
Class 1 Histone Deacetylases and Ataxia-Telangiectasia Mutated Kinase Control the Survival of Murine Pancreatic Cancer Cells upon dNTP Depletion

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs and Chemicals
2.2. Murine PDAC Cell Lines
2.3. Immunoblot
2.4. Flow Cytometry
2.5. Statistics
3. Results
3.1. Class 1 HDACs Promote Apoptosis Induction by Hydroxyurea in PDAC Cells
3.2. Hydroxyurea Induces Apoptosis More Effectively Than Irinotecan in the PDAC Cell Panel
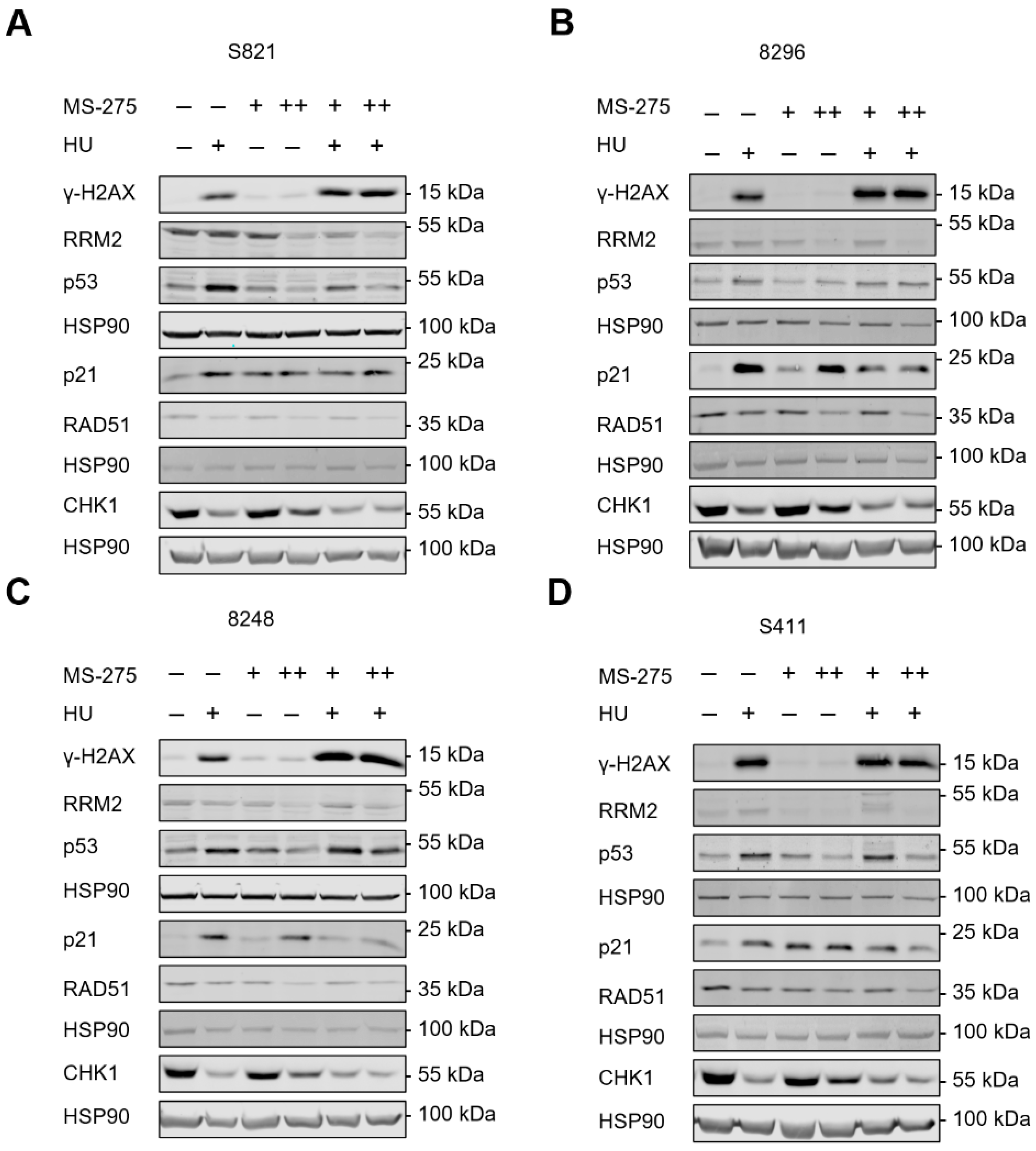
3.3. Hydroxyurea and Entinostat Dysregulate Proteins That Control the Cell Cycle and DNA Repair
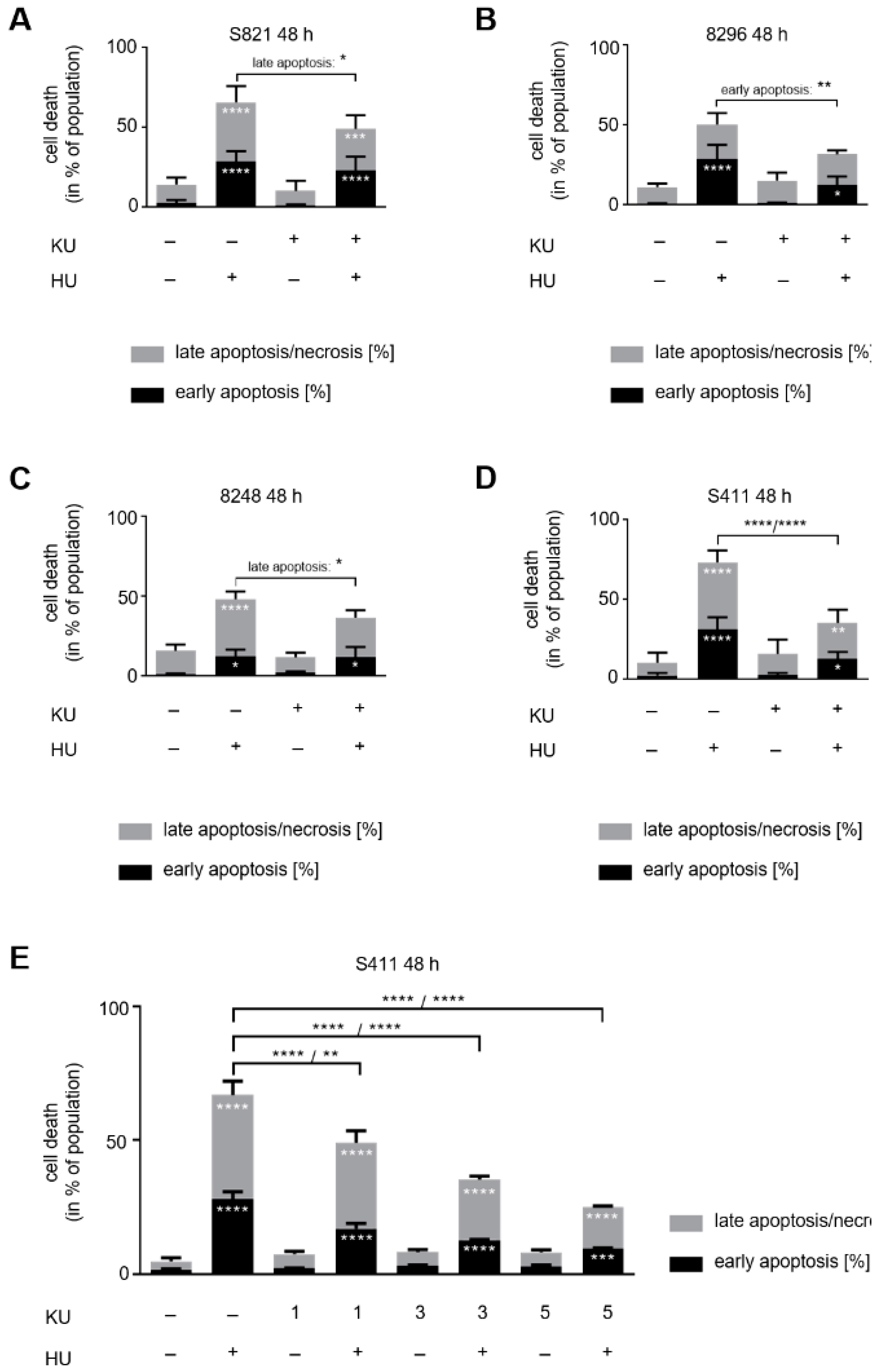
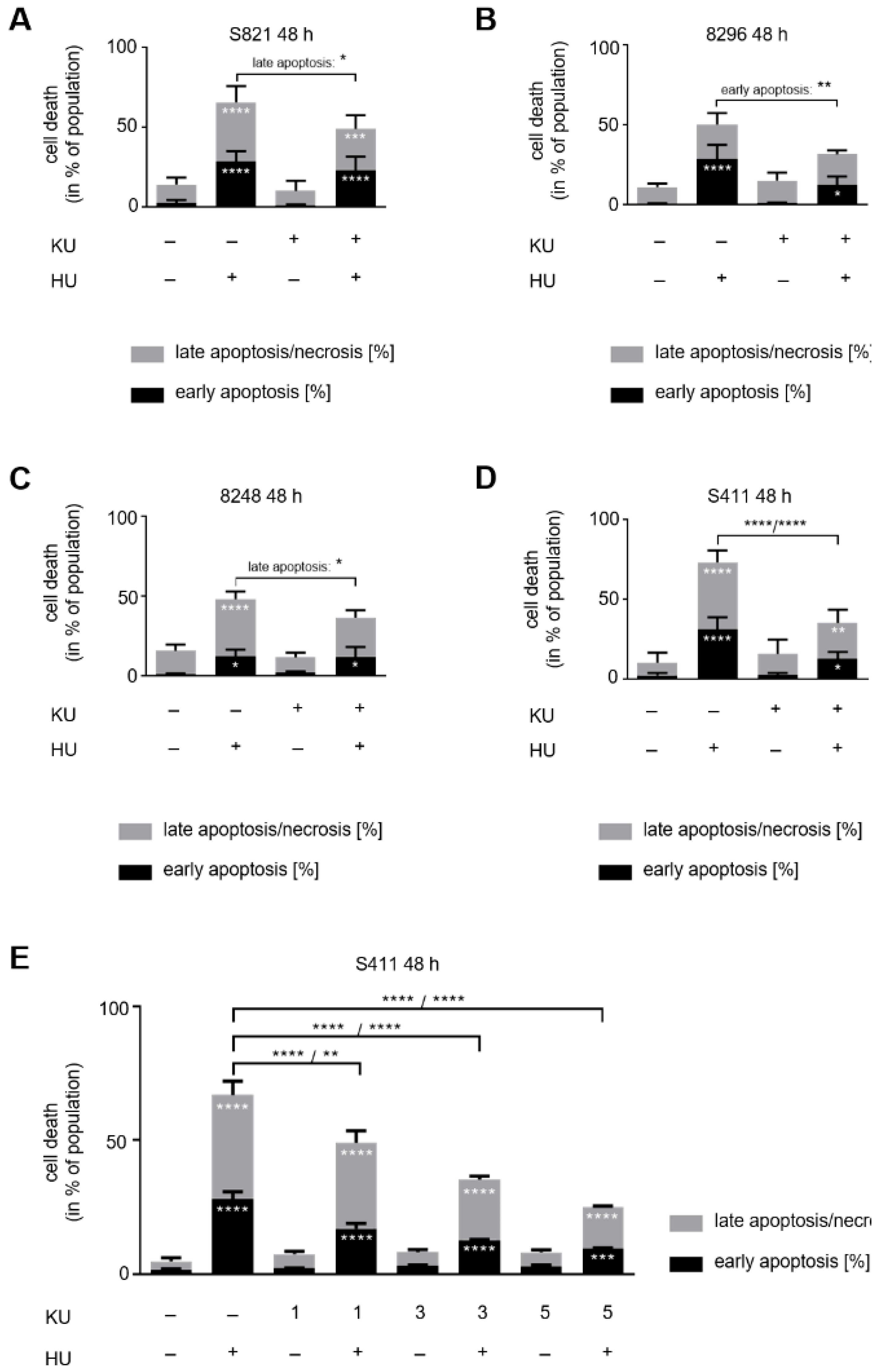
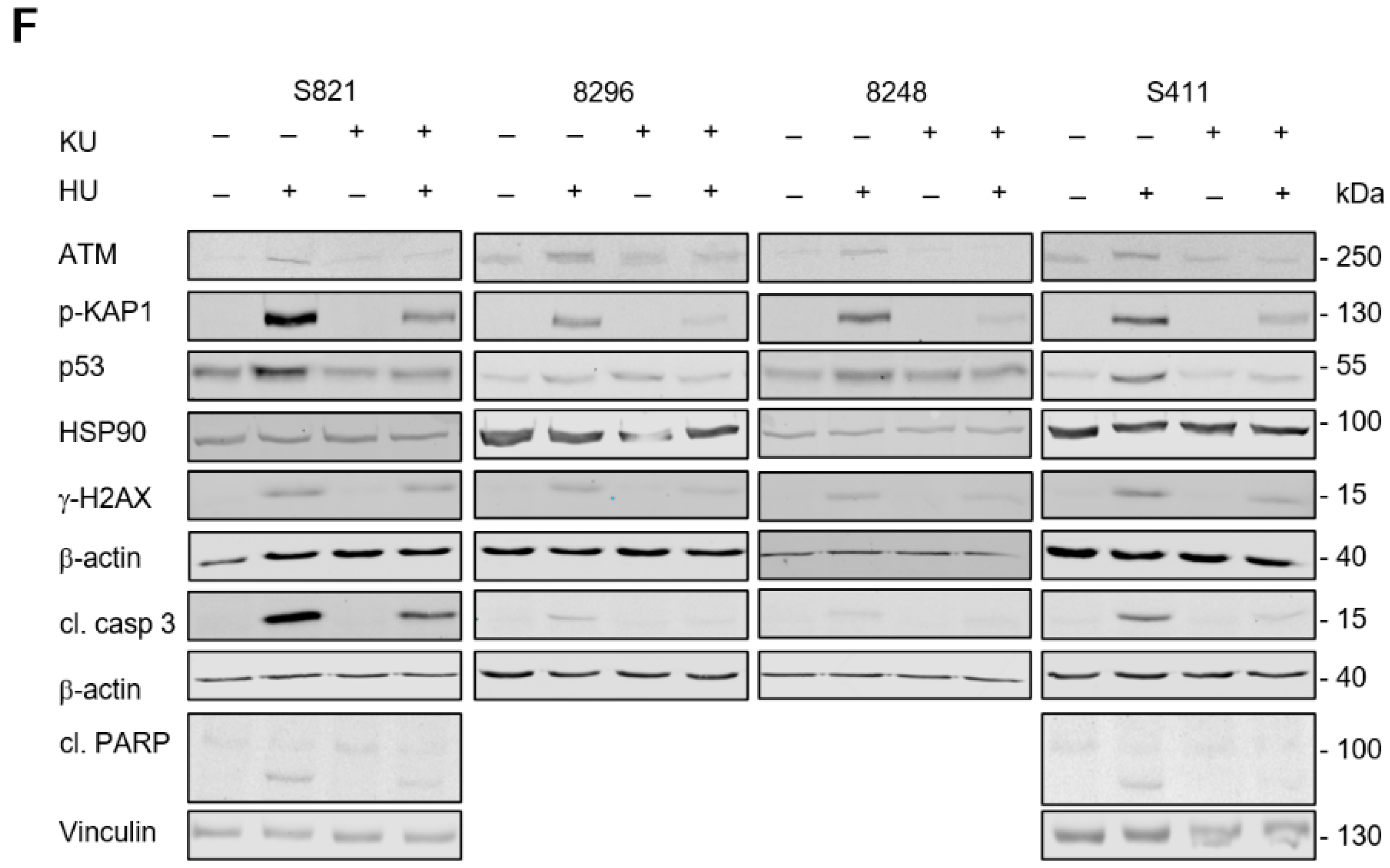
3.4. ATM Signaling Promotes Apoptotic DNA-Fragmentation in Hydroxyurea Treated PDAC Cells
3.5. ATM Signaling Is Necessary for Apoptosis Induction in Hydroxyurea Treated PDAC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wei, K.; Hackert, T. Surgical Treatment of Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 1971. [Google Scholar] [CrossRef]
- Elsayed, M.; Abdelrahim, M. The Latest Advancement in Pancreatic Ductal Adenocarcinoma Therapy: A Review Article for the Latest Guidelines and Novel Therapies. Biomedicines 2021, 9, 389. [Google Scholar] [CrossRef]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.; Kiweler, N.; Mahboobi, S.; Krämer, O.H. How to Distinguish Between the Activity of HDAC1-3 and HDAC6 with Western Blot. Methods Mol. Biol. 2017, 1510, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Kulka, L.A.M.; Fangmann, P.V.; Panfilova, D.; Olzscha, H. Impact of HDAC Inhibitors on Protein Quality Control Systems: Consequences for Precision Medicine in Malignant Disease. Front Cell Dev. Biol. 2020, 8, 425. [Google Scholar] [CrossRef]
- Schneider, G.; Krämer, O.H.; Fritsche, P.; Schuler, S.; Schmid, R.M.; Saur, D. Targeting histone deacetylases in pancreatic ductal adenocarcinoma. J. Cell Mol. Med. 2010, 14, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Muerkoster, S.S.; Schafer, H. Targeting apoptosis pathways in pancreatic cancer. Cancer Lett. 2013, 332, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Krämer, O.H.; Schmid, R.M.; Saur, D. Acetylation as a transcriptional control mechanism-HDACs and HATs in pancreatic ductal adenocarcinoma. J. Gastrointest Cancer 2011, 42, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Schuler, S.; Fritsche, P.; Diersch, S.; Arlt, A.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 attenuates TRAIL-induced apoptosis of pancreatic cancer cells. Mol. Cancer 2010, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, P.; Seidler, B.; Schuler, S.; Schnieke, A.; Göttlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, O.H.; Zhu, P.; Ostendorff, H.P.; Golebiewski, M.; Tiefenbach, J.; Peters, M.A.; Brill, B.; Groner, B.; Bach, I.; Heinzel, T.; et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. Embo J. 2003, 22, 3411–3420. [Google Scholar] [CrossRef] [Green Version]
- De Dosso, S.; Siebenhuner, A.R.; Winder, T.; Meisel, A.; Fritsch, R.; Astaras, C.; Szturz, P.; Borner, M. Treatment landscape of metastatic pancreatic cancer. Cancer Treat. Rev. 2021, 96, 102180. [Google Scholar] [CrossRef]
- Ammerpohl, O.; Trauzold, A.; Schniewind, B.; Griep, U.; Pilarsky, C.; Grutzmann, R.; Saeger, H.D.; Janssen, O.; Sipos, B.; Kloppel, G.; et al. Complementary effects of HDAC inhibitor 4-PB on gap junction communication and cellular export mechanisms support restoration of chemosensitivity of PDAC cells. Br. J. Cancer 2007, 96, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, S.E.; Ding, L.Y.; Mo, X.; Bekaii-Saab, T.; Kulp, S.K.; Chen, C.S.; Huang, P.H. Suppression of Tumor Growth and Muscle Wasting in a Transgenic Mouse Model of Pancreatic Cancer by the Novel Histone Deacetylase Inhibitor AR-42. Neoplasia 2016, 18, 765–774. [Google Scholar] [CrossRef] [Green Version]
- Laschanzky, R.S.; Humphrey, L.E.; Ma, J.; Smith, L.M.; Enke, T.J.; Shukla, S.K.; Dasgupta, A.; Singh, P.K.; Howell, G.M.; Brattain, M.G.; et al. Selective Inhibition of Histone Deacetylases 1/2/6 in Combination with Gemcitabine: A Promising Combination for Pancreatic Cancer Therapy. Cancers 2019, 11, 1327. [Google Scholar] [CrossRef] [Green Version]
- Dovzhanskiy, D.I.; Arnold, S.M.; Hackert, T.; Oehme, I.; Witt, O.; Felix, K.; Giese, N.; Werner, J. Experimental in vivo and in vitro treatment with a new histone deacetylase inhibitor belinostat inhibits the growth of pancreatic cancer. BMC Cancer 2012, 12, 226. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.H.; Xu, X.G.; Yan, S.L.; Sun, Z.; Ying, Y.; Wang, B.K.; Tu, Y.X. Depletion of HDAC1, 7 and 8 by Histone Deacetylase Inhibition Confers Elimination of Pancreatic Cancer Stem Cells in Combination with Gemcitabine. Sci. Rep. 2018, 8, 1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edderkaoui, M.; Xu, S.; Chheda, C.; Morvaridi, S.; Hu, R.W.; Grippo, P.J.; Mascarinas, E.; Principe, D.R.; Knudsen, B.; Xue, J.; et al. HDAC3 mediates smoking-induced pancreatic cancer. Oncotarget 2016, 7, 7747–7760. [Google Scholar] [CrossRef] [Green Version]
- Shouksmith, A.E.; Shah, F.; Grimard, M.L.; Gawel, J.M.; Raouf, Y.S.; Geletu, M.; Berger-Becvar, A.; de Araujo, E.D.; Luchman, H.A.; Heaton, W.L.; et al. Identification and Characterization of AES-135, a Hydroxamic Acid-Based HDAC Inhibitor That Prolongs Survival in an Orthotopic Mouse Model of Pancreatic Cancer. J. Med. Chem. 2019, 62, 2651–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalla Pozza, E.; Manfredi, M.; Brandi, J.; Buzzi, A.; Conte, E.; Pacchiana, R.; Cecconi, D.; Marengo, E.; Donadelli, M. Trichostatin A alters cytoskeleton and energy metabolism of pancreatic adenocarcinoma cells: An in depth proteomic study. J. Cell Biochem. 2018, 119, 2696–2707. [Google Scholar] [CrossRef]
- Kumar, D.; Sarma, P.; Bhadra, M.P.; Tangutur, A.D. Impact of Hybrid-polar Histone Deacetylase Inhibitor m-Carboxycinnamic Acid bis-Hydroxyamide on Human Pancreatic Adenocarcinoma Cells. Anticancer Agents Med. Chem. 2019, 19, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Xu, S.; Cui, J.; Poddar, S.; Le, T.M.; Hayrapetyan, H.; Li, L.; Wu, N.; Moore, A.M.; Zhou, L.; et al. Histone deacetylase inhibition is synthetically lethal with arginine deprivation in pancreatic cancers with low argininosuccinate synthetase 1 expression. Theranostics 2020, 10, 829–840. [Google Scholar] [CrossRef]
- Ponath, V.; Frech, M.; Bittermann, M.; Al Khayer, R.; Neubauer, A.; Brendel, C.; Pogge von Strandmann, E. The Oncoprotein SKI Acts as A Suppressor of NK Cell-Mediated Immunosurveillance in PDAC. Cancers 2020, 12, 2857. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Arlinghaus, L.R.; Cardin, D.B.; Goff, L.; Berlin, J.D.; Parikh, A.; Abramson, R.G.; Yankeelov, T.E.; Hiebert, S.; Merchant, N.; et al. Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother. Oncol. 2016, 119, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.; Chiorean, E.G.; O’Dwyer, P.J.; Gabrail, N.Y.; Alcindor, T.; Potvin, D.; Chao, R.; Hurwitz, H. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother. Pharm. 2018, 81, 355–364. [Google Scholar] [CrossRef]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zegar, T.; Weiser, T.; Hamdan, F.H.; Berger, B.T.; Lucas, R.; Balourdas, D.I.; Ladigan, S.; Cheung, P.F.; Liffers, S.T.; et al. Characterization of a dual BET/HDAC inhibitor for treatment of pancreatic ductal adenocarcinoma. Int. J. Cancer 2020, 147, 2847–2861. [Google Scholar] [CrossRef]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Shimada, S.; Akiyama, Y.; Ishikawa, Y.; Ogura, T.; Ogawa, K.; Ono, H.; Mitsunori, Y.; Ban, D.; Kudo, A.; et al. Loss of KDM6A characterizes a poor prognostic subtype of human pancreatic cancer and potentiates HDAC inhibitor lethality. Int. J. Cancer 2019, 145, 192–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzulko, M.; Pons, M.; Henke, A.; Schneider, G.; Krämer, O.H. The PP2A subunit PR130 is a key regulator of cell development and oncogenic transformation. Biochim. Biophys Acta Rev. Cancer 2020, 1874, 188453. [Google Scholar] [CrossRef]
- Bug, G.; Ritter, M.; Wassmann, B.; Schoch, C.; Heinzel, T.; Schwarz, K.; Romanski, A.; Krämer, O.H.; Kampfmann, M.; Hoelzer, D.; et al. Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia. Cancer 2005, 104, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Göder, A.; Emmerich, C.; Nikolova, T.; Kiweler, N.; Schreiber, M.; Kuhl, T.; Imhof, D.; Christmann, M.; Heinzel, T.; Schneider, G.; et al. HDAC1 and HDAC2 integrate checkpoint kinase phosphorylation and cell fate through the phosphatase-2A subunit PR130. Nat. Commun. 2018, 9, 764. [Google Scholar] [CrossRef] [Green Version]
- Krämer, O.H.; Knauer, S.K.; Zimmermann, D.; Stauber, R.H.; Heinzel, T. Histone deacetylase inhibitors and hydroxyurea modulate the cell cycle and cooperatively induce apoptosis. Oncogene 2008, 27, 732–740. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, C.; Göder, A.; Beyer, M.; Kiweler, N.; Mahendrarajah, N.; Rauch, A.; Nikolova, T.; Stojanovic, N.; Wieczorek, M.; Reich, T.R.; et al. Class I histone deacetylases regulate p53/NF-κB crosstalk in cancer cells. Cell. Signal. 2017, 29, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Amaru Calzada, A.; Pedrini, O.; Finazzi, G.; Leoni, F.; Mascagni, P.; Introna, M.; Rambaldi, A.; Golay, J.; Associazione Italiana per la Ricerca sul Cancro-Gruppo Italiano Malattie Mieloproliferative Investigators. Givinostat and hydroxyurea synergize in vitro to induce apoptosis of cells from JAK2(V617F) myeloproliferative neoplasm patients. Exp. Hematol. 2013, 41, 253–260.e2. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.W.; Thomas, A.; Murai, J.; Trepel, J.B.; Bates, S.E.; Rajapakse, V.N.; Pommier, Y. Overcoming Resistance to DNA-Targeted Agents by Epigenetic Activation of Schlafen 11 (SLFN11) Expression with Class I Histone Deacetylase Inhibitors. Clin. Cancer Res. 2018, 24, 1944–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finazzi, G.; Vannucchi, A.M.; Martinelli, V.; Ruggeri, M.; Nobile, F.; Specchia, G.; Pogliani, E.M.; Olimpieri, O.M.; Fioritoni, G.; Musolino, C.; et al. A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br. J. Haematol. 2013, 161, 688–694. [Google Scholar] [CrossRef]
- Leitch, C.; Osdal, T.; Andresen, V.; Molland, M.; Kristiansen, S.; Nguyen, X.N.; Bruserud, O.; Gjertsen, B.T.; McCormack, E. Hydroxyurea synergizes with valproic acid in wild-type p53 acute myeloid leukaemia. Oncotarget 2016, 7, 8105–8118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, G.; Henrich, A.; Greiner, G.; Wolf, V.; Lovas, A.; Wieczorek, M.; Wagner, T.; Reichardt, S.; von Werder, A.; Schmid, R.M.; et al. Cross talk between stimulated NF-κB and the tumor suppressor p53. Oncogene 2010, 29, 2795–2806. [Google Scholar] [CrossRef] [Green Version]
- Schneider, G.; Krämer, O.H. NFκB/p53 crosstalk-a promising new therapeutic target. Biochim. Biophys. Acta 2011, 1815, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, N.; Hassan, Z.; Wirth, M.; Wenzel, P.; Beyer, M.; Schafer, C.; Brand, P.; Kroemer, A.; Stauber, R.H.; Schmid, R.M.; et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 2017, 36, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Stauber, R.H.; Knauer, S.K.; Habtemichael, N.; Bier, C.; Unruhe, B.; Weisheit, S.; Spange, S.; Nonnenmacher, F.; Fetz, V.; Ginter, T.; et al. A combination of a ribonucleotide reductase inhibitor and histone deacetylase inhibitors downregulates EGFR and triggers BIM-dependent apoptosis in head and neck cancer. Oncotarget 2012, 3, 31–43. [Google Scholar] [CrossRef]
- Bradner, J.E.; Mak, R.; Tanguturi, S.K.; Mazitschek, R.; Haggarty, S.J.; Ross, K.; Chang, C.Y.; Bosco, J.; West, N.; Morse, E.; et al. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc. Natl. Acad. Sci. USA 2010, 107, 12617–12622. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Su, L.; Hu, S.; Hu, W.; Yip, M.L.; Wu, J.; Gaur, S.; Smith, D.L.; Yuan, Y.C.; Synold, T.W.; et al. A small-molecule blocking ribonucleotide reductase holoenzyme formation inhibits cancer cell growth and overcomes drug resistance. Cancer Res. 2013, 73, 6484–6493. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.C.; Zhou, B.; Zhang, K.; Yuan, Y.C.; Un, F.; Hu, S.; Chou, C.M.; Chen, C.H.; Wu, J.; Wang, Y.; et al. The Novel Ribonucleotide Reductase Inhibitor COH29 Inhibits DNA Repair In Vitro. Mol. Pharm. 2015, 87, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Biederstadt, A.; Hassan, Z.; Schneeweis, C.; Schick, M.; Schneider, L.; Muckenhuber, A.; Hong, Y.; Siegers, G.; Nilsson, L.; Wirth, M.; et al. SUMO pathway inhibition targets an aggressive pancreatic cancer subtype. Gut 2020, 69, 1472–1482. [Google Scholar] [CrossRef] [Green Version]
- Von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009, 137, 361–371.e5. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Ollinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönhuber, N.; Seidler, B.; Schuck, K.; Veltkamp, C.; Schachtler, C.; Zukowska, M.; Eser, S.; Feyerabend, T.B.; Paul, M.C.; Eser, P.; et al. A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat. Med. 2014, 20, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Pons, M.; Reichardt, C.M.; Hennig, D.; Nathan, A.; Kiweler, N.; Stocking, C.; Wichmann, C.; Christmann, M.; Butter, F.; Reichardt, S.; et al. Loss of Wilms tumor 1 protein is a marker for apoptosis in response to replicative stress in leukemic cells. Arch. Toxicol. 2018, 92, 2119–2135. [Google Scholar] [CrossRef]
- Kiweler, N.; Wünsch, D.; Wirth, M.; Mahendrarajah, N.; Schneider, G.; Stauber, R.H.; Brenner, W.; Butter, F.; Krämer, O.H. Histone deacetylase inhibitors dysregulate DNA repair proteins and antagonize metastasis-associated processes. J. Cancer Res. Clin. Oncol 2020, 146, 343–356. [Google Scholar] [CrossRef] [Green Version]
- Marx-Blümel, L.; Marx, C.; Kühne, M.; Sonnemann, J. Assessment of HDACi-Induced Cytotoxicity. Methods Mol. Biol. 2017, 1510, 23–45. [Google Scholar] [CrossRef]
- Øvrebø, J.I.; Edgar, B.A. Polyploidy in tissue homeostasis and regeneration. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Kopp, B.; Khoury, L.; Audebert, M. Validation of the gammaH2AX biomarker for genotoxicity assessment: A review. Arch. Toxicol. 2019, 93, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Grundy, M.K.; Buckanovich, R.J.; Bernstein, K.A. Regulation and pharmacological targeting of RAD51 in cancer. Nar. Cancer 2020, 2, zcaa024. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, M.; Nagel, G.; Zeyn, Y.; Beyer, M.; Laguna, T.; Brachetti, T.; Sellmer, A.; Mahboobi, S.; Conradi, R.; Butter, F.; et al. Human platelet lysate as validated replacement for animal serum to assess chemosensitivity. Altex 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckerman, R.; Donner, A.J.; Mattia, M.; Peart, M.J.; Manley, J.L.; Espinosa, J.M.; Prives, C. A role for Chk1 in blocking transcriptional elongation of p21 RNA during the S-phase checkpoint. Genes Dev. 2009, 23, 1364–1377. [Google Scholar] [CrossRef] [Green Version]
- Gottifredi, V.; Karni-Schmidt, O.; Shieh, S.S.; Prives, C. p53 down-regulates CHK1 through p21 and the retinoblastoma protein. Mol. Cell Biol. 2001, 21, 1066–1076. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulos, A.; Altmeyer, M. The Hammer and the Dance of Cell Cycle Control. Trends Biochem. Sci. 2021, 46, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Alvino, G.M.; Collingwood, D.; Murphy, J.M.; Delrow, J.; Brewer, B.J.; Raghuraman, M.K. Replication in hydroxyurea: It’s a matter of time. Mol. Cell Biol. 2007, 27, 6396–6406. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.A.; Oehler, H.; Voigt, A.; Dalic, D.; Freiwald, A.; Serve, H.; Beli, P. ATR inhibition rewires cellular signaling networks induced by replication stress. Proteomics 2016, 16, 402–416. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.E.; Ferraz Franco, C.; Su, L.I.; Corbin, E.K.; Perkins, S.; Kalyuzhnyy, A.; Jones, A.R.; Brownridge, P.J.; Perkins, N.D.; Eyers, C.E. Temporal modulation of the NF-κB RelA network in response to different types of DNA damage. Biochem. J. 2021, 478, 533–551. [Google Scholar] [CrossRef]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, S.; Hombach-Klonisch, S.; Droge, P.; Klonisch, T. HMGA2 inhibits apoptosis through interaction with ATR-CHK1 signaling complex in human cancer cells. Neoplasia 2013, 15, 263–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oo, Z.Y.; Proctor, M.; Stevenson, A.J.; Nazareth, D.; Fernando, M.; Daignault, S.M.; Lanagan, C.; Walpole, S.; Bonazzi, V.; Skalamera, D.; et al. Combined use of subclinical hydroxyurea and CHK1 inhibitor effectively controls melanoma and lung cancer progression, with reduced normal tissue toxicity compared to gemcitabine. Mol. Oncol. 2019, 13, 1503–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.C.; Siu, W.Y.; Lau, A.; Chan, W.M.; Arooz, T.; Poon, R.Y. Stalled replication induces p53 accumulation through distinct mechanisms from DNA damage checkpoint pathways. Cancer Res. 2006, 66, 2233–2241. [Google Scholar] [CrossRef] [Green Version]
- Stiff, T.; Walker, S.A.; Cerosaletti, K.; Goodarzi, A.A.; Petermann, E.; Concannon, P.; O’Driscoll, M.; Jeggo, P.A. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. Embo J. 2006, 25, 5775–5782. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, T.; Kiweler, N.; Krämer, O.H. Interstrand Crosslink Repair as a Target for HDAC Inhibition. Trends Pharmacol. Sci. 2017, 38, 822–836. [Google Scholar] [CrossRef] [PubMed]
- Bowry, A.; Kelly, R.D.W.; Petermann, E. Hypertranscription and replication stress in cancer. Trends Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Perkhofer, L.; Gout, J.; Roger, E.; Kude de Almeida, F.; Baptista Simoes, C.; Wiesmuller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2021, 70, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Guthle, M.; Zenke, M.; et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat. Commun. 2015, 6, 7677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkhofer, L.; Schmitt, A.; Romero Carrasco, M.C.; Ihle, M.; Hampp, S.; Ruess, D.A.; Hessmann, E.; Russell, R.; Lechel, A.; Azoitei, N.; et al. ATM Deficiency Generating Genomic Instability Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Therapy-Induced DNA Damage. Cancer Res. 2017, 77, 5576–5590. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, A.; Dzulko, M.; Murr, J.; Yen, Y.; Schneider, G.; Krämer, O.H. Class 1 Histone Deacetylases and Ataxia-Telangiectasia Mutated Kinase Control the Survival of Murine Pancreatic Cancer Cells upon dNTP Depletion. Cells 2021, 10, 2520. https://doi.org/10.3390/cells10102520
Nguyen A, Dzulko M, Murr J, Yen Y, Schneider G, Krämer OH. Class 1 Histone Deacetylases and Ataxia-Telangiectasia Mutated Kinase Control the Survival of Murine Pancreatic Cancer Cells upon dNTP Depletion. Cells. 2021; 10(10):2520. https://doi.org/10.3390/cells10102520
Chicago/Turabian StyleNguyen, Alexandra, Melanie Dzulko, Janine Murr, Yun Yen, Günter Schneider, and Oliver H. Krämer. 2021. "Class 1 Histone Deacetylases and Ataxia-Telangiectasia Mutated Kinase Control the Survival of Murine Pancreatic Cancer Cells upon dNTP Depletion" Cells 10, no. 10: 2520. https://doi.org/10.3390/cells10102520
APA StyleNguyen, A., Dzulko, M., Murr, J., Yen, Y., Schneider, G., & Krämer, O. H. (2021). Class 1 Histone Deacetylases and Ataxia-Telangiectasia Mutated Kinase Control the Survival of Murine Pancreatic Cancer Cells upon dNTP Depletion. Cells, 10(10), 2520. https://doi.org/10.3390/cells10102520