CrossRef]
- Chi, J.; Zhu, Y.; Fu, Y.; Liu, Y.; Zhang, X.; Han, L.; Yin, X.; Zhao, D. Cyclosporin A Induces Apoptosis in H9c2 Cardiomyoblast Cells through Calcium-Sensing Receptor-Mediated Activation of the ERK MAPK and P38 MAPK Pathways. Mol. Cell. Biochem. 2012, 367, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Kurebayashi, J.; Kanomata, N.; Yamashita, T.; Kozuka, Y.; Moriya, T.; Sonoo, H. Antitumor and Anticancer Stem Cell Activity of a Poly ADP-Ribose Polymerase Inhibitor Olaparib in Breast Cancer Cells. Breast Cancer-Tokyo 2014, 21, 75–85. [Google Scholar] [CrossRef]
- Svojgr, K.; Kalina, T.; Kanderova, V.; Skopcova, T.; Brdicka, T.; Zuna, J. The Adaptor Protein NTAL Enhances Proximal Signaling and Potentiates Corticosteroid-Induced Apoptosis in T-ALL. Exp. Hematol. 2012, 40, 379–385. [Google Scholar] [CrossRef]
- Lin, H.-P.; Chang, J.-Y.; Lin, S.-R.; Lee, M.-H.; Huang, S.-S.; Hsu, L.-J.; Chang, N.-S. Identification of an In Vivo MEK/WOX1 Complex as a Master Switch for Apoptosis in T Cell Leukemia. Genes Cancer 2011, 2, 550–562. [Google Scholar] [CrossRef][Green Version]
- Banerjee, C.; Goswami, R.; Datta, S.; Rajagopal, R.; Mazumder, S. Arsenic-Induced Alteration in Intracellular Calcium Homeostasis Induces Head Kidney Macrophage Apoptosis Involving the Activation of Calpain-2 and ERK in Clarias Batrachus. Toxicol. Appl. Pharm. 2011, 256, 44–51. [Google Scholar] [CrossRef]
- Yang, H.; Dou, Y.; Zheng, X.; Tan, Y.; Cheng, J.; Li, L.; Du, Y.; Zhu, D.; Lou, Y. Cysteinyl Leukotrienes Synthesis Is Involved in Aristolochic Acid I-Induced Apoptosis in Renal Proximal Tubular Epithelial Cells. Toxicology 2011, 287, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Govind, S.; Sajankila, S.P.; Mi, L.; Roy, R.; Chung, F. Phenethyl Isothiocyanate Sensitizes Human Cervical Cancer Cells to Apoptosis Induced by Cisplatin. Mol. Nutr. Food Res. 2011, 55, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-Y.; Chang, G.-C.; Chen, K.-C.; Hung, H.-W.; Hsu, K.-H.; Sheu, G.-T.; Hsu, S.-L. Sustained Activation of ERK and Cdk2/Cyclin-A Signaling Pathway by Pemetrexed Leading to S-Phase Arrest and Apoptosis in Human Non-Small Cell Lung Cancer A549 Cells. Eur. J. Pharmacol. 2011, 663, 17–26. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, X.; Meng, J.; Wang, Z.-Y. An Anticancer Agent Icaritin Induces Sustained Activation of the Extracellular Signal-Regulated Kinase (ERK) Pathway and Inhibits Growth of Breast Cancer Cells. Eur. J. Pharmacol. 2011, 658, 114–122. [Google Scholar] [CrossRef]
- Snyder, A.; Alsauskas, Z.C.; Leventhal, J.S.; Rosenstiel, P.E.; Gong, P.; Chan, J.J.; Barley, K.; He, J.C.; Klotman, M.E.; Ross, M.J.; et al. HIV-1 Viral Protein r Induces ERK and Caspase-8-Dependent Apoptosis in Renal Tubular Epithelial Cells. Aids 2010, 24, 1107–1119. [Google Scholar] [CrossRef]
- Lu, K.; Liang, C.; Liliang, P.; Yang, C.; Cho, C.; Weng, H.; Tsai, Y.; Wang, K.; Chen, H. Inhibition of Extracellular Signal-regulated Kinases 1/2 Provides Neuroprotection in Spinal Cord Ischemia/Reperfusion Injury in Rats: Relationship with the Nuclear Factor-κB-regulated Anti-apoptotic Mechanisms. J. Neurochem. 2010, 114, 237–246. [Google Scholar] [CrossRef]
- Negrín, G.; Eiroa, J.L.; Morales, M.; Triana, J.; Quintana, J.; Estévez, F. Naturally Occurring Asteriscunolide A Induces Apoptosis and Activation of Mitogen-activated Protein Kinase Pathway in Human Tumor Cell Lines. Mol. Carcinog. 2010, 49, 488–499. [Google Scholar] [CrossRef]
- Huang, C.-F.; Liu, S.-H.; Su, C.-C.; Fang, K.-M.; Yen, C.-C.; Yang, C.-Y.; Tang, F.-C.; Liu, J.-M.; Wu, C.-C.; Lee, K.-I.; et al. Roles of ERK/Akt Signals in Mitochondria-Dependent and Endoplasmic Reticulum Stress-Triggered Neuronal Cell Apoptosis Induced by 4-Methyl-2,4-Bis(4-Hydroxyphenyl)Pent-1-Ene, a Major Active Metabolite of Bisphenol A. Toxicology 2021, 455, 152764. [Google Scholar] [CrossRef]
- Paul, M.; Manikanta, K.; Hemshekhar, M.; Sundaram, M.S.; Naveen, S.; Ramesh, T.N.; Kemparaju, K.; Girish, K.S. Bisdemethoxycurcumin Promotes Apoptosis in Human Platelets via Activation of ERK Signaling Pathway. Toxicol. In Vitro 2020, 63, 104743. [Google Scholar] [CrossRef]
- Sreenivasulu, R.; Reddy, K.T.; Sujitha, P.; Kumar, C.G.; Raju, R.R. Synthesis, Antiproliferative and Apoptosis Induction Potential Activities of Novel Bis(Indolyl)Hydrazide-Hydrazone Derivatives. Bioorgan. Med. Chem. 2019, 27, 1043–1055. [Google Scholar] [CrossRef]
- Lee, J.S.; Lee, M.S.; Cha, E.Y.; Thuong, P.T.; Sul, J.Y.; Park, J.B.; Ko, Y.B. A Natural Ent-Kaurane Diterpenoid Induces Antiproliferation in Ovarian Cancer Cells via ERK1/2 Regulation and Inhibition of Cellular Migration and Invasion. Mol. Med. Rep. 2018, 18, 3898–3906. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Li, J.; Cai, S.; He, S.; Liu, Q.; Jiang, J.; Chen, S.; Long, Y.; Chen, Y.; Chen, P.; et al. Notch1 Regulates Endothelial Apoptosis via the ERK Pathway in Chronic Obstructive Pulmonary Disease. Am. J. Physiol.-Cell Physiol. 2018, 315, C330–C340. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Gao, H.; Ju, P.; Gao, M.; Yuan, Y.; Chen, X.; Liu, K.; Han, Y.; Han, Z. Hispidulin Inhibits Hepatocellular Carcinoma Growth and Metastasis through AMPK and ERK Signaling Mediated Activation of PPARγ. Biomed. Pharmacother. 2018, 103, 272–283. [Google Scholar] [CrossRef]
- Huang, K.; Chen, Y.; Zhang, R.; Wu, Y.; Ma, Y.; Fang, X.; Shen, S. Honokiol Induces Apoptosis and Autophagy via the ROS/ERK1/2 Signaling Pathway in Human Osteosarcoma Cells in Vitro and in Vivo. Cell Death Dis. 2018, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.K.; Kim, S.-J. Desipramine Induces Apoptosis in Hepatocellular Carcinoma Cells. Oncol. Rep. 2017, 38, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Bastola, T.; An, R.; Kim, Y.-C.; Kim, J.; Seo, J. Cearoin Induces Autophagy, ERK Activation and Apoptosis via ROS Generation in SH-SY5Y Neuroblastoma Cells. Molecules 2017, 22, 242. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, S.; Yuan, X.; Hu, Z.; Li, H.; Wu, M.; Yuan, J.; Zhao, Z.; Su, J.; Wang, X.; et al. Valproic Acid Promotes Human Glioma U87 Cells Apoptosis and Inhibits Glycogen Synthase Kinase-3β Through ERK/Akt Signaling. Cell. Physiol. Biochem. 2016, 39, 2173–2185. [Google Scholar] [CrossRef]
- Teng, H.; Huang, Q.; Chen, L. Inhibition of Cell Proliferation and Triggering of Apoptosis by Agrimonolide through MAP Kinase (ERK and P38) Pathways in Human Gastric Cancer AGS Cells. Food Funct. 2016, 7, 4605–4613. [Google Scholar] [CrossRef]
- Jang, H.J.; Hong, E.M.; Park, S.W.; Byun, H.W.; Koh, D.H.; Choi, M.H.; Kae, S.H.; Lee, J. Statin Induces Apoptosis of Human Colon Cancer Cells and Downregulation of Insulin-like Growth Factor 1 Receptor via Proapoptotic ERK Activation. Oncol. Lett. 2016, 12, 250–256. [Google Scholar] [CrossRef]
- Zhang, D.-X.; Ma, D.-Y.; Yao, Z.-Q.; Fu, C.-Y.; Shi, Y.-X.; Wang, Q.-L.; Tang, Q.-Q. ERK1/2/P53 and NF-ΚB Dependent-PUMA Activation Involves in Doxorubicin-Induced Cardiomyocyte Apoptosis. Eur. Rev. Med. Pharmacol. 2016, 20, 2435–2442. [Google Scholar]
- Huang, F.; Liu, Q.; Xie, S.; Xu, J.; Huang, B.; Wu, Y.; Xia, D. Cypermethrin Induces Macrophages Death through Cell Cycle Arrest and Oxidative Stress-Mediated JNK/ERK Signaling Regulated Apoptosis. Int. J. Mol. Sci. 2016, 17, 885. [Google Scholar] [CrossRef]
- Feng, D.; Wang, B.; Ma, Y.; Shi, W.; Tao, K.; Zeng, W.; Cai, Q.; Zhang, Z.; Qin, H. The Ras/Raf/Erk Pathway Mediates the Subarachnoid Hemorrhage-Induced Apoptosis of Hippocampal Neurons Through Phosphorylation of P53. Mol. Neurobiol. 2015, 53, 5737–5748. [Google Scholar] [CrossRef]
- Yeh, P.-S.; Wang, W.; Chang, Y.-A.; Lin, C.-J.; Wang, J.-J.; Chen, R.-M. Honokiol Induces Autophagy of Neuroblastoma Cells through Activating the PI3K/Akt/MTOR and Endoplasmic Reticular Stress/ERK1/2 Signaling Pathways and Suppressing Cell Migration. Cancer Lett. 2016, 370, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ruan, H.; Gu, P.; Ding, W.; Luo, X.; Huang, R.; Zhao, W.; Gao, L. The Roles of P38 MAPK and ERK1/2 in Coplanar Polychlorinated Biphenyls-Induced Apoptosis of Human Extravillous Cytotrophoblast-Derived Transformed Cells. Cell. Physiol. Biochem. 2015, 36, 2418–2432. [Google Scholar] [CrossRef]
- Baek, S.H.; Kim, C.; Lee, J.H.; Nam, D.; Lee, J.; Lee, S.-G.; Chung, W.-S.; Jang, H.-J.; Kim, S.-H.; Ahn, K.S. Cinobufagin Exerts Anti-Proliferative and pro-Apoptotic Effects through the Modulation ROS-Mediated MAPKs Signaling Pathway. Immunopharm. Immunot. 2015, 37, 265–273. [Google Scholar] [CrossRef]
- Meng, G.; Wang, W.; Chai, K.; Yang, S.; Li, F.; Jiang, K. Combination Treatment with Triptolide and Hydroxycamptothecin Synergistically Enhances Apoptosis in A549 Lung Adenocarcinoma Cells through PP2A-Regulated ERK, P38 MAPKs and Akt Signaling Pathways. Int. J. Oncol. 2015, 46, 1007–1017. [Google Scholar] [CrossRef]
- Pathania, A.S.; Kumar, S.; Guru, S.K.; Bhushan, S.; Sharma, P.R.; Aithagani, S.K.; Singh, P.P.; Vishwakarma, R.A.; Kumar, A.; Malik, F. The Synthetic Tryptanthrin Analogue Suppresses STAT3 Signaling and Induces Caspase Dependent Apoptosis via ERK Up Regulation in Human Leukemia HL-60 Cells. PLoS ONE 2014, 9, e110411. [Google Scholar] [CrossRef]
- Zerin, T.; Song, H.-Y.; Kim, Y.-S. Extracellular Signal-Regulated Kinase Pathway Play Distinct Role in Acetochlor-Mediated Toxicity and Intrinsic Apoptosis in A549 Cells. Toxicol. In Vitro 2015, 29, 85–92. [Google Scholar] [CrossRef]
- Lee, Y.J.; Choi, S.-Y.; Yang, J.H. NMDA Receptor-Mediated ERK 1/2 Pathway Is Involved in PFHxS-Induced Apoptosis of PC12 Cells. Sci. Total Environ. 2014, 491–492, 227–234. [Google Scholar] [CrossRef]
- Lee, S.H.; Kang, Y.J.; Sung, B.; Kim, D.H.; Lim, H.S.; Kim, H.R.; Kim, S.J.; Yoon, J.-H.; Moon, H.R.; Chung, H.Y.; et al. MHY-449, a Novel Dihydrobenzofuro[4,5-b][1,8] Naphthyridin-6-One Derivative, Induces Apoptotic Cell Death through Modulation of Akt/FoxO1 and ERK Signaling in PC3 Human Prostate Cancer Cells. Int. J. Oncol. 2014, 44, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.-H.; Tseng, T.-J.; Su, C.-C.; Tang, F.-C.; Yen, C.-C.; Liu, Y.-Y.; Yang, C.-Y.; Wu, C.-C.; Chen, K.-L.; Hung, D.-Z.; et al. Arsenic Induces Reactive Oxygen Species-Caused Neuronal Cell Apoptosis through JNK/ERK-Mediated Mitochondria-Dependent and GRP 78/CHOP-Regulated Pathways. Toxicol. Lett. 2014, 224, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xu, L.; Fan, Y.; Li, C.; Zhang, Y.; Zheng, H.; Hou, K.; Qu, X.; Liu, Y. Interferon-α Enhances 5′-Deoxy-5-Fluorouridine-Induced Apoptosis by ERK-Dependant Upregulation of Thymidine Phosphorylase. Biomed Res. Int. 2013, 2013, 1–8. [Google Scholar] [CrossRef][Green Version]
- Chien, C.-S.; Ma, K.-H.; Lee, H.-S.; Liu, P.-S.; Li, Y.-H.; Huang, Y.-S.; Chueh, S.-H. Dual Effect of Capsaicin on Cell Death in Human Osteosarcoma G292 Cells. Eur. J. Pharmacol. 2013, 718, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Zheng, T.; Zhang, M.; Wang, D.; Du, S.; Li, X.; Fang, J.; Cao, X. Static Mechanical Stress Induces Apoptosis in Rat Endplate Chondrocytes through MAPK and Mitochondria-Dependent Caspase Activation Signaling Pathways. PLoS ONE 2013, 8, e69403. [Google Scholar] [CrossRef]
- Pan, T.-L.; Wang, P.-W. Explore the Molecular Mechanism of Apoptosis Induced by Tanshinone IIA on Activated Rat Hepatic Stellate Cells. Evid.-Based Complement. Altern Med. Ecam. 2012, 2012, 734987. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, H.-G.; Yang, J.-H. Perfluorooctane Sulfonate-Induced Apoptosis of Cerebellar Granule Cells Is Mediated by ERK 1/2 Pathway. Chemosphere 2013, 90, 1597–1602. [Google Scholar] [CrossRef]
- Bhattarai, G.; Lee, Y.-H.; Lee, N.-H.; Lee, I.-K.; Yun, B.-S.; Hwang, P.-H.; Yi, H.-K. Fomitoside-K from Fomitopsis Nigra Induces Apoptosis of Human Oral Squamous Cell Carcinomas (YD-10B) via Mitochondrial Signaling Pathway. Biol. Pharm. Bull. 2012, 35, 1711–1719. [Google Scholar] [CrossRef][Green Version]
- Lee, C.-C.; Lin, Y.-H.; Chang, W.-H.; Wu, Y.-C.; Chang, J.-G. The Small Molecule Calactin Induces DNA Damage and Apoptosis in Human Leukemia Cells. Eur. J. Cancer Prev. 2012, 21, 467–473. [Google Scholar] [CrossRef]
- Lin, M.-W.; Lin, A.-S.; Wu, D.-C.; Wang, S.S.W.; Chang, F.-R.; Wu, Y.-C.; Huang, Y.-B. Euphol from Euphorbia Tirucalli Selectively Inhibits Human Gastric Cancer Cell Growth through the Induction of ERK1/2-Mediated Apoptosis. Food Chem. Toxicol. 2012, 50, 4333–4339. [Google Scholar] [CrossRef]
- Jo, J.-R.; Park, J.S.; Park, Y.-K.; Chae, Y.Z.; Lee, G.-H.; Park, G.-Y.; Jang, B.-C. Pinus Densiflora Leaf Essential Oil Induces Apoptosis via ROS Generation and Activation of Caspases in YD-8 Human Oral Cancer Cells. Int. J. Oncol. 2011, 40, 1238–1245. [Google Scholar] [CrossRef]
- Arbon, K.S.; Christensen, C.M.; Harvey, W.A.; Heggland, S.J. Cadmium Exposure Activates the ERK Signaling Pathway Leading to Altered Osteoblast Gene Expression and Apoptotic Death in Saos-2 Cells. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2011, 50, 198–205. [Google Scholar] [CrossRef]
- Yang, L.; Ling, Y.; Zhang, Z.; Zhao, Q.; Tang, J.; Ji, H.; Zhang, Y. ZL11n Is a Novel Nitric Oxide-Releasing Derivative of Farnesylthiosalicylic Acid That Induces Apoptosis in Human Hepatoma HepG2 Cells via MAPK/Mitochondrial Pathways. Biochem. Biophys. Res. Commun. 2011, 409, 752–757. [Google Scholar] [CrossRef]
- Liu, H.; Li, H.; Xu, A.; Kan, Q.; Liu, B. Role of Phosphorylated ERK in Amygdala Neuronal Apoptosis in Single-Prolonged Stress Rats. Mol. Med. Rep. 2010, 3, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.; Kanaujiya, J.K.; Lochab, S.; Tripathi, S.B.; Bhatt, M.L.B.; Singh, P.K.; Sanyal, S.; Trivedi, A.K. 2-D Gel Electrophoresis-Based Proteomic Analysis Reveals That Ormeloxifen Induces G0-G1 Growth Arrest and ERK-Mediated Apoptosis in Chronic Myeloid Leukemia Cells K562. Proteomics 2011, 11, 1517–1529. [Google Scholar] [CrossRef]
- Iwayama, H.; Sakamoto, T.; Nawa, A.; Ueda, N. Crosstalk between Smad and Mitogen-Activated Protein Kinases for the Regulation of Apoptosis in Cyclosporine A-Induced Renal Tubular Injury. Nephron Extra 2011, 1, 178–189. [Google Scholar] [CrossRef] [PubMed]
- An, H.-X.; Jin, Z.-F.; Ge, X.-F.; Wu, J.; Chen, C.; Zhang, F.-M.; Qu, W.; Liu, X.-G.; Liu, S.-Y. Parathyroid Hormone(1-34)-Induced Apoptosis in Neuronal Rat PC12 Cells: Implications for Neurotoxicity. Pathol.-Res. Pract. 2010, 206, 821–827. [Google Scholar] [CrossRef]
- Guo, D.; Guo, C.; Fang, L.; Sang, T.; Wang, Y.; Wu, K.; Guo, C.; Wang, Y.; Pan, H.; Chen, R.; et al. Qizhen Capsule Inhibits Colorectal Cancer by Inducing NAG-1/GDF15 Expression That Mediated via MAPK/ERK Activation. J. Ethnopharmacol. 2021, 273, 113964. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, S.H.; Kim, Y.M.; Kim, S.H.; Yoo, E.S.; Woo, J.S.; Jung, G.H.; Jung, S.H.; Kim, B.S.; Jung, J.Y. Shikonin Inhibits Proliferation of Melanoma Cells by MAPK Pathway-Mediated Induction of Apoptosis. Biosci. Rep. 2021, 41. [Google Scholar] [CrossRef]
- Ghosh, J.; Das, J.; Manna, P.; Sil, P.C. Hepatotoxicity of Di-(2-Ethylhexyl)Phthalate Is Attributed to Calcium Aggravation, ROS-Mediated Mitochondrial Depolarization, and ERK/NF-ΚB Pathway Activation. Free Radic Biol. Med. 2010, 49, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, H.J.; Kwon, C.H.; Kim, J.H.; Woo, J.S.; Jung, J.S.; Kim, J.M. Role of ERK Activation in Cisplatin-Induced Apoptosis in OK Renal Epithelial Cells. J. Appl. Toxicol. 2005, 25, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Park, H.J.; Lee, B.I.; Ahn, Y.H.; Kim, S.U.; Choi, K.S. Bcl-2 Blocks Cisplatin-Induced Apoptosis by Suppression of ERK-Mediated P53 Accumulation in B104 Cells. Mol. Brain Res. 2001, 93, 18–26. [Google Scholar] [CrossRef]
- Park, B.G.; Yoo, C.I.; Kim, H.T.; Kwon, C.H.; Kim, Y.K. Role of Mitogen-Activated Protein Kinases in Hydrogen Peroxide-Induced Cell Death in Osteoblastic Cells. Toxicology 2005, 215, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Boutahar, N.; Reynaud, E.; Lassabliere, F.; Borg, J. Timing Differences of Signaling Response in Neuron Cultures Activated by Glutamate Analogue or Free Radicals. Brain Res. 2008, 1191, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.-K.; Cho, W.Y.; Sung, S.A.; Kim, H.K.; Won, N.H. MEK Inhibitor, U0126, Attenuates Cisplatin-Induced Renal Injury by Decreasing Inflammation and Apoptosis. Kidney Int. 2005, 67, 458–466. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Wu, D.-C.; Huang, F.-P.; Yang, G.-Y. Inhibition of MEK/ERK 1/2 Pathway Reduces pro-Inflammatory Cytokine Interleukin-1 Expression in Focal Cerebral Ischemia. Brain Res. 2004, 996, 55–66. [Google Scholar] [CrossRef]
- Sinha, D.; Bannergee, S.; Schwartz, J.H.; Lieberthal, W.; Levine, J.S. Inhibition of Ligand-Independent ERK1/2 Activity in Kidney Proximal Tubular Cells Deprived of Soluble Survival Factors Up-Regulates Akt and Prevents Apoptosis. J. Biol. Chem. 2004, 279, 10962–10972. [Google Scholar] [CrossRef]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Grant, S. Cotargeting Survival Signaling Pathways in Cancer. J. Clin. Investig. 2008, 118, 3003–3006. [Google Scholar] [CrossRef]
- Tang, D.; Wu, D.; Hirao, A.; Lahti, J.M.; Liu, L.; Mazza, B.; Kidd, V.J.; Mak, T.W.; Ingram, A.J. ERK Activation Mediates Cell Cycle Arrest and Apoptosis after DNA Damage Independently of P53. J. Biol. Chem. 2002, 277, 12710–12717. [Google Scholar] [CrossRef]
- Billecke, C.; Finniss, S.; Tahash, L.; Miller, C.; Mikkelsen, T.; Farrell, N.P.; Bögler, O. Polynuclear Platinum Anticancer Drugs Are More Potent than Cisplatin and Induce Cell Cycle Arrest in Glioma. Neuro-Oncology 2006, 8, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. P53, the Cellular Gatekeeper for Growth and Division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis—the P53 Network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. P53 Has a Direct Apoptogenic Role at the Mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; McGuirk, M.; Hockenberry, T.N.; Wu, Q.; Ashar, H.; Black, S.; Wen, S.F.; Wang, L.; Kirschmeier, P.; Bishop, W.R.; et al. Human Survivin Is Negatively Regulated by Wild-Type P53 and Participates in P53-Dependent Apoptotic Pathway. Oncogene 2002, 21, 2613–2622. [Google Scholar] [CrossRef]
- Wang, S.; Shi, X. Mechanisms of Cr(VI)-Induced P53 Activation: The Role of Phosphorylation, Mdm2 and ERK. Carcinogenesis 2001, 22, 757–762. [Google Scholar] [CrossRef]
- Yeh, P.Y.; Chuang, S.-E.; Yeh, K.-H.; Song, Y.C.; Chang, L.L.-Y.; Cheng, A.-L. Phosphorylation of P53 on Thr55 by ERK2 Is Necessary for Doxorubicin-Induced P53 Activation and Cell Death. Oncogene 2004, 23, 3580–3588. [Google Scholar] [CrossRef]
- Liu, J.; Mao, W.; Ding, B.; Liang, C. ERKs/P53 Signal Transduction Pathway Is Involved in Doxorubicin-Induced Apoptosis in H9c2 Cells and Cardiomyocytes. Am. J. Physiol.-Heart C 2008, 295, H1956–H1965. [Google Scholar] [CrossRef]
- Singh, S.; Upadhyay, A.K.; Ajay, A.K.; Bhat, M.K. P53 Regulates ERK Activation in Carboplatin Induced Apoptosis in Cervical Carcinoma: A Novel Target of P53 in Apoptosis. FEBS Lett. 2006, 581, 289–295. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Shin, S.J.; Kim, H.-S. ERK1/2 Activation Mediated by the Nutlin-3-Induced Mitochondrial Translocation of P53. Int. J. Oncol. 2013, 42, 1027–1035. [Google Scholar] [CrossRef]
- Heo, J.-I.; Oh, S.-J.; Kho, Y.-J.; Kim, J.-H.; Kang, H.-J.; Park, S.-H.; Kim, H.-S.; Shin, J.-Y.; Kim, M.-J.; Kim, M.; et al. ATM Mediates Interdependent Activation of P53 and ERK through Formation of a Ternary Complex with P-P53 and p-ERK in Response to DNA Damage. Mol. Biol. Rep. 2012, 39, 8007–8014. [Google Scholar] [CrossRef]
- Ozben, T. Oxidative Stress and Apoptosis: Impact on Cancer Therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, Y.; Kawai, Y.; Kohda, Y.; Gemba, M. Involvement of Activation of NADPH Oxidase and Extracellular Signal-Regulated Kinase (ERK) in Renal Cell Injury Induced by Zinc. J. Toxicol. Sci. 2005, 30, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Ramachandiran, S.; Huang, Q.; Dong, J.; Lau, S.S.; Monks, T.J. Mitogen-Activated Protein Kinases Contribute to Reactive Oxygen Species-Induced Cell Death in Renal Proximal Tubule Epithelial Cells. Chem. Res. Toxicol. 2002, 15, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shan, P.; Sasidhar, M.; Chupp, G.L.; Flavell, R.A.; Choi, A.M.K.; Lee, P.J. Reactive Oxygen Species and Extracellular Signal-Regulated Kinase 1/2 Mitogen-Activated Protein Kinase Mediate Hyperoxia-Induced Cell Death in Lung Epithelium. Am. J. Resp. Cell Mol. 2003, 28, 305–315. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, Y.-Z.; Kagan, E.; Bonner, J.C. Peroxynitrite Targets the Epidermal Growth Factor Receptor, Raf-1, and MEK Independently to Activate MAPK*. J. Biol. Chem. 2000, 275, 22479–22486. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Cho, H.-N.; Soh, J.-W.; Jhon, G.J.; Cho, C.-K.; Chung, H.-Y.; Bae, S.; Lee, S.-J.; Lee, Y.-S. Oxidative Stress-Induced Apoptosis Is Mediated by ERK1/2 Phosphorylation. Exp. Cell Res. 2003, 291, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Nabeyrat, E.; Jones, G.E.; Fenwick, P.S.; Barnes, P.J.; Donnelly, L.E. Mitogen-Activated Protein Kinases Mediate Peroxynitrite-Induced Cell Death in Human Bronchial Epithelial Cells. Am. J. Physiol.-Lung C 2003, 284, L1112–L1120. [Google Scholar] [CrossRef]
- Kohda, Y.; Hiramatsu, J.; Gemba, M. Involvement of MEK/ERK Pathway in Cephaloridine-Induced Injury in Rat Renal Cortical Slices. Toxicol. Lett. 2003, 143, 185–194. [Google Scholar] [CrossRef]
- Shin, H.-J.; Kwon, H.-K.; Lee, J.-H.; Anwar, M.A.; Choi, S. Etoposide Induced Cytotoxicity Mediated by ROS and ERK in Human Kidney Proximal Tubule Cells. Sci. Rep.-UK 2016, 6, 34064. [Google Scholar] [CrossRef]
- McCubrey, J.A.; LaHair, M.M.; Franklin, R.A. Reactive Oxygen Species-Induced Activation of the MAP Kinase Signaling Pathways. Antioxid Redox Sign 2006, 8, 1775–1789. [Google Scholar] [CrossRef]
- Torres, M.; Forman, H.J. Redox Signaling and the MAP Kinase Pathways. Biofactors 2003, 17, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Nox Proteins in Signal Transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Verbon, E.H.; Post, J.A.; Boonstra, J. The Influence of Reactive Oxygen Species on Cell Cycle Progression in Mammalian Cells. Gene 2012, 511, 1–6. [Google Scholar] [CrossRef] [PubMed]
- León-Buitimea, A.; Rodríguez-Fragoso, L.; Lauer, F.T.; Bowles, H.; Thompson, T.A.; Burchiel, S.W. Ethanol-Induced Oxidative Stress Is Associated with EGF Receptor Phosphorylation in MCF-10A Cells Overexpressing CYP2E1. Toxicol. Lett. 2012, 209, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Lander, H.M.; Milbank, A.J.; Tauras, J.M.; Hajjar, D.P.; Hempstead, B.L.; Schwartz, G.D.; Kraemer, R.T.; Mirza, U.A.; Chait, B.T.; Burk, S.C.; et al. Redox Regulation of Cell Signalling. Nature 1996, 381, 380–381. [Google Scholar] [CrossRef]
- Deora, A.A.; Hajjar, D.P.; Lander, H.M. Recruitment and Activation of Raf-1 Kinase by Nitric Oxide-Activated Ras †. Biochem.-US 2000, 39, 9901–9908. [Google Scholar] [CrossRef]
- Hoyos, B.; Imam, A.; Korichneva, I.; Levi, E.; Chua, R.; Hammerling, U. Activation of C-Raf Kinase by Ultraviolet Light. J. Biol. Chem. 2002, 277, 23949–23957. [Google Scholar] [CrossRef]
- Nowak, G. Protein Kinase C-α and ERK1/2 Mediate Mitochondrial Dysfunction, Decreases in Active Na+ Transport, and Cisplatin-Induced Apoptosis in Renal Cells. J. Biol. Chem. 2002, 277, 43377–43388. [Google Scholar] [CrossRef]
- Basu, A.; Tu, H. Activation of ERK during DNA Damage-Induced Apoptosis Involves Protein Kinase Cδ. Biochem. Biophys. Res. Commun. 2005, 334, 1068–1073. [Google Scholar] [CrossRef]
- Zhou, J.-Y.; Liu, Y.; Wu, G.S. The Role of Mitogen-Activated Protein Kinase Phosphatase-1 in Oxidative Damage–Induced Cell Death. Cancer Res. 2006, 66, 4888–4894. [Google Scholar] [CrossRef]
- Moosavi, S.M.; Prabhala, P.; Ammit, A.J. Role and Regulation of MKP-1 in Airway Inflammation. Respir. Res. 2017, 18, 154. [Google Scholar] [CrossRef]
- Chiarugi, P.; Fiaschi, T.; Taddei, M.L.; Talini, D.; Giannoni, E.; Raugei, G.; Ramponi, G. Two Vicinal Cysteines Confer a Peculiar Redox Regulation to Low Molecular Weight Protein Tyrosine Phosphatase in Response to Platelet-Derived Growth Factor Receptor Stimulation. J. Biol. Chem. 2001, 276, 33478–33487. [Google Scholar] [CrossRef]
- Sugiura, R.; Toda, T.; Shuntoh, H.; Yanagida, M.; Kuno, T. Pmp1+, a Suppressor of Calcineurin Deficiency, Encodes a Novel MAP Kinase Phosphatase in Fission Yeast. EMBO J. 1998, 17, 140–148. [Google Scholar] [CrossRef]
- Unni, A.M.; Lockwood, W.W.; Zejnullahu, K.; Lee-Lin, S.-Q.; Varmus, H. Evidence That Synthetic Lethality Underlies the Mutual Exclusivity of Oncogenic KRAS and EGFR Mutations in Lung Adenocarcinoma. Elife 2015, 4, e06907. [Google Scholar] [CrossRef]
- Unni, A.M.; Harbourne, B.; Oh, M.H.; Wild, S.; Ferrarone, J.R.; Lockwood, W.W.; Varmus, H. Hyperactivation of ERK by Multiple Mechanisms Is Toxic to RTK-RAS Mutation-Driven Lung Adenocarcinoma Cells. Elife 2018, 7, e33718. [Google Scholar] [CrossRef]
- Park, J.-W.; Wollmann, G.; Urbiola, C.; Fogli, B.; Florio, T.; Geley, S.; Klimaschewski, L. Sprouty2 Enhances the Tumorigenic Potential of Glioblastoma Cells. Neuro-Oncology 2018, 20, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Arkun, Y.; Yasemi, M. Dynamics and Control of the ERK Signaling Pathway: Sensitivity, Bistability, and Oscillations. PLoS ONE 2018, 13, e0195513. [Google Scholar] [CrossRef]
- Chen, J.-R.; Plotkin, L.I.; Aguirre, J.I.; Han, L.; Jilka, R.L.; Kousteni, S.; Bellido, T.; Manolagas, S.C. Transient Versus Sustained Phosphorylation and Nuclear Accumulation of ERKs Underlie Anti-Versus Pro-Apoptotic Effects of Estrogens. J. Biol. Chem. 2005, 280, 4632–4638. [Google Scholar] [CrossRef]
- Fehrenbacher, N.; Bastholm, L.; Kirkegaard-Sørensen, T.; Rafn, B.; Bøttzauw, T.; Nielsen, C.; Weber, E.; Shirasawa, S.; Kallunki, T.; Jäättelä, M. Sensitization to the Lysosomal Cell Death Pathway by Oncogene-Induced Down-Regulation of Lysosome-Associated Membrane Proteins 1 and 2. Cancer Res. 2008, 68, 6623–6633. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Yan, Y.; Daubert, R.A.; Han, J.; Schnellmann, R.G. ERK Promotes Hydrogen Peroxide-Induced Apoptosis through Caspase-3 Activation and Inhibition of Akt in Renal Epithelial Cells. Am. J. Physiol.-Ren. 2007, 292, F440–F447. [Google Scholar] [CrossRef]
- Zhuang, S.; Kinsey, G.R.; Yan, Y.; Han, J.; Schnellmann, R.G. Extracellular Signal-Regulated Kinase Activation Mediates Mitochondrial Dysfunction and Necrosis Induced by Hydrogen Peroxide in Renal Proximal Tubular Cells. J. Pharmacol. Exp. Ther. 2008, 325, 732–740. [Google Scholar] [CrossRef] [PubMed]
- An, H.-J.; Maeng, O.; Kang, K.-H.; Lee, J.-O.; Kim, Y.-S.; Paik, S.-G.; Lee, H. Activation of Ras Up-Regulates Pro-Apoptotic BNIP3 in Nitric Oxide-Induced Cell Death*. J. Biol. Chem. 2006, 281, 33939–33948. [Google Scholar] [CrossRef]
- Yang, R.; Piperdi, S.; Gorlick, R. Activation of the RAF/Mitogen-Activated Protein/Extracellular Signal-Regulated Kinase Kinase/Extracellular Signal-Regulated Kinase Pathway Mediates Apoptosis Induced by Chelerythrine in Osteosarcoma. Clin. Cancer Res. 2008, 14, 6396–6404. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann-Zeh, A.; Rodriguez-Viciana, P.; Ulrich, E.; Gilbert, C.; Coffer, P.; Downward, J.; Evan, G. Suppression of C-Myc-Induced Apoptosis by Ras Signalling through PI(3)K and PKB. Nature 1997, 385, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Benchimol, S. The Involvement of MAPK Signaling Pathways in Determining the Cellular Response to P53 Activation. J. Biol. Chem. 2006, 281, 3832–3840. [Google Scholar] [CrossRef]
- Chen, C.-H.; Wang, W.-J.; Kuo, J.-C.; Tsai, H.-C.; Lin, J.-R.; Chang, Z.-F.; Chen, R.-H. Bidirectional Signals Transduced by DAPK?ERK Interaction Promote the Apoptotic Effect of DAPK. EMBO J. 2004, 24, 294–304. [Google Scholar] [CrossRef]
- Zheng, A.; Kallio, A.; Härkönen, P. Tamoxifen-Induced Rapid Death of MCF-7 Breast Cancer Cells Is Mediated via Extracellularly Signal-Regulated Kinase Signaling and Can Be Abrogated by Estrogen. Endocrinology 2007, 148, 2764–2777. [Google Scholar] [CrossRef]
- Choi, J.; Yip-Schneider, M.; Albertin, F.; Wiesenauer, C.; Wang, Y.; Schmidt, C.M. The Effect of Doxorubicin on MEK-ERK Signaling Predicts Its Efficacy in HCC. J. Surg. Res. 2008, 150, 219–226. [Google Scholar] [CrossRef]
- Bermudez, O.; Pagès, G.; Gimond, C. The Dual-Specificity MAP Kinase Phosphatases: Critical Roles in Development and Cancer. Am. J. Physiol. Cell Physiol. 2010, 299, C189–C202. [Google Scholar] [CrossRef]
- Lee, M.; Kim, S.Y.; Kim, J.; Kim, H.-S.; Kim, S.-M.; Kim, E.J. Mitogen-Activated Protein Kinase Phosphatase-1 Inhibition and Sustained Extracellular Signal-Regulated Kinase 1/2 Activation in Camptothecin-Induced Human Colon Cancer Cell Death. Cancer Biol. Ther. 2013, 14, 1007–1015. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kidger, A.M.; Rushworth, L.K.; Stellzig, J.; Davidson, J.; Bryant, C.J.; Bayley, C.; Caddye, E.; Rogers, T.; Keyse, S.M.; Caunt, C.J. Dual-Specificity Phosphatase 5 Controls the Localized Inhibition, Propagation, and Transforming Potential of ERK Signaling. Proc. Natl. Acad Sci USA 2017, 114, E317–E326. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Nakayama, K.; Yamamoto, T.; Nishida, E. Regulatory Mechanisms and Function of ERK MAP Kinases. J. Biochem. 2004, 136, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L. “Location, Location, Location”: Activation and Targeting of MAP Kinases by G Protein-Coupled Receptors. J. Mol. Endocrinol. 2003, 30, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Formstecher, E.; Ramos, J.W.; Fauquet, M.; Calderwood, D.A.; Hsieh, J.C.; Canton, B.; Nguyen, X.T.; Barnier, J.V.; Camonis, J.; Ginsberg, M.H.; et al. PEA-15 Mediates Cytoplasmic Sequestration of ERK MAP Kinase. Dev. Cell 2001, 1, 239–250. [Google Scholar] [CrossRef]
- Mebratu, Y.A.; Dickey, B.F.; Evans, C.; Tesfaigzi, Y. The BH3-Only Protein Bik/Blk/Nbk Inhibits Nuclear Translocation of Activated ERK1/2 to Mediate IFNgamma-Induced Cell Death. J. Cell Biol. 2008, 183, 429–439. [Google Scholar] [CrossRef]
- Mizrak, S.C.; Renault-Mihara, F.; Párraga, M.; Bogerd, J.; van de Kant, H.J.G.; López-Casas, P.P.; Paz, M.; del Mazo, J.; de Rooij, D.G. Phosphoprotein Enriched in Astrocytes-15 Is Expressed in Mouse Testis and Protects Spermatocytes from Apoptosis. Reproduction 2007, 133, 743–751. [Google Scholar] [CrossRef]
- Hartman, M.L. Non-Apoptotic Cell Death Signaling Pathways in Melanoma. Int. J. Mol. Sci. 2020, 21, 2980. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular Senescence in Cancer and Aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- Ogier-Denis, E.; Pattingre, S.; Benna, J.E.; Codogno, P. Erk1/2-Dependent Phosphorylation of Gα-Interacting Protein Stimulates Its GTPase Accelerating Activity and Autophagy in Human Colon Cancer Cells. J. Biol. Chem. 2000, 275, 39090–39095. [Google Scholar] [CrossRef]
- Pattingre, S.; Bauvy, C.; Codogno, P. Amino Acids Interfere with the ERK1/2-Dependent Control of Macroautophagy by Controlling the Activation of Raf-1 in Human Colon Cancer HT-29 Cells. J. Biol. Chem. 2003, 278, 16667–16674. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.A.; Berhow, M.A.; Singletary, K.W. Inhibition of Akt Signaling and Enhanced ERK1/2 Activity Are Involved in Induction of Macroautophagy by Triterpenoid B-Group Soyasaponins in Colon Cancer Cells. Carcinogenesis 2005, 27, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Hernández-Tiedra, S.; Torres, S.; Lorente, M.; Guzmán, M.; Velasco, G. Detecting Autophagy in Response to ER Stress Signals in Cancer. Methods Enzym. 2011, 489, 297–317. [Google Scholar] [CrossRef]
- Li, X.; Feng, K.; Li, J.; Yu, D.; Fan, Q.; Tang, T.; Yao, X.; Wang, X. Curcumin Inhibits Apoptosis of Chondrocytes through Activation ERK1/2 Signaling Pathways Induced Autophagy. Nutrients 2017, 9, 414. [Google Scholar] [CrossRef]
- Bartholomeusz, C.; Rosen, D.; Wei, C.; Kazansky, A.; Yamasaki, F.; Takahashi, T.; Itamochi, H.; Kondo, S.; Liu, J.; Ueno, N.T. PEA-15 Induces Autophagy in Human Ovarian Cancer Cells and Is Associated with Prolonged Overall Survival. Cancer Res. 2008, 68, 9302–9310. [Google Scholar] [CrossRef]
- Kim, B.-W.; Lee, E.-R.; Min, H.-M.; Jeong, H.-S.; Ahn, J.-Y.; Kim, J.-H.; Choi, H.-Y.; Choi, H.; Kim, E.Y.; Park, S.P.; et al. Sustained ERK Activation Is Involved in the Kaempferol-Induced Apoptosis of Breast Cancer Cells and Is More Evident under 3-D Culture Condition. Cancer Biol. Ther. 2008, 7, 1080–1089. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Tran, E.; Nguyen, T.H.; Do, P.T.; Huynh, T.H.; Huynh, H. The Role of Activated MEK-ERK Pathway in Quercetin-Induced Growth Inhibition and Apoptosis in A549 Lung Cancer Cells. Carcinogenesis 2004, 25, 647–659. [Google Scholar] [CrossRef]
- Ota, Y.; Itoh, Y.; Kaise, A.; Ohta, K.; Endo, Y.; Masuda, M.; Sowa, Y.; Sakai, T.; Suzuki, T. Targeting Cancer with PCPA-Drug Conjugates: LSD1 Inhibition-Triggered Release of 4-Hydroxytamoxifen. Angew. Chem. Int. Ed. 2016, 55, 16115–16118. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).