Abstract
In chronic hepatitis B and C virus infections persistently elevated antigen levels drive CD8+ T cells toward a peculiar differentiation state known as T cell exhaustion, which poses crucial constraints to antiviral immunity. Available evidence indicates that T cell exhaustion is associated with a series of metabolic and signaling deregulations and with a very peculiar epigenetic status which all together lead to reduced effector functions. A clear mechanistic network explaining how intracellular metabolic derangements, transcriptional and signaling alterations so far described are interconnected in a comprehensive and unified view of the T cell exhaustion differentiation profile is still lacking. Addressing this issue is of key importance for the development of innovative strategies to boost host immunity in order to achieve viral clearance. This review will discuss the current knowledge in HBV and HCV infections, addressing how innate immunity, metabolic derangements, extensive stress responses and altered epigenetic programs may be targeted to restore functionality and responsiveness of virus-specific CD8 T cells in the context of chronic virus infections.
1. Introduction
Therapy for chronic hepatitis B (CHB) relies on direct acting antiviral drugs, that suppress virus production but do not eradicate HBV from the liver, requiring lifelong treatments [1]. Therefore, there is a pressing need to find new therapeutic strategies achieving a definitive cure for CHB. Conversely, direct acting antiviral drugs (DAA) currently available for chronic hepatitis C lead to a sustained virologic response in 85–99% of cases, although a large population of treatment failures still exists [2]. For these reasons, the present review is aimed at addressing the emerging concepts of immune modulation for both chronic hepatitis B and C deriving from the most recent advances in viral pathogenesis.
In chronic hepatitis B and C, virus-specific T-lymphocytes have been described to be deeply dysfunctional, with severity of their hyporesponsive state at least in part depending upon viremia and antigenemia levels [3] (Figure 1).
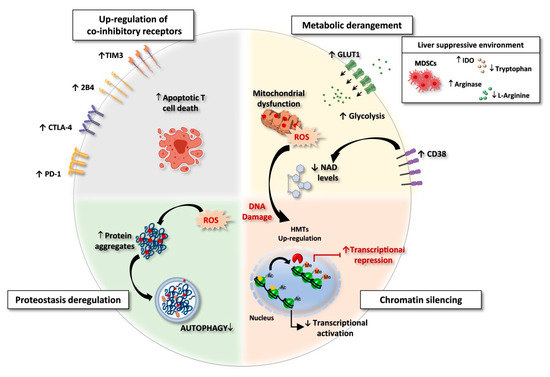
Figure 1.
Exhausted CD8 T cells in HBV and HCV chronic infections. Features of exhausted T cells: (i) Upregulation of multiple co-inhibitory receptors; (ii) Metabolic derangement with depolarized mitochondria and ROS accumulation. A suppressive intrahepatic microenvironment enriched with amino acids degrading enzymes (e.g., arginase and IDO/TDO) causes reduction of amino acid availability. Lower respiratory capacity and amino acid starvation favor glycolysis as a compensatory mechanism, with upregulation of the glucose transporter GLUT-1. In addition, over-expression of CD38 can lead to increased intracellular NAD consumption, exacerbating mitochondrial dysfunction. (iii) Chromatin silencing due to histone methyltransferases (HMTs) upregulation and to the reduced availability of acetyl-CoA, as a consequence of low NAD levels, fundamental co-factor for histone acetylation. (iv) Proteostasis deregulation. High ROS levels lead to oxidation and damage of proteins and cell organelles. Exhausted CD8 T cells express a deregulated proteostasis with lower autophagy rates, leading to a defective clearance of aggregated proteins and damaged organelles. Created with BioRender.com, access on 1 September 2021.
Such a dysfunctional state, called “T cell exhaustion”, is characterized by the progressive loss of T cell effector functions, due to the repeated triggering of CD8 T cells caused by persistent exposure to high antigen concentrations [4]. First observed in chronic LCMV infection in mice, it has subsequently been described in different models of animal and human chronic virus infections and in cancer [4]. Different degrees of functional T cell impairment have been identified, with IL-2 production, cytolytic activity, cytokine polyfunctionality and proliferative capacity lost first, followed by TNF-α and IFN-γ secretion, up to physical T cell deletion at the most terminal stages of exhaustion [5]. Besides persistent antigenic stimulation, further extrinsic factors, such as increased regulatory T cell activity, inhibitory cytokines, and the loss of CD4 T cell help have been shown to promote T cell dysfunction [6,7,8,9].
Comparisons of gene expression profiles of virus-specific CD8 T cells derived from acutely or chronically LCMV-infected mice led to the discovery of a high expression of the co-inhibitory receptor PD-1 in chronic infection, and to its role in promoting a dysfunctional phenotype [10]. Later, the co-expression of other up-regulated inhibitory checkpoints has been associated with the exhausted CD8 T cell phenotype [6,11,12,13]. Typically, the higher is the number of inhibitory receptors co-expressed by exhausted T cells, the more severe is the exhaustion state [11]. Additional features of exhausted T cells are an altered transcriptional program, a deregulated metabolism, a loss of memory markers, an up-regulation of some transcription factors, such as Eomes and Blimp-1 [14], with severe T cell exhaustion associated to high Eomes expression [15]. However, it has recently been demonstrated that not only Eomes expression, but also its subcellular localization influences the degree of T cell exhaustion: when present within the nucleus, Eomes regulates PD-1 expression by competing with the transcription factor Tbet for binding to Tbox sequences. Thus, a high Eomes:Tbet ratio can enhance PD-1 expression, since Eomes is a weaker PD-1 repressor than Tbet [16].
Starting from early observations showing that terminally exhausted PD-1hi and less differentiated PD-1int coexist within exhausted LCMV-specific CD8 T cells, it has progressively become clear that exhausted virus-specific CD8 T cells cannot be considered a homogeneously dysfunctional population [17]. Indeed, T cell subsets with different degrees of functionality and different sensitivity to immune modulatory interventions have been detected in different models of chronic viral infections. In addition, recent single cell (sc) RNA-seq data support the concept of a tissue-associated heterogeneity, showing that the transcriptomic profile of exhausted virus-specific CD8 T cells can be shaped by the specific microenvironment where they differentiate [18]. A subset of exhausted T cells expressing the TCF1 transcription factor has been identified as endowed with memory-like characteristics and a higher sensitivity to inhibitory checkpoint blockade than their TCF1-negative counterparts, that is composed of more terminally exhausted T cells [19,20]. Also in chronic hepatitis B, subsets of differentially exhausted virus-specific CD8 T cells have been observed [21,22], among which terminally exhausted cells do not represent the majority of the virus-specific population both at peripheral (personal communication) and at intrahepatic/intratumor level in CHB patients with hepatocellular carcinoma [23]. Moreover, various degrees of T cell dysfunction have been identified for HBV-specific CD8 T cells that recognize different HBV antigens [24]. Notably, PD-1 and CD127 expression levels can allow to identify a subset of PD-1low/-CD127high CD8 T cells, which are more sensitive to functional restoration, predicting the in vitro response to immune-modulatory treatments (personal communication) and thus allowing to identify those patients who are more likely to benefit from immune-modulation. Co-expression of CD127, PD-1 and TCF1 has been reported to identify also in chronic hepatitis C a subset of virus-specific CD8 T cells with residual proliferative potential [25]. This memory-like T cell subset is maintained after spontaneous or antiviral therapy-induced resolution of chronic infection, showing that recovery after a long-lasting exposure to HCV cannot lead to a complete reacquisition of phenotypic and function features of canonical memory T cells [25]. In line with this observation, virus-specific T cells maintain a “molecular scar” after resolution of chronic HBV and HCV infections which doesn’t persist following spontaneous control of acute hepatitis [26,27,28,29,30].
Although severe exhaustion is likely incompletely reversible even after complete antigen clearance, functional restoration can apparently be achieved with less terminally exhausted T cells, suggesting that immune-modulation may still be a possible strategy to fight chronic HBV and HCV infections.
Based on these premises, this review will focus on the proposed immune-modulatory approaches that have been tested in vitro or in vivo in chronic hepatitis B and C (Figure 2 and Table 1 and Table 2), targeting:
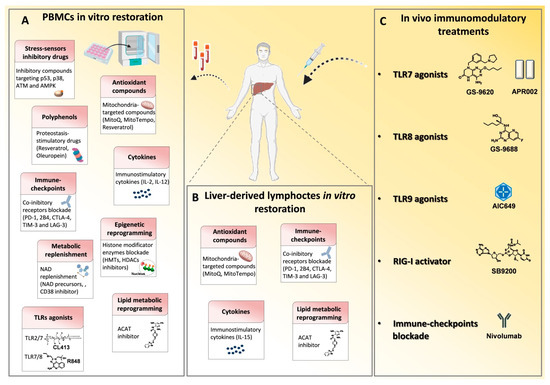
Figure 2.
Immune based approaches in viral hepatitis. Many drugs that modulate the immune system have been employed in chronic HBV and HCV infections in order to reconstitute antiviral T cell responses and achieve viral clearance. (A) In vitro T cell modulatory treatments tested on patient-derived PBMCs and (B) on liver-derived lymphocytes to restore signaling deregulations, cytokine levels, metabolic functions and epigenetic landscape and to block inhibitory receptor signals. (C) In vivo T cell function reconstitution interventions are summarized. They target the activation of innate immunity receptors, such as toll like receptors (TLRs) and retinoic acid-inducible gene 1 (RIG-I) by specific agonists and through checkpoint blockade, such as with an anti-PD-1 monoclonal antibody (Nivolumab). p53, tumor-suppressor protein; p38, mitogen-activated protein kinase; ATM, Ataxia Telangiectasia Mutated; AMPK, AMP-activated protein kinase; MitoQ, mitoquinone; MitoTempo, mitochondria-targeted superoxide dismutase; PD-1, programmed cell death 1; 2B4, Natural Killer Cell Receptor 2B4; CTLA4, cytotoxic T lymphocyte antigen 4; TIM3, T cell immunoglobulin mucin receptor 3; LAG3, lymphocyte activation gene 3 protein; HMTs, histone methyltransferases; HDACs, histone deacetylases; NAD, Nicotinamide adenine dinucleotide; NMN, nicotinamide mononucleotide; ACAT, Acetyl-CoA Acetyltransferase.

Table 1.
Novel immuno-therapeutic approaches for HBV and HCV chronic infections.
Table 2.
Combination Therapies based on immune-modulation for T cell restoration.
- -
- innate immunity receptors
- -
- co-inhibitory molecules
- -
- metabolic pathways
- -
- cytokine functions
- -
- epigenetic control of DNA transcription
2. Stimulation of Innate Immunity Receptors as an Immunomodulatory Strategy to Overcome HBV-Specific T Cell Exhaustion
Among the possible strategies to develop novel therapies for chronic HBV infection, the reconstitution of the anti-viral T cell function by stimulation of innate immunity receptors, such as toll like receptors (TLRs), retinoic acid-inducible gene 1 (RIG-I) and stimulator of interferon genes (STING), represents a potential approach to rescue protective immunity in CHB patients [71] (Figure 2 and Table 1 and Table 2). These agents are designed to stimulate pattern recognition receptors ultimately leading to the production of cytokines with antiviral activity (e.g., IFNs), with the final purpose of activating host immunity in a non-antigen-specific manner. TLRs are located on the cell surface or within the endosome where they sense molecular patterns associated with viruses and bacteria.
Many attempts to stimulate innate immunity through TLR7 activation have recently been described [31]. TLR7 is predominantly expressed on the endosomal membrane of B lymphocytes and plasmacytoid dendritic cells and its stimulation results in the activation of transcription factors NFkB and IRF7.
Recent studies showed a sustained antiviral response in chimpanzees and woodchucks displayed by the TLR7 agonist GS-9620 [32,33]. A further study conducted in humans by Boni et al. demonstrated that TLR7 stimulation can enhance NK and T cell responses in chronic patients with inhibition of HBV replication induced by nucleos(t)ide analogues (NUC) [34]. Although TLR7 stimulation improved the effect of NUC on T cell responses, this was not sufficient to permit the acquisition of an optimal immune reactivity adequate to provide complete control of infection, as detectable after spontaneous control of self-limited acute infections. This lack of therapeutic efficacy in CHB patients could be widely explained by the very low dose of drug used in humans to avoid possible toxic effects. Recently, a new TLR-7 activating agent, named APR002, has been used alone or in combination with Entecavir (ETV) in the woodchuck model of infection. Importantly, this compound displayed a preferential intrahepatic delivery due to the active uptake by the organic-transporting polypeptide 1B1/3 transporters, that are highly expressed on hepatocytes. Thanks to this liver selectivity, APR002 was highly localized in the woodchuck liver minimizing systemic exposure and promoting a sustained viral control in animals with chronic hepatitis [35,36].
TLR8 is another member of the pattern recognition receptor family, which is preferentially expressed in myeloid dendritic cells (DCs), monocytes, macrophages and Tregs. Recently, much interest was directed to GS-9688 (Selgantolimod), an agonist of endosomal TLR8, whose stimulation leads to the production of immunomodulatory (e.g., IL-12, IL-18) and pro-inflammatory cytokines (e.g., IL-1β, TNF-α), ultimately triggering a strong IFN-γ production by liver resident cells. Through these effects, agonist-induced activation of TLR-8 demonstrated antiviral efficacy both in vitro in HBV-infected primary human hepatocytes and in vivo in woodchucks chronically infected with woodchuck hepatitis virus (WHV) [37,38]. Further insights into the mechanism of action of GS-9688 were provided by investigating its effect in vitro on human immune cells of healthy subjects and CHB patients. TLR8 stimulation activated DCs and mononuclear phagocytes to produce IL-12 and TNF-α, increased the frequency of functional HBV-specific CD8 T cells in a proportion of CHB patients, activated NK cells, mucosal-associated invariant T cells (MAITs), and reduced the frequency of Tregs and myeloid-derived suppressor cells (MDSC) [39]. The safety, pharmacokinetics and pharmacodynamics of Selgantolimod was evaluated in CHB patients in a phase 1b study, in which a good tolerability and a transient dose-dependent elevation of serum IL-12 and IL-1RA were shown, suggesting possible effects on immune responses [40].
Interestingly, dual-acting TLR7/8 (R848) and TLR2/7 (CL413) agonists were described as potent inducers of a broad spectrum of antiviral cytokines able to efficiently inhibit HBV production in HBV-infected primary human hepatocytes, thus providing the rationale for a poly-specific TLR agonist therapy, as a novel approach for HBV cure [41].
More recently, the new immunomodulatory AIC649, an inactivated parapoxvirus-based particle able to stimulate innate immunity via the TLR9 pathway, was shown in combination with Entecavir to significantly decrease viral DNA and WHsAg in the woodchuck model of chronic hepatitis. Suppression of viral replication in the liver correlated with the intrahepatic expression of type I and II IFNs [42]. The importance of the TLR9 signaling as a new target for stimulatory approaches was also emphasized by a recent report demonstrating that host conditioning with the CpG oligonucleotide could restore exhausted intrahepatic CXCR5+ CD8 T cells, via upregulation of co-stimulatory molecules CD80/86 and OX40 on myeloid cells with subsequent increase of CD28-mediated signals on T cells [72].
RIG-I is an RNA sensor present in the cytoplasm, that induces IRF3, IRF7 and NF-kB activation leading to the production of type I and III IFNs, ISGs and pro-inflammatory cytokines. SB 9200 is an oral small dinucleotide that activates RIG-I and nucleotide binding oligomerization domain containing protein 2 (NOD2), resulting in the stimulation of the IFN signaling pathway. Pretreatment with SB 9200 followed by Entecavir administration demonstrated a marked antiviral efficacy and increased the expression of type I IFNs and ISGs in blood and liver in woodchucks chronically infected with WHV [43]. SB 9200 showed antiviral efficacy also against several HCV genotypes and against HCV derived from chronically infected patients who failed to respond to current antiviral treatments [44]. Remarkably, a recent study showed the efficacy of a small IRF3 agonist, called F7, and of a RIG-I agonist derived from a PAMP motif within the HBV genome comprising 50ppp and the poly-U/UC region of viral RNA in suppressing cccDNA in HBV-infected hepatocytes [73]. However, RIG-I stimulation does not represent at present a close therapeutic perspective for chronic hepatitis B since, despite some promising antiviral effects shown in animal models, a phase IIb trial with a RIG-I agonist has been discontinued due to unexpected serious adverse events.
3. Boosting Adaptive Immune Response by Blocking Co-Inhibitory Pathways
The role of co-inhibitory molecules in HBV-specific CD8 T cell exhaustion has extensively been described by many in vitro studies on both peripheral and intrahepatic virus-specific CD8 T cells (Figure 2, Table 1 and Table 2). Indeed, in chronic HBV infection, circulating virus-specific CD8 T cells are characterized by the up-regulation of several inhibitory receptors such as PD-1, 2B4, CTLA-4, TIM-3 and LAG-3 with maximal expression of PD-1 in liver-infiltrating HBV-specific T lymphocytes [45,46,53,54,55,56] (Figure 1).
As previously reported in other chronic viral infections, such as LCMV and HCV [4,19,20,25,26,74,75], also exhausted HBV-specific CD8 T cells are a heterogeneous population composed of different subsets with distinct levels of exhaustion according to different HBV antigen specificity that coexist together [21,22,24,27,76]. Due to this heterogeneity, virus-specific CD8 T cells have been reported in other models of T cell exhaustion to display variable susceptibility to immune restoration treatments [19,20,77,78,79,80]. Therefore, identifying those patients who are more likely to benefit from immune-therapy on the basis of their degree of dysfunction may represent a useful strategy to select optimal candidates for functional T cell restoration treatments.
PD-1 is the most widely studied inhibitory receptor in both tumors and chronic viral infections and at present, in the field of chronic HBV infection, strategies aimed at blocking the PD-1/PD-L1 pathway represent the most investigated therapeutic option in vitro, in animal models and in chronically infected patients. Agents that block the PD-1/PD-L1 axis have been described to enhance HBV-specific T cell responses in many in vitro studies, showing a higher level of improvement on the intrahepatic than on the peripheral compartment [46,47,53]. However, T cell functional restoration induced by PD-1/PD-L1 blockade, also in association with the modulation of other co-inhibitory or co-stimulatory pathways, such as CTLA-4, CD137, TIM-3, 2B4, was observed only in a limited percentage of CHB patients with a wide heterogeneity of T cell responsiveness [45,54,55,56]. Interestingly, an increased secretion of IFN-γ and IL-21 producing HBV-specific CD4 T cells has been observed upon OX40 stimulation in combination with PD-L1 blockade, suggesting that immunotherapeutic approaches can also improve HBV-specific CD4 T cell responses [57].
A triple combination therapy of ETV treatment, DNA vaccination and in vivo PD-L1 blockade has been reported in woodchucks infected with WHV to boost virus-specific T cell responses leading to loss of WHsAg and decrease of viral replication [48,49]. In a more recent study, WHV-infected woodchucks were treated with a combination of anti-PD-L1 antibody and NUC. This combinatory approach improved control of viremia and antigenemia compared to ETV treatment alone, but with efficacy restricted to a proportion of animals [50]. The first evidence for a potential benefit of PD-1/PD-L1 blockade in the treatment of chronically infected HBV patients comes from the first clinical trial of anti-PD-1 (Nivolumab) in patients with advanced hepatocellular carcinoma, some of them with HBV infection under antiviral therapy [81]. Indeed, the PD-1 inhibitor nivolumab showed a safety profile devoid of severe hepatic toxicity, with no reactivation of HBV among the cohort of CHB patients. Interestingly, 6% of subjects with chronic HBV infection revealed a substantial decline of HBsAg upon nivolumab treatment.
A recent report extensively reviewed PD-1/PD-L1 blockade immunotherapy clinical trials to investigate the safety and efficacy of this new immunomodulatory strategy in the particular setting of HBV-infected HCC patients. The authors reported acceptable toxicity similar to that of non-infected HCC subjects and promising outcomes. However, due to the standard anti-viral therapies routinely administered in these trials, the antiviral efficacy of anti-PD-1/PD-L1 treatment was not evaluated [51].
Recently, in a phase 1 clinical trial a single low dose of Nivolumab was given to virally suppressed HBeAg negative chronic HBV patients with or without the HBV therapeutic vaccine GS-4774. Of note, significant HBsAg declines were observed in a limited proportion of patients, while one subject showed undetectable HBsAg levels associated with an increase of core- and surface-antigen specific T cell responses [52]. No serious adverse effects were reported likely due to the very low dose (ten times lower than the approved dose for tumors) and the short time of anti-PD-1 administration. Despite these encouraging results, however, the major concern related to PD-1/PD-L1 pathway blockade in chronic HBV infection remains the possibility of severe liver inflammation and hepatitis exacerbation [70]. In addition, the evidence that sustained responses upon PD-1 blockade treatment are detectable only in a limited proportion of HCC and chronic hepatitis B patients suggests the need of developing predictive parameters of response to treatment in order to select the patient cohorts that can benefit of check-point inhibition and to design further modulatory approaches to combine with PD-1 blockade to be used in alternative to it. Along with this line of possible strategies, inhibition of acyl-CoA:cholesterol acyltransferase (ACAT) has recently been reported to significantly improve in vitro HBV-specific CD8 T cell responsiveness to PD-1 blockade pointing to the possibility to combine metabolic and immune checkpoint modulation for the functional cure of HBV infection and HBV-related HCC [69].
4. Targeting Metabolism to Restore T Cell Exhaustion
4.1. Early Chronic Infections
TCR stimulation elicits multiple intracellular pathways required for metabolic reprogramming and effector function engagement. During T cell activation, glycolysis and mitochondrial dynamics are central for fine-tuning metabolic switching to meet the energetic requirements of T cells in response to nutrients. The metabolic plasticity is a key feature of T cell activation and it is fundamental for the responsiveness to changes in the tissue microenvironment [82,83].
During acute infection, CD8 T cells shift from mitochondrial oxidative phosphorylation distinctive of the quiescent state of naïve T cells, to a glycolysis-driven energetic metabolism necessary for activated effector T cells. This remarkable metabolic reprogramming, known as Warburg effect or aerobic glycolysis, is mandatory to meet the initial proliferative burst and it is due to phosphoinositide 3-kinase (PI3K) and mTOR signaling pathways triggered by TCR and CD28 co-stimulation [82,84,85]. After completion of the effector phase, developing memory T cells need to shift back to oxidative metabolism guided by mitochondrial fatty acid degradation. On the other hand, during chronic viral infection, long-term antigen exposure drives a specific T cell differentiation program [82,83,86,87].
Transcriptional and functional profiles of developing exhausted T cells display metabolic derangements, including suppressed glycolytic functions, reduced cellular respiration associated with diminished glucose uptake, with a final marked decline in ATP generation. This early scenario is evolutionarily maintained in early exhausted T cells in both murine (i.e., LCMV) and human (i.e., chronically-evolving acute HCV infection) virus infection, but the transcriptional dysregulated signature is less conserved in established exhausted T cells across different species and different models of infection, including HCV [64,88,89], where functional enriched modules have been associated to cell cycle replication, RNA processing, intracellular signaling and histone methylation processes [64].
Therefore, when severe exhaustion is not established yet, CD8 T cells appear to be unable to meet the energy requirement for effector function maturation. Indeed, they show reduced glucose utilization associated with high anabolic signal levels, as indicated by the strong upregulation of genes involved in class I PI3K–AKT-mTOR signaling linked to CD28 upregulation [88], which may cause TCR-signaling defects resulting in a high expression of the PD-1 co-inhibitory receptor. This derangement skews precursors of exhausted T cells to an altered metabolic profile susceptible to mitochondrial depolarization and high reactive oxygen species (ROS) production that contribute to the activation of stress sensor signaling pathways associated with the inhibition of TCR signalosome and of telomerase activity [64].
Several deregulated signaling pathways and molecules are involved in these early metabolic defects, including the peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC-1α) [88], which is a key transcriptional regulator of mitochondrial biogenesis-related genes [4,90], as well as DNA-repair and metabolic effectors, such as p53/ATM and AMPK/p38, reported in HCV infection [64]. Indeed, PGC-1α expression is significantly reduced in early exhausted virus-specific CD8 T cells, which express high levels of PD-1 and display depolarized mitochondria with a distinctive morphology resulting from mitochondria accumulation and fusion which lead to ROS overproduction [88]. In vivo, PGC-1α overexpression corrected the dysregulated mitochondrial phenotype and significantly increased polyfunctionality of CD8 T cells, by improving the metabolic cell fitness in chronic LCMV infection [88].
During early acute HCV infection two different studies described transcriptional derangements related to DNA repair and histone methylation processes in early exhausted T cells [64,89]. Due to the high antigenemia, a continuous TCR stimulation activates proliferative signals which require a sustained DNA repair response to fix the resulting DNA damages. In an effort to elucidate the mechanisms behind the poor metabolic fitness described in early exhausted CD8 T cells, different regulatory pathways responsible for metabolic alterations have been described allowing to define possible targeting strategies for their correction. For instance, a specific inhibition of downstream hyper-expressed effectors of the activation signaling cascade in HCV-specific CD8 T cells elicits a functional metabolic and antiviral T cell reconstitution in the majority of tested patients [64]. This CD8 T cell restoration effect can be achieved in chronically evolving acute HCV infection by blocking p53, a master regulator of cell-cycle arrest and metabolic reprogramming, activated in response to replicative stress, which can ultimately cause cellular apoptosis in case of irreparable insult [64,91,92]. Similar effects have been reported by inhibiting the upstream and downstream effectors of p53 [64,93], including ataxia telangiectasia mutated kinase (ATM), the major activator of the DNA damage response [94], activated during energetic stress, and p38 MAPK, a final effector molecule which can inhibit TCR-signaling and telomerase functionality [64,95,96] (Figure 2 and Table 1 and Table 2).
Signaling deregulation in the acute phase of chronically evolving HCV infection is possibly guided by ROS overproduction resulting from an oxidative metabolic reprogramming modulated by oxidative-instructive signals on mitochondrial activity that have been reported by both PD-1 and p53 [91,97]. Indeed, lymphocyte treatment with anti-oxidant molecules, such as resveratrol and N-acetyl-L-cysteine (NAC), can reduce intracellular ROS levels with a resulting activity decline of the upstream replicative-stress sensor ATM kinase [64]. Thus, the energy unbalance associated with low glycolytic levels and mitochondrial derangement, can increase DNA damage through ROS over-production, cell-cycle arrest, ultimately resulting either in apoptosis of early exhausted lymphocytes or in the survival of terminally exhausted poorly functional CD8 T cells.
Mitochondrial ROS-induced DNA damage accumulation results in the persistent activation of DNA repair mechanisms, including PARP enzymes [98], as suggested by the up-regulation of PARP-2 and PARP-1 transcripts in chronically evolving as compared with self-limiting HCV infections [64]. PARPs utilize NAD molecules as donors of poly-(ADP)-ribose monomers to catalyse the formation of poly-(ADP)-ribose chains that mediate early recruitment of DNA repair factors in correspondence of the DNA breaks [99]. Persistent PARP activation, induced by persistent DNA damage, can lead to a profound consumption of the cellular NAD pool, which can affect different metabolic and signaling pathways (see below).
4.2. Established Chronic Infections
Comparison of early- and late-stages of chronic HCV and HBV infections by transcriptomic analyses of virus-specific CD8 cells [26,64,65,89] revealed completely divergent profiles of early and late exhausted CD8 T cells: upregulated genes were highly enriched in early exhausted cells derived from the acute phase of HCV infection, while a massive gene downregulation was prevalent in late exhausted cells of the established chronic phase of both infections [64,65].
Indeed, in long term chronic infections, an extensive transcriptional reprogramming with global repression in gene expression has been described in both hepatitis B and C (Figure 1), as a likely survival strategy which can allow exhausted lymphocytes to endure continuous antigenic stimulation that leads to the progressive loss of effector functions.
In both chronic infections the most significantly downregulated cellular pathways and functions are relative to mitochondria, proteostasis, DNA repair and transcription [64,65] (Figure 1). Interestingly, these altered processes are mechanistically interconnected and, at least in part, attributable to NAD shortage (personal communication). Indeed, in addition to hyper-stimulation of PARPs activity by accumulated DNA damage, that can take place in the acute phase of infection, CD38, a major NAD+ hydrolyzing enzyme, is expected to play a central role in NAD consumption and depletion [100], since it is overexpressed in both HBV- and HCV-specific CD8 T cells in the acute and chronic stages of infection [64,65] (Figure 1). NAD depletion can cause reduced activity of sirtuins, which are NAD-dependent deacetylases with multiple regulatory roles [101]. In addition, NAD shortage can influence epigenetic control of DNA transcription both by limiting sirtuins’ activity [102,103,104] and by metabolic modifications. Indeed, lowered NAD/NADH ratio can induce a preferential conversion of pyruvate to lactate and decreased formation of acetyl-CoA available for histone acetylation [105], leading to chromatin condensation and gene silencing, in line with the global gene expression downregulation observed in chronic hepatitis B and C [64,65] (Figure 1). Along this line, CD38 overexpression causing NAD shortage and sirtuin 1 activity reduction has been demonstrated to cause the increased activity of the methyltransferase EZH2 in dysfunctional T cells from patients with systemic lupus erythematosus, leading to enhanced histone methylation [104]. This mechanism is likely to be active also in virus-specific CD8 T cells during chronic hepatitis C, where an enhanced histone methylation by EZH2 has been observed [64].
Reduced sirtuin activity also contributes to mitochondrial dysfunction, through increased mitochondrial ROS and impaired mitophagy and may affect directly several DNA repair pathways, generating a vicious cycle leading to persistent DNA damage and NAD consumption [106].
In addition, generation of the sirtuin-inhibitor nicotinamide as a product of NAD hydrolysis by CD38 [107], can lead to loss of sirtuin function, further exacerbating DNA damage and mitochondrial dysfunction [106,108].
Given the key role suggested by these findings for NAD depletion in CD8 T cell exhaustion pathogenesis, replenishment of the depleted NAD pool may represent a possible strategy to be pursued in the future to restore the anti-viral function of exhausted CD8 T cells.
In the persistently infected liver different inhibitory signals originate from the peculiar intrahepatic environment, contributing to further repress exhausted T cells by influencing T cell metabolism and function.
Amino acid starvation is one of the mechanisms described to inhibit T cell proliferation, since damaged hepatocytes and other infiltrating immune cells, can release enzymes able to degrade amino acids, like arginine or tryptophan. Indeed, hepatocytes constitutively express tryptophan-2,3-deoxygenase (TDO), that catalyzes the first limiting step of tryptophan degradation oriented to the biosynthesis of NAD, producing toxic intermediates collectively known as kynurenines [109] (Figure 1). Hepatocyte damage can lead to tryptophan depletion together with the massive production of kynurenines, that have been demonstrated to inhibit T and NK cell proliferation. T cell expansion at the intrahepatic level can be critically affected also by shortage of another essential amino acid, L-arginine, due to an excess of arginase derived from damaged hepatocytes [109], leading to T cell arrest in the G0/G1 phase and CD3ζ downregulation (Figure 1). In chronic HBV infection a subset of arginase-expressing myeloid-derived suppressor cells (MDSC) has been found particularly expanded in the liver of patients with high viral load and low liver inflammation [68]. In these patients, arginine depletion leads to compensative up-regulation of amino acid transporters on T cells, to enhance essential amino acids up-take. The relevance of these mechanisms is confirmed by the evidence that arginine replenishment, as well as the addition of an arginase inhibitor, can partially restore the T cell function [68].
Moreover, the high intrahepatic cholesterol concentration is a factor likely able to influence T cell metabolism and function, as in immune cells lipid accumulation has been reported to affect the cellular function.
In line with this, in chronic hepatitis B inhibition of cholesterol esterification by the acyl-CoA:cholesterol acyltransferase (ACAT), that catalyzes the esterification of excess intracellular cholesterol for storage in lipid droplets, enhanced TCR signaling upon CD8 T cell stimulation, by reducing lipid droplet formation and diverting cholesterol to the cell membrane [69] (Figure 2, Table 1 and Table 2). This strategy has been shown to be active both to enhance HBV- and HCC- specific T cell proliferation and effector functions, by optimizing cells bioenergetics, and to exert an antiviral effect by reducing HBV virions and viral particles formation in vitro [69].
The intrahepatic environment can also influence autophagy up-regulation in tissue-resident T cells, which is needed to counterbalance mitochondria depolarization and thus maintain mitochondrial fitness and T cell functionality in such a condition of high oxidative stress, as autophagy inhibition was shown to induce depolarized mitochondria accumulation [58]. Indeed, HBV-specific CD8 T cells in chronic hepatitis B have been shown by both transcriptional and functional validation analysis to display defective autophagy and dysfunctional mitochondria with low membrane potential and high ROS production [65] (Figure 1). Interestingly, IL-15 and the natural polyphenolic compounds resveratrol and oleuropein not only increased the cellular proteostasis, but could also restore the mitochondrial function, thus leading to an improved antiviral T cell function in CHB patients [58,66] (Figure 2, Table 1 and Table 2).
The presence of depolarized mitochondria is indeed a typical feature of exhausted T cells, detected both in chronic hepatitis B and C, which involves a lower respiratory capacity for energy generation [64,65]. As a consequence, in chronic HBV infection HBV-specific CD8 T cells rely upon a compensatory glycolytic function, with up-regulation of the glucose transporter GLUT-1, in an attempt to counterbalance dysfunctional mitochondria [59,65] (Figure 1). Administration of the cytokine IL-12 could reverse mitochondria depolarization and glycolysis dependence, finally improving the antiviral T cell function [59] (Figure 2, Table 1 and Table 2). Also mitochondria-targeted (mt-)antioxidant compounds led to an improved mitochondrial fitness and T cell cytokine production in T cells isolated from patients with chronic hepatitis B by neutralizing excess ROS production in vitro [65,66]. The mt-antioxidant MitoQ has been tested also in vivo in patients with chronic HCV infection not responding to PEG-interferon plus ribavirin treatment, in a phase II study. It showed lack of toxicity and induced a decrease in ALT levels, showing liver protection activity, although not reducing the viral burden [67] (Figure 2, Table 1 and Table 2).
5. Cytokine Fueling for T Cell Restoration
An important aspect of cytokine therapeutic usage relies on targeting both intracellular signaling and metabolic dysfunctions detected in T cell exhaustion, with the possibility of metabolic reprogramming (Figure 2, Table 1 and Table 2).
IL-2 secretion from CD8 T cells occurs early after TCR engagement. IL-2 boosts AKT1 and mTORC1 activation through IL-2Rα (CD25) triggering, thereby conveying signals to promote glucose and amino acid uptake as well as protein and lipid biosynthesis, required for proper T cell effector functions [110,111,112,113].
In vivo treatment with recombinant IL-2 was recently reported to induce better T cell effector differentiation of HBV-specific CD8+ T cells. Studies were conducted in Iannacone’s lab through the generation of unique mice models which express HBV antigens either selectively in hepatocytes (MUP-core mice), causing defective antigen presentation and lack of differentiation in functional effector CD8 T cells, or only in Kupffer cells and hepatic dendritic cells (HDCs) with efficient antigen presentation leading to proper and functional differentiation of effector CD8 T cells [61]. Through IL-2 supply, defective virus-specific CD8+ T cells restored the ability to proliferate and differentiate in IFN-γ producing cells, whilst anti-PD-L1 alone administration was not effective [61]. Moreover, IL-2 treatment was able to restore the transcriptional program of the dysfunctional virus-specific CD8+ T cells from MUP-core mice livers, as revealed by RNA-seq experiments [61]. The therapeutic efficacy was also confirmed by IL-2 treatment in HBV replication-competent transgenic mice, providing evidence of cytotoxic and IFN-γ secreting core-specific CD8+ T cell differentiation in the liver. Results in this mouse pre-clinical model were then assessed in different HBV infected patient categories. Only HBV-specific T cells derived from immune-tolerant patients, which more closely resemble T cells that have been primed by hepatocytes, were in fact rescued by IL-2 addition in vitro, acquiring proper IFN-γ production levels observed in immune-active patients, which were not boosted by IL-2 addition [61]. These conclusions indicate that the preclinical mouse model developed in Iannacone’s lab nicely reflects the pathogenetic mechanisms involved in neonatal infection and point to the relevance of conducting specific trials in selected cohorts of chronic HBV patients identified on the basis of the degree of exhaustion. In vivo use of IL-2 is, however, not recommended for systemic toxicity [112]. Therefore, a modified version of IL-2 (named IL-2c), previously shown to have reduced toxic systemic effects and able to selectively target CD8 T cells [112], has been employed as a new immunotherapeutic tool in the context of chronic HBV infection [61].
Another therapeutic option is provided by the pro-inflammatory cytokine IL-12 that has been shown to be able to improve the function of HBV-specific CD8 T cells with an augmented effect when tested in combination with PD-1 blockade. IL-12 alone was effective in restoring both polyfunctional secretion of multiple cytokines and cytotoxic activity in HBV-specific CD8+ T cells due to the increased expression of the cytokine-regulatory transcription factor Tbet, along with decline of PD-1 and of the pro-apoptotic Bim expression, with the consequent reduction in Bim-mediated apoptosis [60]. In addition, IL-12 could efficiently enhance mitochondrial fitness with better control of mitochondrial membrane potential and reduction in the percentage of depolarized mitochondria and with restoration of IFN-γ production in HBV-specific CD8 T cells [59].
In vivo systemic IL-12 administration, such as in cancer trials, has poorly been tolerated but the use of TLR-8 agonists (e.g., GS-9688), which stimulate the production of pro-inflammatory cytokines, including IL-12, is an attractive alternative in combination with a therapeutic DNA vaccine for CHB patient treatment [114,115,116].
The magnitude of T cell effector responses is augmented when CD8 cells are activated by the combination of IL-12 and IL-2 by virtue of a functional synergy on STAT1 and STAT3 phosphorylation observed in activated T cells but this combination has never been tested in CHB patients [117].
A promising molecular target for exhausted T cells in CHB patients is represented by IL-15, which has been reported in vitro to increase the autophagic activity of tissue resident memory lymphocytes [58]. Given the deficiency of lysosome-mediated autophagy observed in HBV-specific CD8 T cells [66], IL-15 use in chronic HBV patients deserves to be tested for its capacity to restore the anti-viral CD8 T cell function.
In CHB patients an interesting immune approach for metabolic T cell reconstitution consists of PGE2 signaling blockade. PGE2 has been shown to contribute to CD8 T cell exhaustion development during viral infections both in mice and in humans [63]. Indeed, blockade of PGE2 signaling in the AAV-HBV1.2 mice model reduced TIM3 expression on total CD8 T cells and induced restoration of the CD8+ T cell function and control of HBV infection [63]. However, the exact mechanisms for PGE2 regulation on CD8 T cells is still debated in chronic HBV infection and needs to be elucidated.
In the setting of chronic HCV infection IL-7 has been tested in vitro. It was shown to be able to correct the downregulation of the TNF receptor-associated factor 1 (TRAF1), which correlates with the exhaustion phenotype in HCV-specific CD8 T cells. Moreover, IL-7 could improve HCV-specific CD8 T cell proliferation capacity when added to triggering of the positive costimulatory receptor 41BB, and even more when also PD-L1 blocking was associated, particularly in patients with slowly progressing fibrosis [62].
6. T Cell Epigenetic Targets for Therapies during Chronic Infections
A great research effort has been devoted in the last years to understand the relationship between metabolism and T cell differentiation programs [118,119,120]. The comprehension of how metabolic changes can impact on the epigenetic control of gene expression is key to understanding the T cell differentiation process during persistent viral infections. Epigenetic mechanisms are crucial for T cell differentiation by acting directly on gene expression profiles, thereby representing a multilayered regulation between signaling events, metabolic reprogramming and differentiation gene programs in T cells. TCR and IL-2 signalings coordinate effector T cell activation through the induction of the Myc oncogene, a transcription factor which controls expression of genes encoding transporters and enzymes involved in the glycolysis and glutaminolysis pathways, which regulate ribosome biogenesis by amplifying gene expression [119,121,122,123,124]. Moreover, the TCR-inducible transcription factors, SREBPs (sterol binding proteins), can activate expression of components of the lipid biosynthesis pathway that in the early stages of T cell induction is fundamental for generating new cellular membranes which are necessary for rapid T cell division [125,126]. Other key transcription factors are members of the hypoxia-inducible factors (HIFs) which are directly regulated by IL-2-dependent events, with a further positive feedback for genes that encode glycolysis and glutaminolysis components [127,128].
In this perspective, transcription factors which represent signaling mediators, provide regulating decisions fundamental for metabolic gene reprogramming, which depends also on the nutrient supply present in the microenvironment [129]. All these aspects have strong influence on the proper cellular differentiation program during viral infections.
The correct metabolic reprogramming during T cell differentiation is obviously central for the resultant availability of metabolites which could serve as substrates, donors, cofactors and antagonists for epigenetic-modifying complexes and consequently for the overall epigenetic status.
For instance, glycolysis is the natural primary pathway used to generate the acetyl-CoA cofactor during T cell differentiation [130]. The enzyme LDHA (lactate dehydrogenase A) transforms pyruvate in lactate to avoid oxidation of glucose in the mitochondria, leading to accumulation of citrate in the cytosol, which is then converted into acetyl-CoA by the enzyme ATP-citrate lyase, ultimately ending in the enhancement of histone acetylation modifications, such as histone H3 lysine 9 (H3K9Ac) and histone H3 lysine 27 (H3K27Ac) in the IFN-γ locus, to boost T cell effector gene expression [130,131]. This provides a link between glycolysis, acetyl-CoA and histone acetylation in the regulation of T cell differentiation. Thus, understanding the fine tuning of enzymes that modify histones or DNA such as “writers” which add chemical groups (e.g., histone acetyltransferases—HATs, histone methyltransferases—HMTs, and DNA methyltransferases—DNMTs) and “erasers” which remove them (histone deacetylases—HDACs, and Tet enzymes) will be necessary for the design of correct and effective strategies to reconstitute T cell function and fate by chromatin modulation and epigenetic inhibitor strategies for immune-related diseases.
Expanding studies based on assays with high-throughput sequencing for transposase-accessible chromatin (ATAC-seq) and for chromatin immunoprecipitation (ChIP-seq) have been carried out to characterize histone modifications and chromatin accessibility in immune cells at different stages of development [74,75,132,133,134,135]. By genomic profiling, the transcriptional and epigenetic landscape of exhausted T cells has been elucidated and shown to be endowed with a distinct transcriptional program compared to effector and memory T cells. Exhausted CD8 T cells diverge from other CD8 T cells by ~6000 differentially accessible chromatin regions which are linked to differentially expressed genes regulated by distinctive molecular circuits of transcription factors [75,134,136]. Also in HBV infection derangement of transcriptional factors has been identified with the discovery of up-regulation of negative-regulator of transcription and closed chromatin configuration in HBV-specific CD8 T cells from chronic patients [65]. Integrated network analyses of exhausted T cells showed an altered transcriptional connectivity of pivotal transcription factors, such as Tcf-1 (T-box factor) and Eomes, which are expressed both in functional effector/memory T cells and in exhausted T cells with different transcriptional circuits, leading to different gene expression patterns guided by context-specific activities [15,64,89,137,138]. This scenario strengthens the concept that the epigenetic landscape of exhausted T cells is a stable condition, early induced during development of exhaustion, which regulates responsiveness to checkpoint blockade treatment [75,134,139,140]. As already discussed, checkpoint blockade (e.g., on PD-1) can induce functional reinvigoration of CD8 T cells which is associated however with limited changes in the epigenetic state, explaining why cells tend to revert back to the functional and transcriptional features of the prior exhausted condition [75].
Importantly, highly condensed regions of heterochromatin contain few active genes and are characterized by high levels of histone modifications, such as tri-methylation of histone H3 lysine 9 (H3K9me3), tri-methylation of histone H3 lysine 27 (H3K27me3) and low levels of histone acetylation [141].
As previously described, a distinctive transcriptional pattern has been shown in dysfunctional CD8 T cells with a prevalent downregulation of gene expression also in human chronic hepatitis infections [64,65,89]. Conversely, by investigating epigenetic-modifying complexes, early during chronically-evolving acute infections a dysregulated over-expression of histone methyltransferases (e.g., G9a and EZH2 HMTs) has been observed [64]. One of the best-known methyltransferase complexes active in T cell differentiation is the polycomb repressive complex (PRC2) [142], whose catalytic subunits are EZH2 and EZH1, which are responsible for H3K27me3 histone modification and thus for a non-permissive chromatin signature [143].
In response to a euchromatic DNA double-strand break (DSB), such as the DNA breaks occurring as a result of the high proliferation rate of CD8 T cells in early acute viral infections, an ATM-mediated signal is sent to repress transcription in flanking chromatin regions [144] which is catalyzed by polycomb complex proteins [145,146]. Subunits present in PRC complexes are then recruited to the sites of DNA damage to help DNA repair mechanisms, mediating DSB-induced transcriptional silencing [147,148,149,150]. This scenario is similar to that observed in the early phases of persisting HCV infection, where increased expression and activity of ATM, p53 and HMTs have been observed [64].
Moreover, several studies described key roles for PRC2 and G9a HMT in CD4 and CD8 T cell differentiation for effector and memory commitment [132,151,152,153,154].
G9a (EHMT2) is a lysine methyltransferase whose primary function is to di-methylate histone H3 lysine 9 (H3K9me2) by acting primarily through the recruitment of H3K9me2-binding proteins that prevent transcriptional activation [155]. In addition, G9a directly interacts with Blimp-1, a zinc finger protein that has been shown to repress key aspects of normal memory CD8+ T cell differentiation and to promote high expression of inhibitory receptors during T cell exhaustion in chronic viral infection [14]. Moreover, G9a can directly repress type I cytokine gene expression, such as the IFN-γ locus during Th2 differentiation by regulating both H3K9me2 and H3K27me3 [156].
An intriguing study recently showed that CD8+CD38high T cells displayed increased expression of EZH2 compared with CD8+CD38low, by pinpointing the underlying mechanism responsible for a decreased cytotoxicity of the cells primarily due to low NAD levels which alters chromatin organization [104].
A functional inhibition of HMTs by treatment with specific EZH2 and G9a inhibitory compounds, led to a T cell functional reconstitution both as anti-viral cytokine production and as proliferative capacity, with a partial metabolic restoration [64] (Figure 2, Table 1 and Table 2). Although CD8 T cell functional restoration has been observed in both acute and chronic stages of HCV infection, the highest stimulatory effect on HCV-specific CD8 cells was observed when chronicity was already established [64]. Moreover, in tumor-bearing mice a synergistic effect has been seen between several epigenetic inhibitors (e.g., blockade of EZH2) and check-point blockade treatments (such as with anti-CTLA-4 and anti-PD-1 treatment) with respect to their immune-modulatory actions [157,158].
The picture of T cell exhaustion that emerges from HCV and HBV infections, thus, highlights a profound and wide-ranging cellular perturbation centered on metabolic and signaling deregulation that results in a global scenario of chromatin remodeling leading to a complete repression in gene expression characteristics of the established T cell exhaustion (Figure 1). Therefore, epigenetics drugs represent potential and promising candidate for immune modulatory therapies for chronic viral infections which may have the capacity to restore several T cell functions together.
7. Future Perspectives
Stress responses and deregulated metabolic pathways can severely impair T cell immunity during chronic viral hepatitis. The comprehension of these alterations in metabolism, signaling and epigenetics will certainly open new horizons for the definition of novel immunometabolic targets to restore proper T cell differentiation pathways. Particular attention should be devoted to the possible modulation of epigenetic mechanisms and to circumvent the potent tolerogenic effect exerted by the liver microenvironment, where elevated antigen loads can persistently exert high pressure on T cell responses, skewing them towards the activation of inhibitory intracellular circuits.
Also virus-specific T cell heterogeneity should be better decrypted, perhaps making use of the artificial intelligence to discover novel molecular targets for immune modulatory therapies and novel predictive parameters to identify patients more likely responsive to modulatory compounds.
Funding
This work was supported by a grant from Emilia-Romagna Region, Italy (Programma di Ricerca Regione-Università 2010-2012; PRUa1RI-2012-006), by a grant from the Italian Ministry of Health (Ricerca Finalizzata RF 2013-02359333) and by a PRIN (Progetti di Ricerca di Rilevante Interesse Nazionale) grant (2017MPCWPY_002), Ministry of University and Research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zoulim, F.; Lebossé, F.; Levrero, M. Current treatments for chronic hepatitis B virus infections. Curr. Opin. Virol. 2016, 18, 109–116. [Google Scholar] [CrossRef]
- Spearman, C.W.; Dusheiko, G.M.; Hellard, M.; Sonderup, M. Hepatitis C. Lancet 2019, 394, 1451–1466. [Google Scholar] [CrossRef]
- Rehermann, B. Pathogenesis of chronic viral hepatitis: Differential roles of T cells and NK cells. Nat. Med. 2013, 19, 859–868. [Google Scholar] [CrossRef] [Green Version]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.T.; Oldstone, M.B.A. IL-10: Achieving balance during persistent viral infection. Curr. Top. Microbiol. Immunol. 2014, 380, 129–144. [Google Scholar]
- Wilson, E.B.; Brooks, D.G. The Role of IL-10 in Regulating Immunity to Persistent Viral Infections. In Negative Co-Receptors and Ligands; Ahmed, R., Honjo, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 39–65. ISBN 978-3-642-19545-7. [Google Scholar]
- Battegay, M.; Moskophidis, D.; Rahemtulla, A.; Hengartner, H.; Mak, T.W.; Zinkernagel, R.M. Enhanced establishment of a virus carrier state in adult CD4+ T-cell-deficient mice. J. Virol. 1994, 68, 4700–4704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Chew, G.M.; Fujita, T.; Webb, G.M.; Burwitz, B.J.; Wu, H.L.; Reed, J.S.; Hammond, K.B.; Clayton, K.L.; Ishii, N.; Abdel-Mohsen, M.; et al. TIGIT Marks Exhausted T Cells, Correlates with Disease Progression, and Serves as a Target for Immune Restoration in HIV and SIV Infection. PLoS Pathog. 2016, 12, e1005349. [Google Scholar] [CrossRef] [Green Version]
- Tinoco, R.; Carrette, F.; Barraza, M.L.; Otero, D.C.; Magaña, J.; Bosenberg, M.W.; Swain, S.L.; Bradley, L.M. PSGL-1 Is an Immune Checkpoint Regulator that Promotes T Cell Exhaustion. Immunity 2016, 44, 1190–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A Role for the Transcriptional Repressor Blimp-1 in CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLane, L.M.; Ngiow, S.F.; Chen, Z.; Attanasio, J.; Manne, S.; Ruthel, G.; Wu, J.E.; Staupe, R.P.; Xu, W.; Amaravadi, R.K.; et al. Role of nuclear localization in the regulation and function of T-bet and Eomes in exhausted CD8 T cells. Cell Rep. 2021, 35, 109120. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by αPD-L1 blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandu, I.; Cerletti, D.; Oetiker, N.; Borsa, M.; Wagen, F.; Spadafora, I.; Welten, S.P.M.; Stolz, U.; Oxenius, A.; Claassen, M. Landscape of Exhausted Virus-Specific CD8 T Cells in Chronic LCMV Infection. Cell Rep. 2020, 32, 108078. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, D.T.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef] [Green Version]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Li, Y.; Tang, L.; Guo, L.; Chen, C.; Gu, S.; Zhou, Y.; Ye, G.; Li, X.; Wang, W.; Liao, X.; et al. CXCL13-mediated recruitment of intrahepatic CXCR5+CD8+ T cells favors viral control in chronic HBV infection. J. Hepatol. 2020, 72, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Zhu, Y.O.; Becht, E.; Aw, P.; Chen, J.; Poidinger, M.; de Sessions, P.F.; Hibberd, M.L.; Bertoletti, A.; Lim, S.G.; et al. Multifactorial heterogeneity of virus-specific T cells and association with the progression of human chronic hepatitis B infection. Sci. Immunol. 2019, 4, eaau6905. [Google Scholar] [CrossRef]
- Cheng, Y.; Gunasegaran, B.; Singh, H.D.; Dutertre, C.-A.; Loh, C.Y.; Lim, J.Q.; Crawford, J.C.; Lee, H.K.; Zhang, X.; Lee, B.; et al. Non-terminally exhausted tumor-resident memory HBV-specific T cell responses correlate with relapse-free survival in hepatocellular carcinoma. Immunity 2021, 54, 1825–1840.e7. [Google Scholar] [CrossRef]
- Schuch, A.; Salimi Alizei, E.; Heim, K.; Wieland, D.; Kiraithe, M.M.; Kemming, J.; Llewellyn-Lacey, S.; Sogukpinar, Ö.; Ni, Y.; Urban, S.; et al. Phenotypic and functional differences of HBV core-specific versus HBV polymerase-specific CD8+ T cells in chronically HBV-infected patients with low viral load. Gut 2018, 68, 905–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieland, D.; Kemming, J.; Schuch, A.; Emmerich, F.; Knolle, P.; Neumann-Haefelin, C.; Held, W.; Zehn, D.; Hofmann, M.; Thimme, R. TCF1+ hepatitis C virus-specific CD8+ T cells are maintained after cessation of chronic antigen stimulation. Nat. Commun. 2017, 8, 15050. [Google Scholar] [CrossRef] [Green Version]
- Hensel, N.; Gu, Z.; Sagar; Wieland, D.; Jechow, K.; Kemming, J.; Llewellyn-Lacey, S.; Gostick, E.; Sogukpinar, O.; Emmerich, F.; et al. Memory-like HCV-specific CD8+ T cells retain a molecular scar after cure of chronic HCV infection. Nat. Immunol. 2021, 22, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Heim, K.; Binder, B.; Sagar; Wieland, D.; Hensel, N.; Llewellyn-Lacey, S.; Gostick, E.; Price, D.A.; Emmerich, F.; Vingerhoet, H.; et al. TOX defines the degree of CD8+ T cell dysfunction in distinct phases of chronic HBV infection. Gut 2020, 70, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Aregay, A.; Owusu Sekyere, S.; Deterding, K.; Port, K.; Dietz, J.; Berkowski, C.; Sarrazin, C.; Manns, M.P.; Cornberg, M.; Wedemeyer, H. Elimination of hepatitis C virus has limited impact on the functional and mitochondrial impairment of HCV-specific CD8+ T cell responses. J. Hepatol. 2019, 71, 889–899. [Google Scholar] [CrossRef]
- Tonnerre, P.; Wolski, D.; Subudhi, S.; Aljabban, J.; Hoogeveen, R.C.; Damasio, M.; Drescher, H.K.; Bartsch, L.M.; Tully, D.C.; Sen, D.R.; et al. Differentiation of exhausted CD8+ T cells after termination of chronic antigen stimulation stops short of achieving functional T cell memory. Nat. Immunol. 2021, 22, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Yates, K.B.; Tonnerre, P.; Martin, G.E.; Gerdemann, U.; Al Abosy, R.; Comstock, D.E.; Weiss, S.A.; Wolski, D.; Tully, D.C.; Chung, R.T.; et al. Epigenetic scars of CD8+ T cell exhaustion persist after cure of chronic infection in humans. Nat. Immunol. 2021, 22, 1020–1029. [Google Scholar] [CrossRef]
- Fosdick, A.; Zheng, J.; Pflanz, S.; Frey, C.R.; Hesselgesser, J.; Halcomb, R.L.; Wolfgang, G.; Tumas, D.B. Pharmacokinetic and pharmacodynamic properties of gs-9620, a novel toll-like receptor 7 agonist, demonstrate interferon-stimulated gene induction without detectable serum interferon at low oral dosess. J. Pharmacol. Exp. Ther. 2014, 348, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Menne, S.; Tumas, D.B.; Liu, K.H.; Thampi, L.; Aldeghaither, D.; Baldwin, B.H.; Bellezza, C.A.; Cote, P.J.; Zheng, J.; Halcomb, R.; et al. Sustained efficacy and seroconversion with the toll-like receptor 7 agonist GS-9620 in the woodchuck model of chronic hepatitis B. J. Hepatol. 2015, 62, 1237–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanford, R.E.; Guerra, B.; Chavez, D.; Giavedoni, L.; Hodara, V.L.; Brasky, K.M.; Fosdick, A.; Frey, C.R.; Zheng, J.; Wolfgang, G.; et al. GS-9620, an oral agonist of toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013, 144, 1508–1517.e10. [Google Scholar] [CrossRef] [Green Version]
- Boni, C.; Vecchi, A.; Rossi, M.; Laccabue, D.; Giuberti, T.; Alfieri, A.; Lampertico, P.; Grossi, G.; Facchetti, F.; Brunetto, M.R.; et al. TLR7 Agonist Increases Responses of Hepatitis B Virus–Specific T Cells and Natural Killer Cells in Patients with Chronic Hepatitis B Treated With Nucleos(T)Ide Analogues. Gastroenterology 2018, 154, 1764–1777.e7. [Google Scholar] [CrossRef]
- Korolowizc, K.E.; Li, B.; Huang, X.; Yon, C.; Rodrigo, E.; Corpuz, M.; Plouffe, D.M.; Kallakury, B.V.; Suresh, M.; Wu, T.Y.-H.; et al. Liver-Targeted Toll-Like Receptor 7 Agonist Combined with Entecavir Promotes a Functional Cure in the Woodchuck Model of Hepatitis B Virus. Hepatol. Commun. 2019, 3, 1296–1310. [Google Scholar] [CrossRef]
- Bertoletti, A.; Le Bert, N. Fine-Tuning TLR-7-Based Therapy for Functional HBV Cure. Hepatol. Commun. 2019, 3, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Mackman, R.L.; Mish, M.; Chin, G.; Perry, J.K.; Appleby, T.; Aktoudianakis, V.; Metobo, S.; Pyun, P.; Niu, C.; Daffis, S.; et al. Discovery of GS-9688 (Selgantolimod) as a Potent and Selective Oral Toll-Like Receptor 8 Agonist for the Treatment of Chronic Hepatitis B. J. Med. Chem. 2020, 63, 10188–10203. [Google Scholar] [CrossRef]
- Daffis, S.; Balsitis, S.; Chamberlain, J.; Zheng, J.; Santos, R.; Rowe, W.; Ramakrishnan, D.; Pattabiraman, D.; Spurlock, S.; Chu, R.; et al. Toll-Like Receptor 8 Agonist GS-9688 Induces Sustained Efficacy in the Woodchuck Model of Chronic Hepatitis B. Hepatology 2021, 73, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Amin, O.E.; Colbeck, E.J.; Daffis, S.; Khan, S.; Ramakrishnan, D.; Pattabiraman, D.; Chu, R.; Micolochick Steuer, H.; Lehar, S.; Peiser, L.; et al. Therapeutic Potential of TLR8 Agonist GS-9688 (Selgantolimod) in Chronic Hepatitis B: Remodeling of Antiviral and Regulatory Mediators. Hepatology 2021, 74, 55–71. [Google Scholar] [CrossRef]
- Gane, E.J.; Kim, H.J.; Visvanathan, K.; Kim, Y.J.; Nguyen, A.; Wallin, J.J.; Chen, D.Y.; McDonald, C.; Arora, P.; Tan, S.K.; et al. Safety, pharmacokinetics, and pharmacodynamics of the oral TLR8 agonist selgantolimod in chronic hepatitis B. Hepatology 2021, 74, 1737–1749. [Google Scholar] [CrossRef] [PubMed]
- Janovec, V.; Hodek, J.; Clarova, K.; Hofman, T.; Dostalik, P.; Fronek, J.; Chlupac, J.; Chaperot, L.; Durand, S.; Baumert, T.F.; et al. Toll-like receptor dual-acting agonists are potent inducers of PBMC-produced cytokines that inhibit hepatitis B virus production in primary human hepatocytes. Sci. Rep. 2020, 10, 12767. [Google Scholar] [CrossRef]
- Korolowicz, K.E.; Suresh, M.; Li, B.; Huang, X.; Yon, C.; Leng, X.; Kallakury, B.V.; Tucker, R.D.; Menne, S. Treatment with the immunomodulator aic649 in combination with entecavir produces antiviral efficacy in the woodchuck model of chronic hepatitis b. Viruses 2021, 13, 648. [Google Scholar] [CrossRef]
- Suresh, M.; Korolowicz, K.E.; Balarezo, M.; Iyer, R.P.; Padmanabhan, S.; Cleary, D.; Gimi, R.; Sheri, A.; Yon, C.; Kallakury, B.V.; et al. Antiviral efficacy and host immune response induction during sequential treatment with SB 9200 followed by entecavir in woodchucks. PLoS ONE 2017, 12, e0169631. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.; Cunningham, M.E.; Wing, P.; DeSilva, S.; Challa, R.; Sheri, A.; Padmanabhan, S.; Iyer, R.P.; Korba, B.E.; Afdhal, N.; et al. SB 9200, a novel agonist of innate immunity, shows potent antiviral activity against resistant HCV variants. J. Med. Virol. 2017, 89, 1620–1628. [Google Scholar] [CrossRef]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Ravanetti, L.; Urbani, S.; Giuberti, T.; Cavalli, A.; Vandelli, C.; Andreone, P.; et al. Combined blockade of programmed death-1 and activation of CD137 increase responses of human liver T cells against HBV, but not HCV. Gastroenterology 2012, 143, 1576–1585.e4. [Google Scholar] [CrossRef]
- Bengsch, B.; Martin, B.; Thimme, R. Restoration of HBV-specific CD8+ T cell function by PD-1 blockade in inactive carrier patients is linked to T cell differentiation. J. Hepatol. 2014, 61, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral Intrahepatic T-Cell Responses Can Be Restored by Blocking Programmed Death-1 Pathway in Chronic Hepatitis B. Gastroenterology 2010, 138, 682–693.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, E.; Ma, Z.; Wu, W.; Kosinska, A.; Zhang, X.; Möller, I.; Seiz, P.; Glebe, D.; Wang, B.; et al. Enhancing Virus-Specific Immunity In Vivo by Combining Therapeutic Vaccination and PD-L1 Blockade in Chronic Hepadnaviral Infection. PLoS Pathog. 2014, 10, e1003856. [Google Scholar] [CrossRef] [Green Version]
- Kosinska, A.D.; Liu, J.; Lu, M.; Roggendorf, M. Therapeutic vaccination and immunomodulation in the treatment of chronic hepatitis B: Preclinical studies in the woodchuck. Med. Microbiol. Immunol. 2015, 204, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsitis, S.; Gali, V.; Mason, P.J.; Chaniewski, S.; Levine, S.M.; Wichroski, M.J.; Feulner, M.; Song, Y.; Granaldi, K.; Loy, J.K.; et al. Safety and efficacy of anti-PD-L1 therapy in the woodchuck model of HBV infection. PLoS ONE 2018, 13, e0190058. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yan, C.; Zhu, J.; Chen, X.; Fu, Q.; Zhang, H.; Tong, Z.; Liu, L.; Zheng, Y.; Zhao, P.; et al. Anti–PD-1/PD-L1 Blockade Immunotherapy Employed in Treating Hepatitis B Virus Infection–Related Advanced Hepatocellular Carcinoma: A Literature Review. Front. Immunol. 2020, 11, 11. [Google Scholar] [CrossRef]
- Gane, E.; Verdon, D.J.; Brooks, A.E.; Gaggar, A.; Nguyen, A.H.; Subramanian, G.M.; Schwabe, C.; Dunbar, P.R. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: A pilot study. J. Hepatol. 2019, 71, 900–907. [Google Scholar] [CrossRef]
- Boni, C.; Fisicaro, P.; Valdatta, C.; Amadei, B.; Di Vincenzo, P.; Giuberti, T.; Laccabue, D.; Zerbini, A.; Cavalli, A.; Missale, G.; et al. Characterization of Hepatitis B Virus (HBV)-Specific T-Cell Dysfunction in Chronic HBV Infection. J. Virol. 2007, 81, 4215–4225. [Google Scholar] [CrossRef] [Green Version]
- Schurich, A.; Khanna, P.; Lopes, A.R.; Han, K.J.; Peppa, D.; Micco, L.; Nebbia, G.; Kennedy, P.T.F.; Geretti, A.M.; Dusheiko, G.; et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 2011, 53, 1494–1503. [Google Scholar] [CrossRef]
- Nebbia, G.; Peppa, D.; Schurich, A.; Khanna, P.; Singh, H.D.; Cheng, Y.; Rosenberg, W.; Dusheiko, G.; Gilson, R.; ChinAleong, J.; et al. Upregulation of the Tim-3/Galectin-9 Pathway of T Cell Exhaustion in Chronic Hepatitis B Virus Infection. PLoS ONE 2012, 7, e47648. [Google Scholar]
- Raziorrouh, B.; Schraut, W.; Gerlach, T.; Nowack, D.; Grüner, N.H.; Ulsenheimer, A.; Zachoval, R.; Wächtler, M.; Spannagl, M.; Haas, J.; et al. The immunoregulatory role of CD244 in chronic hepatitis B infection and its inhibitory potential on virus-specific CD8+ T-cell function. Hepatology 2010, 52, 1934–1947. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, F.J.; Wild, K.; Smits, M.; Zoldan, K.; Csernalabics, B.; Flecken, T.; Lang, J.; Ehrenmann, P.; Emmerich, F.; Hofmann, M.; et al. OX40 stimulation and PD-L1 blockade synergistically augment HBV-specific CD4 T cells in patients with HBeAg-negative infection. J. Hepatol. 2019, 70, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Swadling, L.; Pallett, L.J.; Diniz, M.O.; Baker, J.M.; Amin, O.E.; Stegmann, K.A.; Burton, A.R.; Schmidt, N.M.; Jeffery-Smith, A.; Zakeri, N.; et al. Human Liver Memory CD8+ T Cells Use Autophagy for Tissue Residence. Cell Rep. 2020, 30, 687–698.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R.; et al. Distinct Metabolic Requirements of Exhausted and Functional Virus-Specific CD8 T Cells in the Same Host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Schurich, A.; Pallett, L.J.; Lubowiecki, M.; Singh, H.D.; Gill, U.S.; Kennedy, P.T.; Nastouli, E.; Tanwar, S.; Rosenberg, W.; Maini, M.K. The Third Signal Cytokine IL-12 Rescues the Anti-Viral Function of Exhausted HBV-Specific CD8 T Cells. PLoS Pathog. 2013, 9, e1003208. [Google Scholar] [CrossRef]
- Bénéchet, A.P.; De Simone, G.; Di Lucia, P.; Cilenti, F.; Barbiera, G.; Le Bert, N.; Fumagalli, V.; Lusito, E.; Moalli, F.; Bianchessi, V.; et al. Dynamics and genomic landscape of CD8+ T cells undergoing hepatic priming. Nature 2019, 574, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cubero, E.; Subirá, D.; Sanz-de-Villalobos, E.; Parra-Cid, T.; Madejón, A.; Miquel, J.; Olveira, A.; González-Praetorius, A.; García-Samaniego, J.; Larrubia, J.-R. According to Hepatitis C Virus (HCV) Infection Stage, Interleukin-7 Plus 4-1BB Triggering Alone or Combined with PD-1 Blockade Increases TRAF1 low HCV-Specific CD8 + Cell Reactivity. J. Virol. 2018, 92, e01443-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.H.; Perry, C.J.; Tsui, Y.C.; Staron, M.M.; Parish, I.A.; Dominguez, C.X.; Rosenberg, D.W.; Kaech, S.M. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat. Med. 2015, 21, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Barili, V.; Fisicaro, P.; Montanini, B.; Acerbi, G.; Filippi, A.; Forleo, G.; Romualdi, C.; Ferracin, M.; Guerrieri, F.; Pedrazzi, G.; et al. Targeting p53 and histone methyltransferases restores exhausted CD8+ T cells in HCV infection. Nat. Commun. 2020, 11, 604. [Google Scholar] [CrossRef] [Green Version]
- Fisicaro, P.; Barili, V.; Montanini, B.; Acerbi, G.; Ferracin, M.; Guerrieri, F.; Salerno, D.; Boni, C.; Massari, M.; Cavallo, M.C.; et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 2017, 23, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Acerbi, G.; Montali, I.; Ferrigno, G.D.; Barili, V.; Schivazappa, S.; Alfieri, A.; Laccabue, D.; Loglio, A.; Borghi, M.; Massari, M.; et al. Functional reconstitution of HBV-specific CD8 T cells by in vitro polyphenol treatment in chronic hepatitis B. J. Hepatol. 2021, 74, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.J.; Murphy, M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef]
- Pallett, L.J.; Gill, U.S.; Quaglia, A.; Sinclair, L.V.; Jover-Cobos, M.; Schurich, A.; Singh, K.P.; Thomas, N.; Das, A.; Chen, A.; et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat. Med. 2015, 21, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, N.M.; Wing, P.A.C.; Diniz, M.O.; Pallett, L.J.; Swadling, L.; Harris, J.M.; Burton, A.R.; Jeffery-Smith, A.; Zakeri, N.; Amin, O.E.; et al. Targeting human Acyl-CoA:cholesterol acyltransferase as a dual viral and T cell metabolic checkpoint. Nat. Commun. 2021, 12, 2814. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M. Liver damage related to immune checkpoint inhibitors. Hepatol. Int. 2019, 13, 248–252. [Google Scholar] [CrossRef]
- Suslov, A.; Wieland, S.; Menne, S. Modulators of innate immunity as novel therapeutics for treatment of chronic hepatitis B. Curr. Opin. Virol. 2018, 30, 9–17. [Google Scholar] [CrossRef]
- Kumashie, K.G.; Cebula, M.; Hagedorn, C.; Kreppel, F.; Pils, M.C.; Koch-Nolte, F.; Rissiek, B.; Wirth, D. Improved Functionality of Exhausted Intrahepatic CXCR5+ CD8+ T Cells Contributes to Chronic Antigen Clearance Upon Immunomodulation. Front. Immunol. 2021, 11, 592328. [Google Scholar] [CrossRef]
- Lee, S.; Goyal, A.; Perelson, A.S.; Ishida, Y.; Saito, T.; Gale, M. Suppression of hepatitis B virus through therapeutic activation of RIG-I and IRF3 signaling in hepatocytes. iScience 2021, 24, 101969. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogeveen, R.C.; Robidoux, M.P.; Schwarz, T.; Heydmann, L.; Cheney, J.A.; Kvistad, D.; Aneja, J.; Melgaço, J.G.; Fernandes, C.A.; Chung, R.T.; et al. Phenotype and function of HBV-specific T cells is determined by the targeted epitope in addition to the stage of infection. Gut 2018, 68, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1 + PD-1 + CD8 + T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef] [Green Version]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255.e6. [Google Scholar] [CrossRef] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Klein Geltink, R.I.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.R.; Herman, C.E.; MacIver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [Green Version]
- Chapman, N.M.; Boothby, M.R.; Chi, H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 2020, 20, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, E.I.; Macal, M.; Lewis, G.M.; Harker, J.A. Innate and Adaptive Immune Regulation during Chronic Viral Infections. Annu. Rev. Virol. 2015, 2, 573–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolski, D.; Foote, P.K.; Chen, D.Y.; Lewis-Ximenez, L.L.; Fauvelle, C.; Aneja, J.; Walker, A.; Tonnerre, P.; Torres-Cornejo, A.; Kvistad, D.; et al. Early Transcriptional Divergence Marks Virus-Specific Primary Human CD8+ T Cells in Chronic versus Acute Infection. Immunity 2017, 47, 648–663.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The Transcription Factor FoxO1 Sustains Expression of the Inhibitory Receptor PD-1 and Survival of Antiviral CD8+ T Cells during Chronic Infection. Immunity 2014, 41, 802–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. P53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Akbar, A.N.; Henson, S.M.; Lanna, A. Senescence of T Lymphocytes: Implications for Enhancing Human Immunity. Trends Immunol. 2016, 37, 866–876. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Berger, N.A. Poly(ADP-Ribose) in the Cellular Response to DNA Damage. Radiat. Res. 1985, 101, 4. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [Green Version]
- Shahgaldi, S.; Kahmini, F.R. A comprehensive review of Sirtuins: With a major focus on redox homeostasis and metabolism. Life Sci. 2021, 282, 119803. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A. Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch-Presegué, L.; Vaquero, A. Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. FEBS J. 2015, 282, 1745–1767. [Google Scholar] [CrossRef]
- Katsuyama, E.; Suarez-Fueyo, A.; Bradley, S.J.; Mizui, M.; Marin, A.V.; Mulki, L.; Krishfield, S.; Malavasi, F.; Yoon, J.; Sui, S.J.H.; et al. The CD38/NAD/SIRTUIN1/EZH2 Axis Mitigates Cytotoxic CD8 T Cell Function and Identifies Patients with SLE Prone to Infections. Cell Rep. 2020, 30, 112–123.e4. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sauve, A.A. NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta Proteins Proteom. 2016, 1864, 1787–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Avalos, J.L.; Bever, K.M.; Wolberger, C. Mechanism of sirtuin nhibition by nicotinamide: Altering the NAD + cosubstrate specificity of a Sir2 enzyme. Mol. Cell 2005, 17, 855–868. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Fisicaro, P.; Barili, V.; Rossi, M.; Montali, I.; Vecchi, A.; Acerbi, G.; Laccabue, D.; Zecca, A.; Penna, A.; Missale, G.; et al. Pathogenetic Mechanisms of T Cell Dysfunction in Chronic HBV Infection and Related Therapeutic Approaches. Front. Immunol. 2020, 11, 849. [Google Scholar] [CrossRef] [PubMed]
- Kalia, V.; Sarkar, S.; Subramaniam, S.; Haining, W.N.; Smith, K.A.; Ahmed, R. Prolonged Interleukin-2Rα Expression on Virus-Specific CD8+ T Cells Favors Terminal-Effector Differentiation In Vivo. Immunity 2010, 32, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipkin, M.E.; Sacks, J.A.; Cruz-Guilloty, F.; Lichtenheld, M.G.; Bevan, M.J.; Rao, A. Interleukin-2 and Inflammation Induce Distinct Transcriptional Programs that Promote the Differentiation of Effector Cytolytic T Cells. Immunity 2010, 32, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Spolski, R.; Li, P.; Leonard, W.J. Biology and regulation of IL-2: From molecular mechanisms to human therapy. Nat. Rev. Immunol. 2018, 18, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Kallies, A. Synchronizing transcriptional control of T cell metabolism and function. Nat. Rev. Immunol. 2015, 15, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Boni, C.; Barili, V.; Acerbi, G.; Rossi, M.; Vecchi, A.; Laccabue, D.; Penna, A.; Missale, G.; Ferrari, C.; Fisicaro, P. HBV Immune-Therapy: From Molecular Mechanisms to Clinical Applications. Int. J. Mol. Sci. 2019, 20, 2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maini, M.K.; Burton, A.R. Restoring, releasing or replacing adaptive immunity in chronic hepatitis B. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.K.; Pallett, L.J. Defective T-cell immunity in hepatitis B virus infection: Why therapeutic vaccination needs a helping hand. Lancet Gastroenterol. Hepatol. 2018, 3, 192–202. [Google Scholar] [CrossRef]
- Gollob, J.A.; Schnipper, C.P.; Murphy, E.A.; Ritz, J.; Frank, D.A. The functional synergy between IL-12 and IL-2 involves p38 mitogen-activated protein kinase and is associated with the augmentation of STAT serine phosphorylation. J. Immunol. 1999, 162, 4472–4481. [Google Scholar]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic Instruction of Immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef]
- Pollizzi, K.N.; Powell, J.D. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 2014, 14, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.; Pinto, A.K.; Curtis, J.D.; Persaud, S.P.; Cella, M.; Lin, C.-C.; Edelson, B.T.; Allen, P.M.; Colonna, M.; Pearce, E.L.; et al. c-Myc-induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nat. Immunol. 2014, 15, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Lochner, M.; Berod, L.; Sparwasser, T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015, 36, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Kidani, Y.; Elsaesser, H.; Hock, M.B.; Vergnes, L.; Williams, K.J.; Argus, J.P.; Marbois, B.N.; Komisopoulou, E.; Wilson, E.B.; Osborne, T.F.; et al. Sterol regulatory element–binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol. 2013, 14, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Carmen, F.C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [Green Version]
- Gnanaprakasam, J.N.R.; Sherman, J.W.; Wang, R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 2017, 35, 63–70. [Google Scholar] [CrossRef]
- Johnson, M.O.; Siska, P.J.; Contreras, D.C.; Rathmell, J.C. Nutrients and the microenvironment to feed a T cell army. Semin. Immunol. 2016, 28, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 6284, 481–485. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.M.; Amezquita, R.A.; Guan, T.; Kleinstein, S.H.; Kaech, S.M. Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8(+) T Cell Terminal Differentiation and Loss of Multipotency. Immunity 2017, 46, 596–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Zhang, K.; Milner, J.J.; Toma, C.; Chen, R.; Scott-Browne, J.P.; Pereira, R.M.; Crotty, S.; Chang, J.T.; Pipkin, M.E.; et al. Epigenetic landscapes reveal transcription factors that regulate CD8(+) T cell differentiation. Nat. Immunol. 2017, 18, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [Green Version]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e8. [Google Scholar] [CrossRef]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Youngblood, B.A.; Austin, J.J.; Mohammed, A.U.R.; Butler, R.; Ahmed, R.; Boss, J.M. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J. Exp. Med. 2014, 211, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef] [Green Version]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Tsukada, Y.I.; Zhang, Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and hox gene silencing. Mol. Cell 2005, 20, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.A.; Van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vissers, J.H.A.; Van Lohuizen, M.; Citterio, E. The emerging role of Polycomb repressors in the response to DNA damage. J. Cell Sci. 2012, 125, 3939–3948. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.L.; Brownlee, P.M.; Durley, S.C.; Beacham, T.; Kent, N.A.; Downs, J.A. The two different isoforms of the RSC chromatin remodeling complex play distinct roles in DNA damage responses. PLoS ONE 2012, 7, e32016. [Google Scholar] [CrossRef]
- Bennett, G.; Papamichos-Chronakis, M.; Peterson, C.L. DNA repair choice defines a common pathway for recruitment of chromatin regulators. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Papamichos-Chronakis, M.; Peterson, C.L. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013, 14, 62–75. [Google Scholar] [CrossRef] [Green Version]
- Kakaradov, B.; Arsenio, J.; Widjaja, C.E.; He, Z.; Aigner, S.; Metz, P.J.; Yu, B.; Wehrens, E.J.; Lopez, J.; Kim, S.H.; et al. Early transcriptional and epigenetic regulation of CD8(+) T cell differentiation revealed by single-cell RNA sequencing. Nat. Immunol. 2017, 18, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Tumes, D.J.; Onodera, A.; Suzuki, A.; Shinoda, K.; Endo, Y.; Iwamura, C.; Hosokawa, H.; Koseki, H.; Tokoyoda, K.; Suzuki, Y.; et al. The Polycomb protein Ezh2 regulates differentiation and plasticity of CD4+ T helper Type 1 and type 2 cells. Immunity 2013, 39, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kinkel, S.; Maksimovic, J.; Bandala-Sanchez, E.; Tanzer, M.C.; Naselli, G.; Zhang, J.G.; Zhan, Y.; Lew, A.M.; Silke, J.; et al. The polycomb repressive complex 2 governs life and death of peripheral T cells. Blood 2014, 124, 737–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karantanos, T.; Chistofides, A.; Barhdan, K.; Li, L.; Boussiotis, V.A. Regulation of T cell differentiation and function by EZH2. Front. Immunol. 2016, 7, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheer, S.; Zaph, C. The lysine methyltransferase G9a in immune cell differentiation and function. Front. Immunol. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Aune, T.M. Dynamic changes in histone-methylation “marks” across the locus encoding interferon-γ during the differentiation of T helper type 2 cells. Nat. Immunol. 2007, 8, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Apostolou, I.; Zhang, J.; Skepner, J.; Anandhan, S.; Zhang, X.; Xiong, L.; Trojer, P.; Aparicio, A.; Subudhi, S.K.; et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J. Clin. Investig. 2018, 128, 3813–3818. [Google Scholar] [CrossRef]
- McGoverne, I.; Dunn, J.; Batham, J.; Tu, W.J.; Chrisp, J.; Rao, S. Epitherapy and immune checkpoint blockade: Using epigenetic reinvigoration of exhausted and dysfunctional T cells to reimburse immunotherapy response. BMC Immunol. 2020, 21, 1–10. [Google Scholar] [CrossRef] [Green Version]
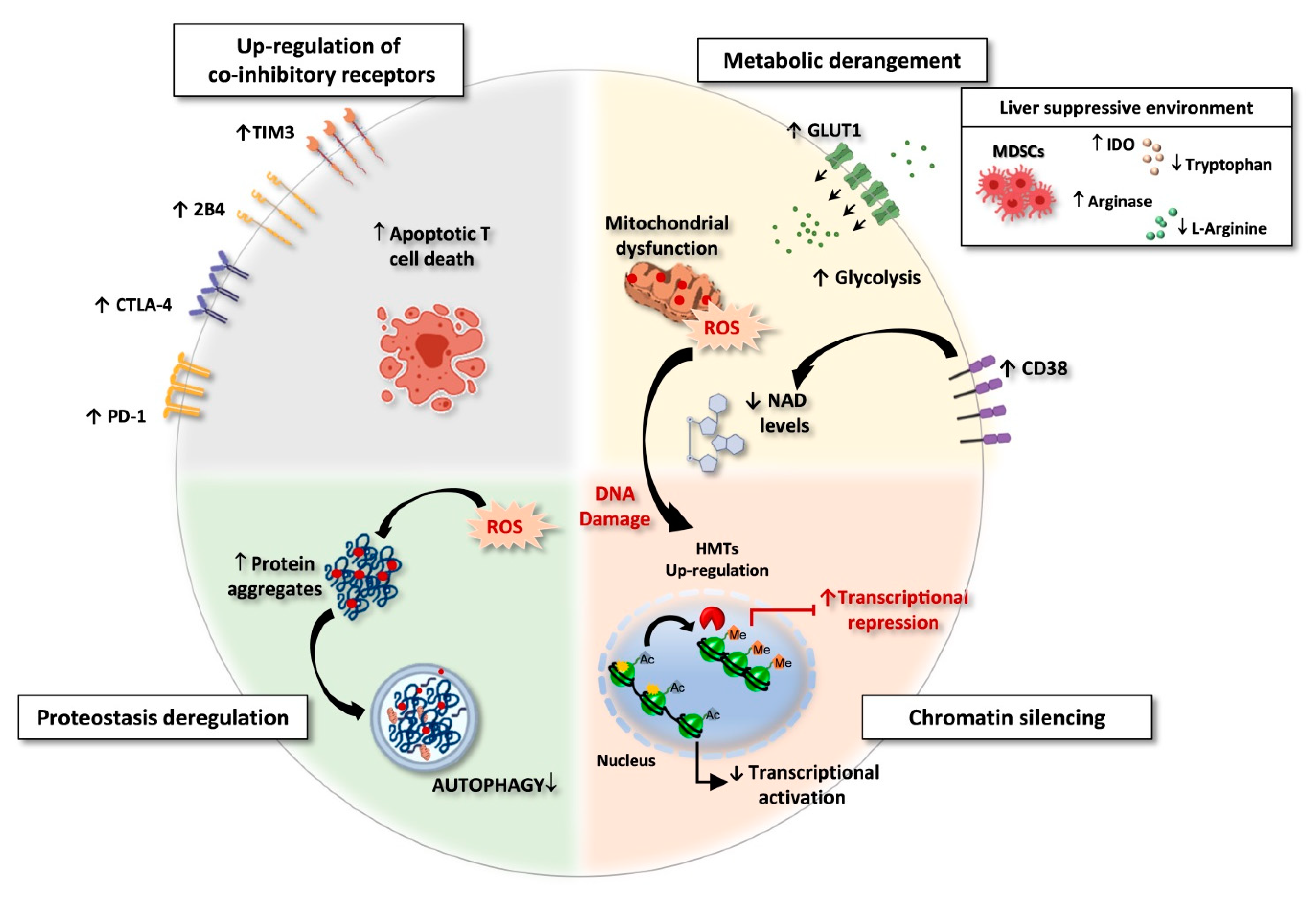
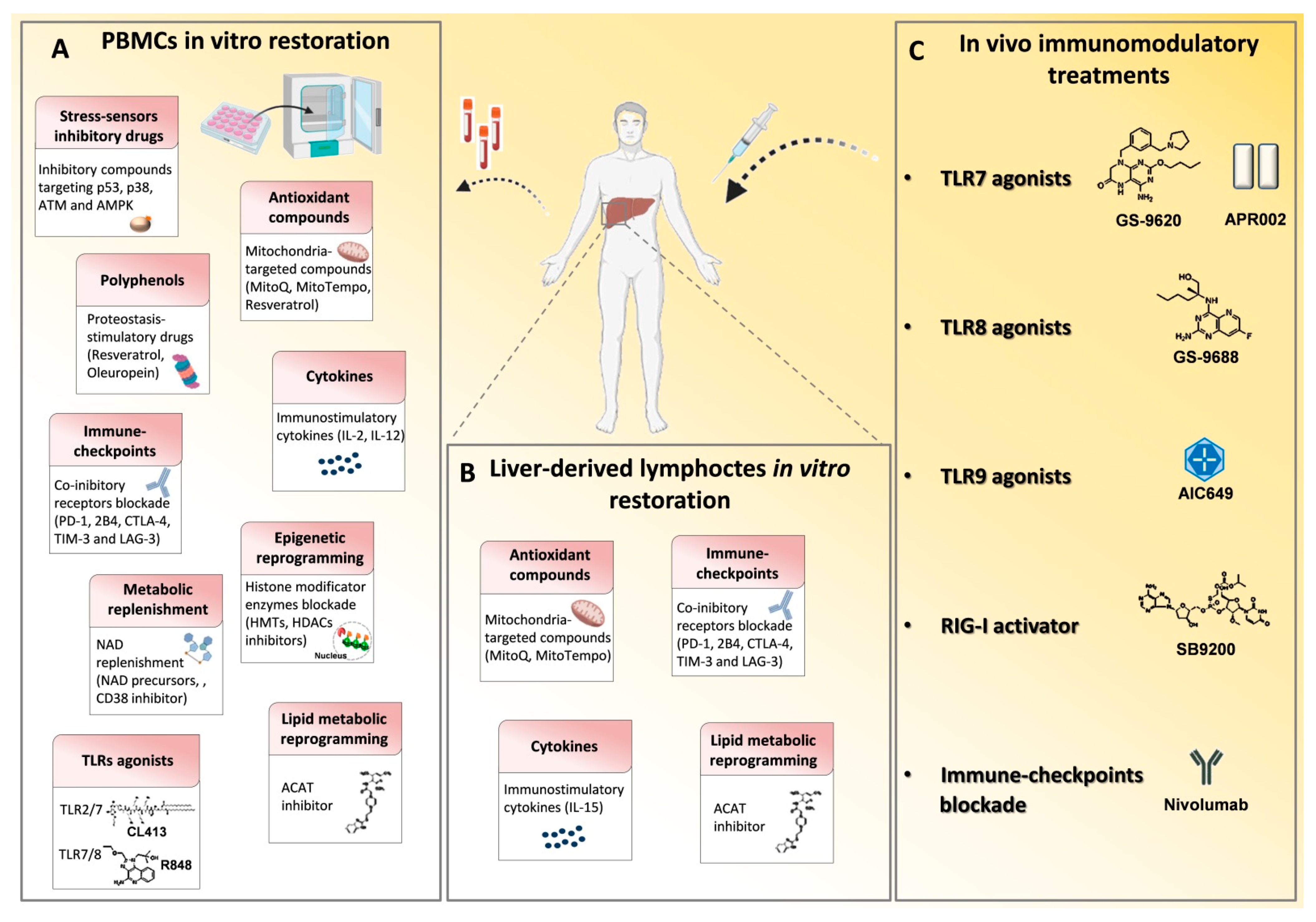
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).