
The Intersection of Purine and Mitochondrial Metabolism in Cancer


Abstract
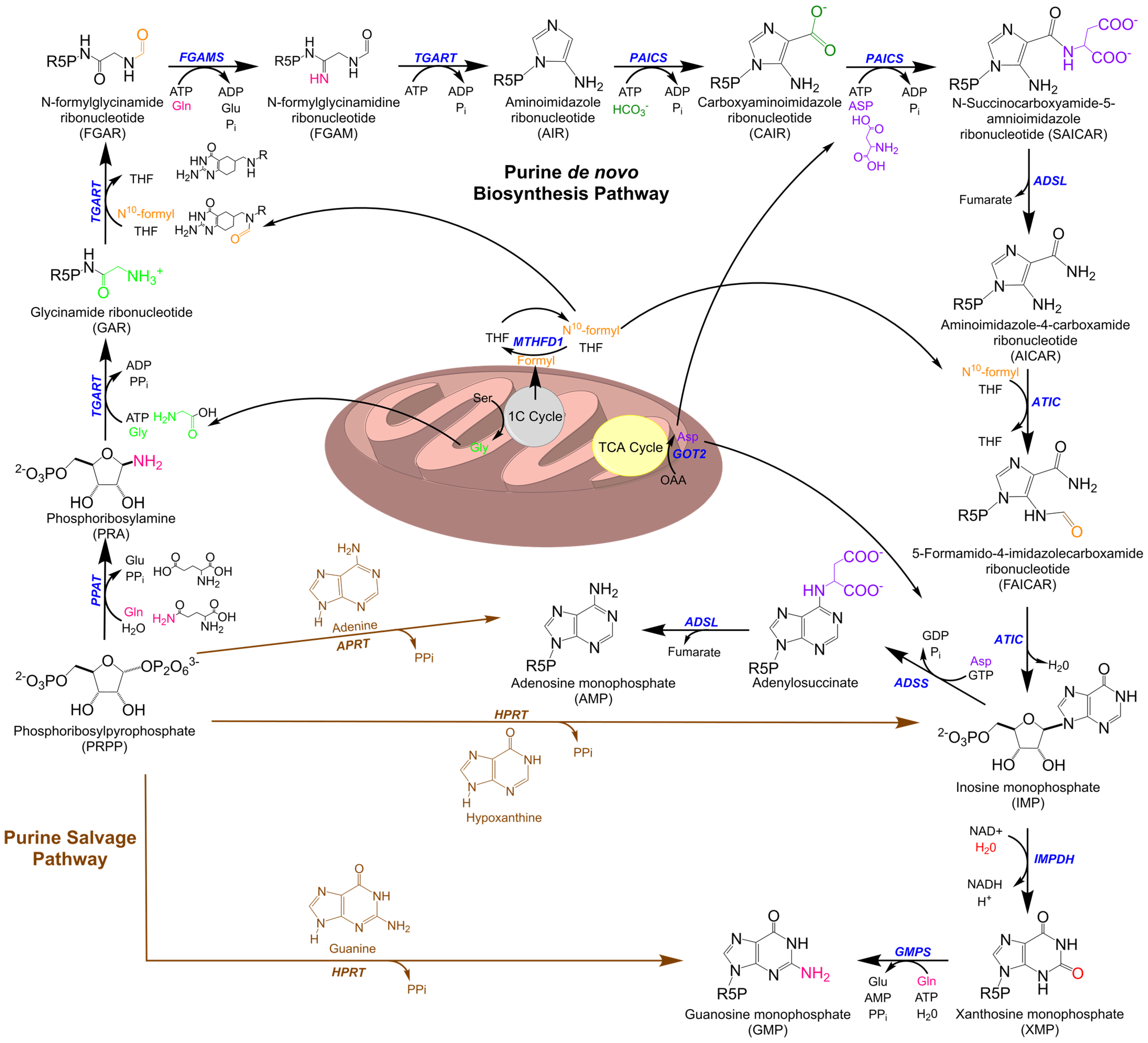
1. Purine Metabolism
2. Purine Metabolism in Cancer
Purine Metabolic Enzymes and Intermediates
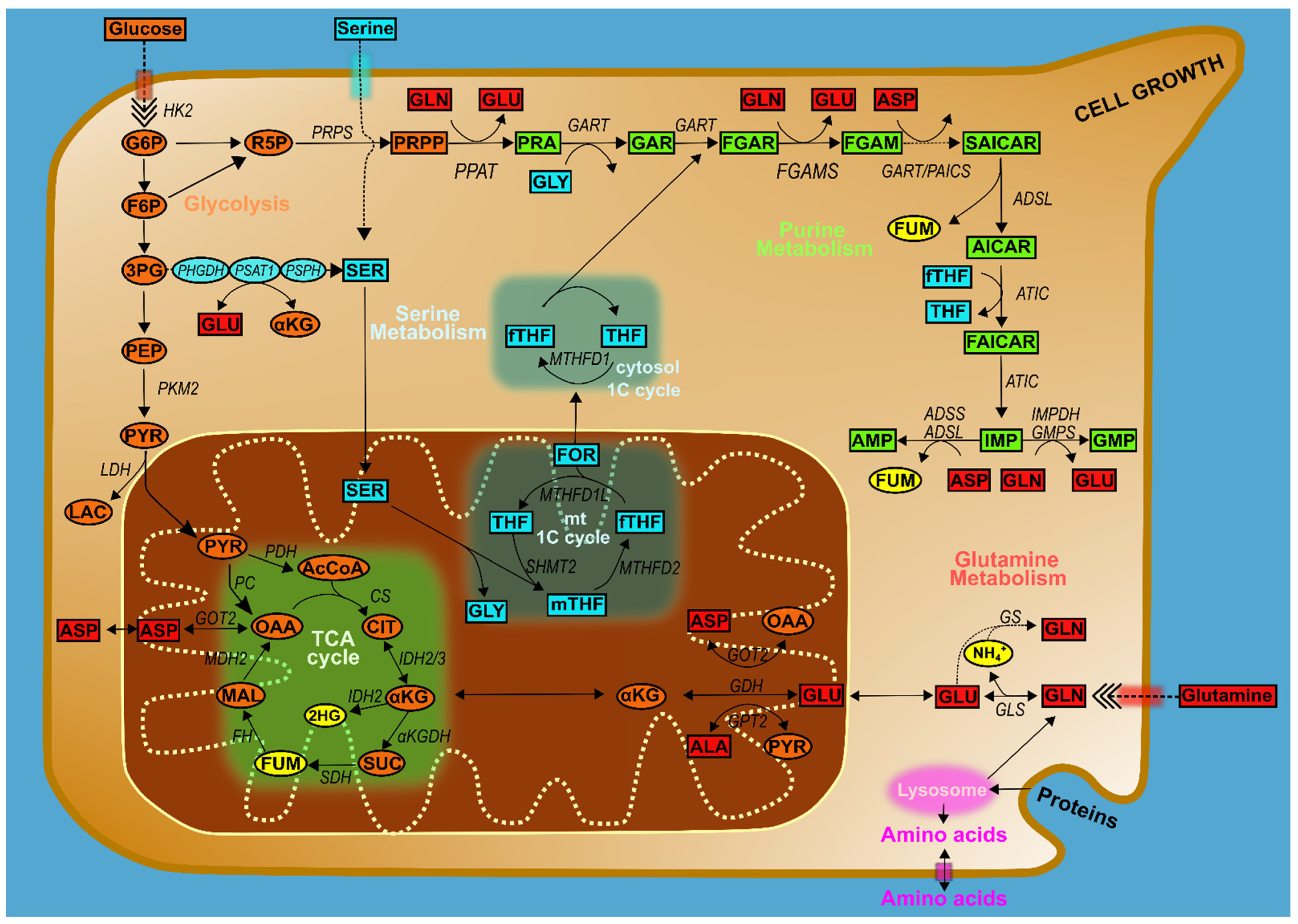
3. Intersections of Purine Metabolism with Broader Cancer Pathways
4. Glucose Metabolism and Purines
4.1. Amino Acids, One-Carbon Metabolism, and Purines
4.2. The Master Regulators of Metabolism
5. Purine Metabolism at the Mitochondria
6. Mitochondria and Purine Metabolism in Ovarian Cancer
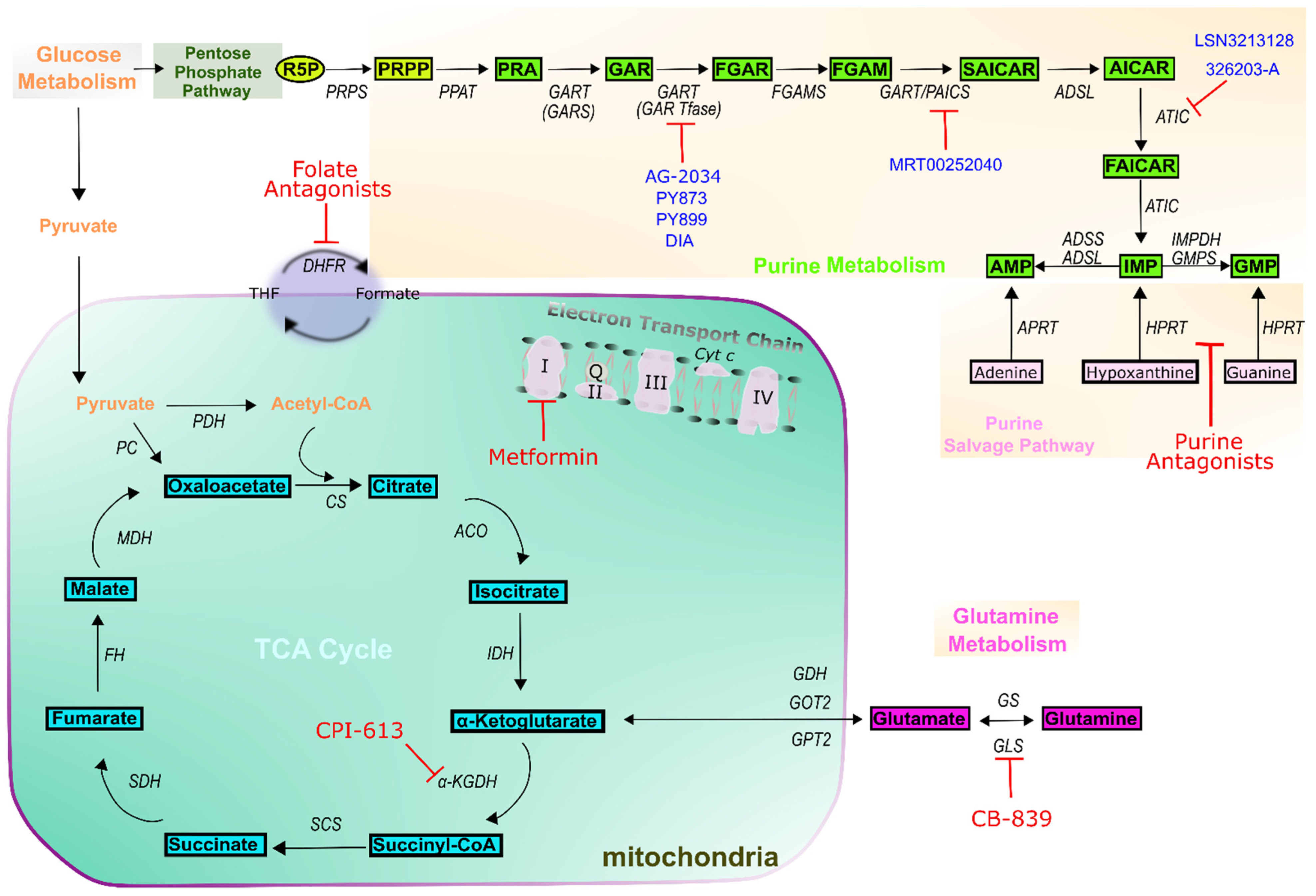
7. Outlook on Novel Purine-Based Cancer Treatment Methodologies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hess, J.R.; Greenberg, N.A. The Role of Nucleotides in the Immune and Gastrointestinal Systems: Potential Clinical Applications. Nutr. Clin. Pr. 2012, 27, 281–294. [Google Scholar] [CrossRef]
- Zhao, H.; Chiaro, C.R.; Zhang, L.; Smith, P.B.; Chan, C.Y.; Pedley, A.M.; Pugh, R.J.; French, J.B.; Patterson, A.D.; Benkovic, S.J. Quantitative Analysis of Purine Nucleotides Indicates That Purinosomes Increase de Novo Purine Biosynthesis. J. Biol. Chem. 2015, 290, 6705–6713. [Google Scholar] [CrossRef] [PubMed]
- Fustin, J.M.; Doi, M.; Yamada, H.; Komatsu, R.; Shimba, S.; Okamura, H. Rhythmic Nucleotide Synthesis in the Liver: Temporal Segregation of Metabolites. Cell Rep. 2012, 1, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Hartman, S.C.; Buchanan, J.M. Nucleic Acids, Purines, Pyrimidines (Nucleotide Synthesis). Annu. Rev. Biochem. 1959, 28, 365–410. [Google Scholar] [CrossRef] [PubMed]
- Camici, M.; Garcia-Gil, M.; Pesi, R.; Allegrini, S.; Tozzi, M.G. Purine-Metabolising Enzymes and Apoptosis in Cancer. Cancers 2019, 11, 1354. [Google Scholar] [CrossRef] [PubMed]
- Mádrová, L.; Krijt, M.; Barešová, V.; Václavík, J.; Friedecký, D.; Dobešová, D.; Součková, O.; Škopová, V.; Adam, T.; Zikánová, M. Mass Spectrometric Analysis of Purine de Novo Biosynthesis Intermediates. PLoS ONE 2018, 13, e0208947. [Google Scholar] [CrossRef] [PubMed]
- Chitrakar, I.; Kim-Holzapfel, D.M.; Zhou, W.; French, J.B. Higher Order Structures in Purine and Pyrimidine Metabolism. J. Struct. Biol. 2017, 197, 354–364. [Google Scholar] [CrossRef]
- Zhao, H.; French, J.B.; Fang, Y.; Benkovic, S.J. The Purinosome, a Multi-Protein Complex Involved in the de Novo Biosynthesis of Purines in Humans. Chem. Commun. Camb. 2013, 49, 4444–4452. [Google Scholar] [CrossRef]
- Yamaoka, T.; Yano, M.; Kondo, M.; Sasaki, H.; Hino, S.; Katashima, R.; Moritani, M.; Itakura, M. Feedback Inhibition of Amidophosphoribosyltransferase Regulates the Rate of Cell Growth via Purine Nucleotide, DNA, and Protein Syntheses. J. Biol. Chem. 2001, 276, 21285–21291. [Google Scholar] [CrossRef]
- Smith, J.L. Glutamine PRPP Amidotransferase: Snapshots of an Enzyme in Action. Curr. Opin. Struct. Biol. 1998, 8, 686–694. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Knudsen, G.M.; Pedley, A.M.; He, J.; Johnson, J.L.; Yaron, T.M.; Cantley, L.C.; Benkovic, S.J. Mapping Post-Translational Modifications of de Novo Purine Biosynthetic Enzymes: Implications for Pathway Regulation. J. Proteome. Res. 2019, 18, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Fridman, A.; Saha, A.; Chan, A.; Casteel, D.E.; Pilz, R.B.; Boss, G.R. Cell Cycle Regulation of Purine Synthesis by Phosphoribosyl Pyrophosphate and Inorganic Phosphate. Biochem. J. 2013, 454, 91–99. [Google Scholar] [CrossRef]
- An, S.; Kumar, R.; Sheets, E.D.; Benkovic, S.J. Reversible Compartmentalization of de Novo Purine Biosynthetic Complexes in Living Cells. Science 2008, 320, 103–106. [Google Scholar] [CrossRef]
- French, J.B.; Jones, S.A.; Deng, H.; Pedley, A.M.; Kim, D.; Chan, C.Y.; Hu, H.; Pugh, R.J.; Zhao, H.; Zhang, Y.; et al. Spatial Colocalization and Functional Link of Purinosomes with Mitochondria. Science 2016, 351, 733–737. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- WARBURG, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Lane, A.N.; Fan, T.W. Regulation of Mammalian Nucleotide Metabolism and Biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef]
- Parker, W.B. Enzymology of Purine and Pyrimidine Antimetabolites Used in the Treatment of Cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar] [CrossRef]
- Elion, G.B. Nobel Lecture. The Purine Path to Chemotherapy. Biosci. Rep. 1989, 9, 509–529. [Google Scholar] [CrossRef]
- Villa, E.; Ali, E.S.; Sahu, U.; Ben-Sahra, I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers 2019, 11, 688. [Google Scholar] [CrossRef]
- Lambie, D.G.; Johnson, R.H. Drugs and Folate Metabolism. Drugs 1985, 30, 145–155. [Google Scholar] [CrossRef]
- Walling, J. From Methotrexate to Pemetrexed and beyond. A Review of the Pharmacodynamic and Clinical Properties of Antifolates. Investig. New Drugs 2006, 24, 37–77. [Google Scholar] [CrossRef]
- Sant, M.E.; Lyons, S.D.; Phillips, L.; Christopherson, R.I. Antifolates Induce Inhibition of Amido Phosphoribosyltransferase in Leukemia Cells. J. Biol. Chem. 1992, 267, 11038–11045. [Google Scholar] [CrossRef]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and Phenformin Deplete Tricarboxylic Acid Cycle and Glycolytic Intermediates during Cell Transformation and NTPs in Cancer Stem Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Romero, I.L.; Litchfield, L.M.; Lengyel, E.; Locasale, J.W. Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab. 2016, 24, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II Clinical Trial of Metformin as a Cancer Stem Cell-Targeting Agent in Ovarian Cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Kodama, M.; Oshikawa, K.; Shimizu, H.; Yoshioka, S.; Takahashi, M.; Izumi, Y.; Bamba, T.; Tateishi, C.; Tomonaga, T.; Matsumoto, M.; et al. A Shift in Glutamine Nitrogen Metabolism Contributes to the Malignant Progression of Cancer. Nat. Commun. 2020, 11, 1320. [Google Scholar] [CrossRef]
- Goswami, M.T.; Chen, G.; Chakravarthi, B.V.; Pathi, S.S.; Anand, S.K.; Carskadon, S.L.; Giordano, T.J.; Chinnaiyan, A.M.; Thomas, D.G.; Palanisamy, N.; et al. Role and Regulation of Coordinately Expressed de Novo Purine Biosynthetic Enzymes PPAT and PAICS in Lung Cancer. Oncotarget 2015, 6, 23445–23461. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine Synthesis Promotes Maintenance of Brain Tumor Initiating Cells in Glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef]
- Chakravarthi, B.V.S.K.; Rodriguez Pena, M.D.C.; Agarwal, S.; Chandrashekar, D.S.; Hodigere Balasubramanya, S.A.; Jabboure, F.J.; Matoso, A.; Bivalacqua, T.J.; Rezaei, K.; Chaux, A.; et al. A Role for De Novo Purine Metabolic Enzyme PAICS in Bladder Cancer Progression. Neoplasia 2018, 20, 894–904. [Google Scholar] [CrossRef]
- Barfeld, S.J.; Fazli, L.; Persson, M.; Marjavaara, L.; Urbanucci, A.; Kaukoniemi, K.M.; Rennie, P.S.; Ceder, Y.; Chabes, A.; Visakorpi, T.; et al. Myc-Dependent Purine Biosynthesis Affects Nucleolar Stress and Therapy Response in Prostate Cancer. Oncotarget 2015, 6, 12587–12602. [Google Scholar] [CrossRef]
- Park, H.; Ohshima, K.; Nojima, S.; Tahara, S.; Kurashige, M.; Hori, Y.; Okuzaki, D.; Wada, N.; Ikeda, J.I.; Morii, E. Adenylosuccinate Lyase Enhances Aggressiveness of Endometrial Cancer by Increasing Killer Cell Lectin-like Receptor C3 Expression by Fumarate. Lab. Investig. 2018, 98, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, G.; Liu, X.; Takada, M.; Fan, C.; Simon, J.M.; Ptacek, T.S.; Rodriguez, J.; von Kriegsheim, A.; Liu, J.; Locasale, J.W.; et al. Prolyl Hydroxylase Substrate Adenylosuccinate Lyase Is an Oncogenic Driver in Triple Negative Breast Cancer. Nat. Commun. 2019, 10, 5177. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.H.Y.; Hsu, C.L.; Tsuei, C.Y.; Kuo, T.T.; Huang, C.T.; Hsu, W.M.; Chung, Y.H.; Wu, H.Y.; Hsu, C.C.; Huang, H.C.; et al. Combinatorial Targeting of MTHFD2 and PAICS in Purine Synthesis as a Novel Therapeutic Strategy. Cell Death Dis. 2019, 10, 786. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, S.; Hirayama, A.; Eberhardt, A.O.; Kawaguchi, R.; Sugiura, Y.; Sampetrean, O.; Ikeda, Y.; Warren, M.; Sakamoto, N.; Kitahara, S.; et al. IMP Dehydrogenase-2 Drives Aberrant Nucleolar Activity and Promotes Tumorigenesis in Glioblastoma. Nat. Cell Biol. 2019, 21, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Ni, M.; Chalishazar, M.D.; Huffman, K.E.; Kim, J.; Cai, L.; Shi, X.; Cai, F.; Zacharias, L.G.; Ireland, A.S.; et al. Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell Metab. 2018, 28, 369–382.e5. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.C.; Toh, T.B.; Chan, Z.; Lin, Q.X.X.; Thng, D.K.H.; Hooi, L.; Ding, Z.; Shuen, T.; Toh, H.C.; Dan, Y.Y.; et al. Targeted Inhibition of Purine Metabolism Is Effective in Suppressing Hepatocellular Carcinoma Progression. Hepatol. Commun. 2020, 4, 1362–1381. [Google Scholar] [CrossRef] [PubMed]
- Emmanuel, N.; Ragunathan, S.; Shan, Q.; Wang, F.; Giannakou, A.; Huser, N.; Jin, G.; Myers, J.; Abraham, R.T.; Unsal-Kacmaz, K. Purine Nucleotide Availability Regulates MTORC1 Activity through the Rheb GTPase. Cell Rep. 2017, 19, 2665–2680. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Wang, X.; Li, X.; Xu, G.; Bai, Y.; Wu, J.; Piao, Y.; Shi, Y.; Xiang, R.; Wang, L. Nucleotide de Novo Synthesis Increases Breast Cancer Stemness and Metastasis via CGMP-PKG-MAPK Signaling Pathway. PLoS Biol. 2020, 18, e3000872. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate Kinase M2 Is a Phosphotyrosine-Binding Protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine Is a Natural Ligand and Allosteric Activator of Pyruvate Kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.E.; Doctor, Z.M.; Dwyer, Z.W.; Lee, Y.S. SAICAR Induces Protein Kinase Activity of PKM2 That Is Necessary for Sustained Proliferative Signaling of Cancer Cells. Mol. Cell 2014, 53, 700–709. [Google Scholar] [CrossRef]
- Keller, K.E.; Tan, I.S.; Lee, Y.S. SAICAR Stimulates Pyruvate Kinase Isoform M2 and Promotes Cancer Cell Survival in Glucose-Limited Conditions. Science 2012, 338, 1069–1072. [Google Scholar] [CrossRef]
- Vara-Ciruelos, D.; Russell, F.M.; Hardie, D.G. The Strange Case of AMPK and Cancer: Dr Jekyll or Mr Hyde? Open Biol. 2019, 9, 190099. [Google Scholar] [CrossRef]
- Vazquez, A.; Kamphorst, J.J.; Markert, E.K.; Schug, Z.T.; Tardito, S.; Gottlieb, E. Cancer Metabolism at a Glance. J. Cell Sci. 2016, 129, 3367–3373. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond Aerobic Glycolysis: Transformed Cells Can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Shaw, R.J. Glucose Metabolism and Cancer. Curr. Opin. Cell Biol. 2006, 18, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Zhao, F.; Thompson, C.B. The Molecular Determinants of de Novo Nucleotide Biosynthesis in Cancer Cells. Curr. Opin. Genet. Dev. 2009, 19, 32–37. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Muralidhar, V.; Hosios, A.M.; Israelsen, W.J.; Gui, D.Y.; Newhouse, L.; Ogrodzinski, M.; Hecht, V.; Xu, K.; Acevedo, P.N.; et al. Pyruvate Kinase Isoform Expression Alters Nucleotide Synthesis to Impact Cell Proliferation. Mol. Cell 2015, 57, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Gui, D.Y.; Lewis, C.A.; Vander Heiden, M.G. Allosteric Regulation of PKM2 Allows Cellular Adaptation to Different Physiological States. Sci. Signal. 2013, 6, pe7. [Google Scholar] [CrossRef] [PubMed]
- Dioguardi, F.S.; Flati, V.; Corsetti, G.; Pasini, E.; Romano, C. Is the Response of Tumours Dependent on the Dietary Input of Some Amino Acids or Ratios among Essential and Non-Essential Amino Acids? All That Glitters Is Not Gold. Int. J. Mol. Sci. 2018, 19, E3631. [Google Scholar] [CrossRef] [PubMed]
- Wiese, E.K.; Hitosugi, T. Tyrosine Kinase Signaling in Cancer Metabolism: PKM2 Paradox in the Warburg Effect. Front. Cell Dev. Biol. 2018, 6, 79. [Google Scholar] [CrossRef]
- Newman, A.C.; Maddocks, O.D.K. One-Carbon Metabolism in Cancer. Br. J. Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing Compartmentalized NADPH Metabolism in the Cytosol and Mitochondria of Mammalian Cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic Enzyme Expression Highlights a Key Role for MTHFD2 and the Mitochondrial Folate Pathway in Cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative Flux Analysis Reveals Folate-Dependent NADPH Production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine Starvation Induces Stress and P53-Dependent Metabolic Remodelling in Cancer Cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef]
- Snell, K.; Natsumeda, Y.; Weber, G. The Modulation of Serine Metabolism in Hepatoma 3924A during Different Phases of Cellular Proliferation in Culture. Biochem. J. 1987, 245, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.; Labuschagne, C.F.; Adams, P.D.; Vousden, K.H. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol. Cell 2016, 61, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Juan, W.; Sahyoun, N.R. Intake and Biomarkers of Folate and Risk of Cancer Morbidity in Older Adults, NHANES 1999-2002 with Medicare Linkage. PLoS ONE 2016, 11, e0148697. [Google Scholar] [CrossRef]
- Hansen, M.F.; Jensen, S.Ø.; Füchtbauer, E.-M.; Martensen, P.M. High Folic Acid Diet Enhances Tumour Growth in PyMT-Induced Breast Cancer. Br. J. Cancer 2017, 116, 752–761. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine Addiction: A New Therapeutic Target in Cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Gaglio, D.; Soldati, C.; Vanoni, M.; Alberghina, L.; Chiaradonna, F. Glutamine Deprivation Induces Abortive S-Phase Rescued by Deoxyribonucleotides in k-Ras Transformed Fibroblasts. PLoS ONE 2009, 4, e4715. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.; Weinstock, A.; Wagner, A.; et al. Glutamine Synthetase Activity Fuels Nucleotide Biosynthesis and Supports Growth of Glutamine-Restricted Glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic Shifts toward Glutamine Regulate Tumor Growth, Invasion and Bioenergetics in Ovarian Cancer. Mol. Syst Biol. 2014, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate Is a Limiting Metabolite for Cancer Cell Proliferation under Hypoxia and in Tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Li, F.; Handler, J.; Huang, C.R.; Xiang, Y.; Neretti, N.; Sedivy, J.M.; Zeller, K.I.; Dang, C.V. Global Regulation of Nucleotide Biosynthetic Genes by C-Myc. PLoS ONE 2008, 3, e2722. [Google Scholar] [CrossRef] [PubMed]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct Role of Nucleotide Metabolism in C-MYC-Dependent Proliferation of Melanoma Cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Fan, T.W.M.; Bruntz, R.C.; Yang, Y.; Song, H.; Chernyavskaya, Y.; Deng, P.; Zhang, Y.; Shah, P.P.; Beverly, L.J.; Qi, Z.; et al. Synthesis of Serine and Glycine Fuels Purine Nucleotide Biosynthesis in Human Lung Cancer Tissues. J. Biol. Chem. 2019, 294, 13464–13477. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc Regulates a Transcriptional Program That Stimulates Mitochondrial Glutaminolysis and Leads to Glutamine Addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. MTORC1 Induces Purine Synthesis through Control of the Mitochondrial Tetrahydrofolate Cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef]
- Asby, D.J.; Cuda, F.; Beyaert, M.; Houghton, F.D.; Cagampang, F.R.; Tavassoli, A. AMPK Activation via Modulation of De Novo Purine Biosynthesis with an Inhibitor of ATIC Homodimerization. Chem. Biol. 2015, 22, 838–848. [Google Scholar] [CrossRef]
- Qian, X.; Li, X.; Tan, L.; Lee, J.H.; Xia, Y.; Cai, Q.; Zheng, Y.; Wang, H.; Lorenzi, P.L.; Lu, Z. Conversion of PRPS Hexamer to Monomer by AMPK-Mediated Phosphorylation Inhibits Nucleotide Synthesis in Response to Energy Stress. Cancer Discov. 2018, 8, 94–107. [Google Scholar] [CrossRef]
- Oizel, K.; Tait-Mulder, J.; Fernandez-de-Cossio-Diaz, J.; Pietzke, M.; Brunton, H.; Lilla, S.; Dhayade, S.; Athineos, D.; Blanco, G.R.; Sumpton, D.; et al. Formate Induces a Metabolic Switch in Nucleotide and Energy Metabolism. Cell Death Dis. 2020, 11, 310. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as Biosynthetic Factories for Cancer Proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- French, J.B.; Zhao, H.; An, S.; Niessen, S.; Deng, Y.; Cravatt, B.F.; Benkovic, S.J. Hsp70/Hsp90 Chaperone Machinery Is Involved in the Assembly of the Purinosome. Proc. Natl. Acad. Sci. USA 2013, 110, 2528–2533. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Kyoung, M.; Allen, J.J.; Shokat, K.M.; Benkovic, S.J. Dynamic Regulation of a Metabolic Multi-Enzyme Complex by Protein Kinase CK2. J. Biol. Chem. 2010, 285, 11093–11099. [Google Scholar] [CrossRef] [PubMed]
- Fesik, S.W. Promoting Apoptosis as a Strategy for Cancer Drug Discovery. Nat. Rev. Cancer 2005, 5, 876–885. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting Mitochondria Metabolism for Cancer Therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking Ovarian Cancer II: Reducing Mortality from High-Grade Serous Ovarian Cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef]
- Iida, Y.; Aoki, K.; Asakura, T.; Ueda, K.; Yanaihara, N.; Takakura, S.; Yamada, K.; Okamoto, A.; Tanaka, T.; Ohkawa, K. Hypoxia Promotes Glycogen Synthesis and Accumulation in Human Ovarian Clear Cell Carcinoma. Int. J. Oncol. 2012, 40, 2122–2130. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.A.; Hong, J.; Asaka, R.; Asaka, S.; Hsu, F.C.; Suryo Rahmanto, Y.; Jung, J.G.; Chen, Y.W.; Yen, T.T.; Tomaszewski, A.; et al. Inhibition of the MYC-Regulated Glutaminase Metabolic Axis Is an Effective Synthetic Lethal Approach for Treating Chemoresistant Ovarian Cancers. Cancer Res. 2020, 80, 4514–4526. [Google Scholar] [PubMed]
- Shorstova, T.; Su, J.; Zhao, T.; Dahabieh, M.; Leibovitch, M.; De Sa Tavares Russo, M.; Avizonis, D.; Rajkumar, S.; Watson, I.R.; Del Rincón, S.V.; et al. Reprogramming of Nucleotide Metabolism Mediates Synergy between Epigenetic Therapy and MAP Kinase Inhibition. Mol. Cancer Ther. 2021, 20, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Hatse, S.; De Clercq, E.; Balzarini, J. Role of Antimetabolites of Purine and Pyrimidine Nucleotide Metabolism in Tumor Cell Differentiation. Biochem. Pharmacol. 1999, 58, 539–555. [Google Scholar] [CrossRef]
- Sessa, C.; de Jong, J.; D’Incalci, M.; Hatty, S.; Pagani, O.; Cavalli, F. Phase I Study of the Antipurine Antifolate Lometrexol (DDATHF) with Folinic Acid Rescue. Clin. Cancer Res. 1996, 2, 1123–1127. [Google Scholar] [PubMed]
- Boritzki, T.J.; Barlett, C.A.; Zhang, C.; Howland, E.F. AG2034: A Novel Inhibitor of Glycinamide Ribonucleotide Formyltransferase. Investig. New Drugs 1996, 14, 295–303. [Google Scholar] [CrossRef]
- Batool, S.; Nawaz, M.S.; Mushtaq, G.; Parvaiz, F.; Kamal, M.A. In Silico Analysis of Glycinamide Ribonucleotide Transformylase Inhibition by PY873, PY899 and DIA. Saudi J. Biol. Sci. 2017, 24, 1155–1161. [Google Scholar] [CrossRef][Green Version]
- DeMartino, J.K.; Hwang, I.; Xu, L.; Wilson, I.A.; Boger, D.L. Discovery of a Potent, Nonpolyglutamatable Inhibitor of Glycinamide Ribonucleotide Transformylase. J. Med. Chem. 2006, 49, 2998–3002. [Google Scholar] [CrossRef]
- Xu, L.; Li, C.; Olson, A.J.; Wilson, I.A. Crystal Structure of Avian Aminoimidazole-4-Carboxamide Ribonucleotide Transformylase in Complex with a Novel Non-Folate Inhibitor Identified by Virtual Ligand Screening. J. Biol. Chem. 2004, 279, 50555–50565. [Google Scholar] [CrossRef]
- Fales, K.R.; Njoroge, F.G.; Brooks, H.B.; Thibodeaux, S.; Torrado, A.; Si, C.; Toth, J.L.; Mc Cowan, J.R.; Roth, K.D.; Thrasher, K.J.; et al. Discovery of N-(6-Fluoro-1-Oxo-1,2-Dihydroisoquinolin-7-Yl)-5-[(3R)-3-Hydroxypyrrolidin-1-Yl]Thiophene-2-Sulfonamide (LSN 3213128), a Potent and Selective Nonclassical Antifolate Aminoimidazole-4-Carboxamide Ribonucleotide Formyltransferase (AICARFT) Inhibitor Effective at Tumor Suppression in a Cancer Xenograft Model. J. Med. Chem. 2017, 60, 9599–9616. [Google Scholar] [PubMed]
- Chakravarthi, B.V.S.K.; Goswami, M.T.; Pathi, S.S.; Dodson, M.; Chandrashekar, D.S.; Agarwal, S.; Nepal, S.; Hodigere Balasubramanya, S.A.; Siddiqui, J.; Lonigro, R.J.; et al. Expression and Role of PAICS, a de Novo Purine Biosynthetic Gene in Prostate Cancer. Prostate 2018, 78, 693–694. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Chen, Y.; Jia, J.; Li, L.; Yang, S. Knockdown of PAICS Inhibits Malignant Proliferation of Human Breast Cancer Cell Lines. Biol. Res. 2018, 51, 24. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Hughes-Hallett, J.; Timson, R.C.; Ilagan, E.; Yuan, M.; Asara, J.M.; Ben-Sahra, I.; Manning, B.D. The MTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell Rep. 2017, 21, 1331–1346. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Chan, C.Y.; Pedley, A.M.; Kim, D.; Xia, C.; Zhuang, X.; Benkovic, S.J. Microtubule-Directed Transport of Purine Metabolons Drives Their Cytosolic Transit to Mitochondria. Proc. Natl. Acad. Sci. USA 2018, 115, 13009–13014. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug Class | Drug Name | Target(s) | Example Indications | Reference |
|---|---|---|---|---|
| Folate antagonists | Aminopterin | Dihydrofolate reductase (DHFR) | Leukemias | [26,27] |
| Methotrexate | DHFR Thymidylate synthase (TS) Bifunctional purine biosynthesis protein PURH (ATIC) Amido phosphoribosyltransferase (PPAT) | Acute lymphoblastic Leukemias (ALL) Lymphoma Brain tumors Osteosarcoma Breast cancer | [26,27] | |
| Pemetrexed | DHFR TS ATIC Trifunctional purine biosynthetic protein adenosine-3 (GART) | Lung cancer Ovarian cancer Head and neck Liver cancer Mesothelioma Advanced cancers | [26,27,28] | |
| Pralatexate | DHFR TS | Multiples Myeloma (MM) | [26,27] | |
| Purine antagonists | 6-mercaptopurine | Hypoxanthine-guanine phosphoribosyltransferase (HGPRTase) PPAT | Leukemias ALL | [23,24] |
| 6-thioguanine | HGPRTase | Leukemias ALL | [23,24] | |
| Fludarabine Cladribine | DNA synthesis DNA repair | Leukemias MM ALL | [23,24] | |
| Pentostatin | Adenosine deaminase (ADA) DNA synthesis | Leukemias ALL Renal cancer | [23,24] | |
| Clofarabine Nelarabine | DNA elongation | Leukemias ALL Recurrent neoplasm | [23,24] | |
| Mitochondria | Metformin | ETC complex I | Ovarian cancer Breast cancer Prostate cancer Endometrial Lung cancer | [29,30,31] |
| CPI-613 | α-ketoglutarate Dehydrogenase (α-KGDH) | Pancreatic cancer AML | [32] | |
| CB-839 | Glutaminase (GLS) | Lung cancer | [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Vitto, H.; Arachchige, D.B.; Richardson, B.C.; French, J.B. The Intersection of Purine and Mitochondrial Metabolism in Cancer. Cells 2021, 10, 2603. https://doi.org/10.3390/cells10102603
De Vitto H, Arachchige DB, Richardson BC, French JB. The Intersection of Purine and Mitochondrial Metabolism in Cancer. Cells. 2021; 10(10):2603. https://doi.org/10.3390/cells10102603
Chicago/Turabian StyleDe Vitto, Humberto, Danushka B. Arachchige, Brian C. Richardson, and Jarrod B. French. 2021. "The Intersection of Purine and Mitochondrial Metabolism in Cancer" Cells 10, no. 10: 2603. https://doi.org/10.3390/cells10102603
APA StyleDe Vitto, H., Arachchige, D. B., Richardson, B. C., & French, J. B. (2021). The Intersection of Purine and Mitochondrial Metabolism in Cancer. Cells, 10(10), 2603. https://doi.org/10.3390/cells10102603