
Metabolic Stress Adaptations Underlie Mammary Gland Morphogenesis and Breast Cancer Progression


{kind=link}
{kind=link}
Abstract
:1. Metabolic Regulation in Mammary Gland Morphogenesis
1.1. Mammary Gland Morphogenesis
1.2. Terminal End Buds
1.3. Terminal End Bud as a Model for Breast Cancer
1.4. Lineage-Specific Stress Signaling in Mammary Epithelium Development
2. Metabolic Regulators Heterogeneously Activated in 3D MCF10A Spheroids
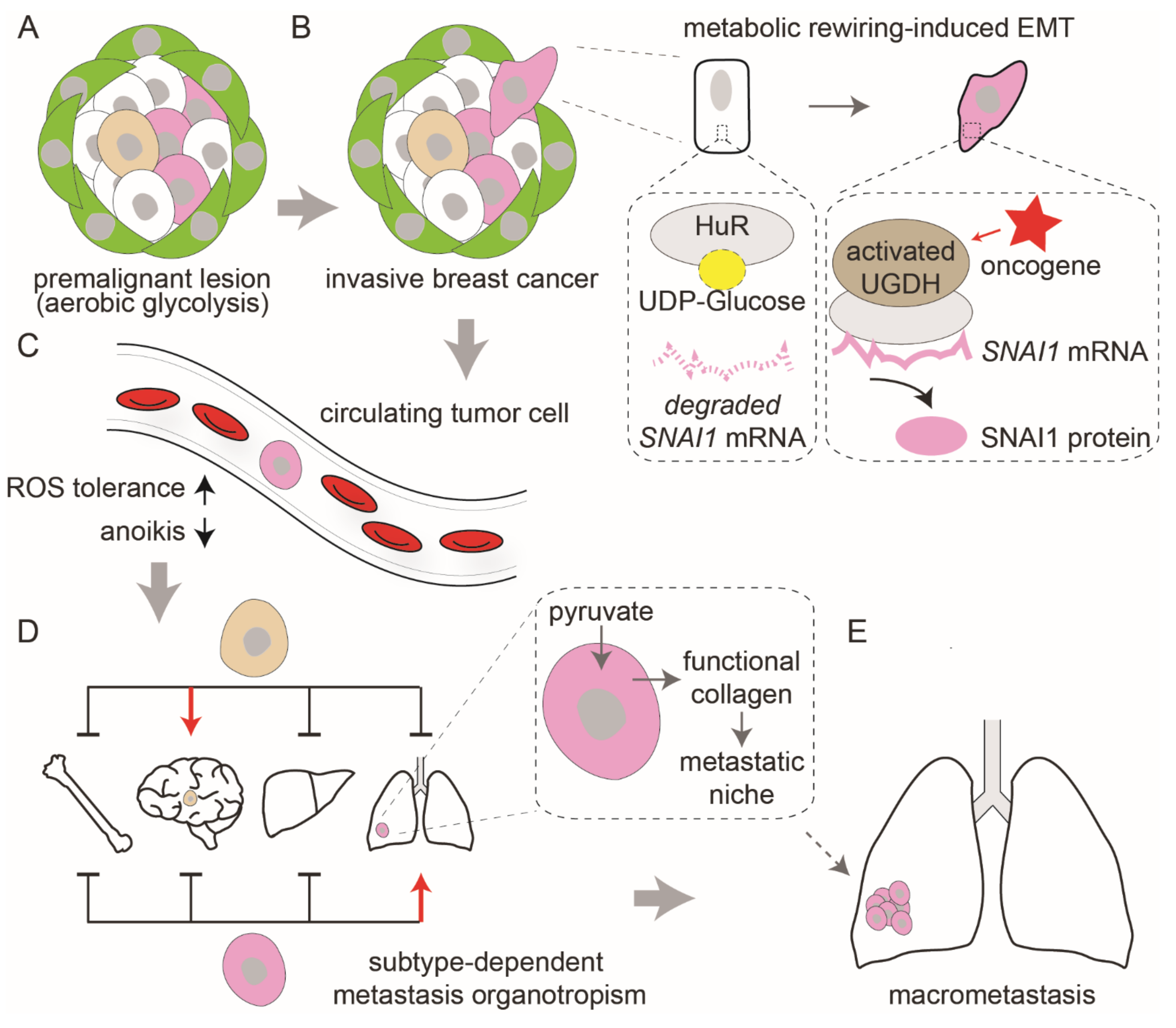
3. Metabolic Reprogramming Governs Cancer Progression
4. Lineage-Specific and Subtype-Selective Stress Regulatory Networks in Mammary Gland and Breast Cancer
4.1. Lineage-Specific Metabolic Identities of Mammary Cells
4.2. The Metabolic Reprogramming and Molecular Diversity of Primary Breast Cancers
4.3. TNBC-Specific Metabolic Signatures
4.4. Metabolic Networks in Luminal Breast Cancer
4.5. Metabolic Networks in HER2 Positive Breast Cancer
5. Future Directions and Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cowin, P.; Wysolmerski, J. Molecular mechanisms guiding embryonic mammary gland development. Cold Spring Harb. Perspect. Biol. 2010, 2, a003251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, D. The mammary gland: A unique organ for the study of development and tumorigenesis. J. Mammary Gland Biol. Neoplasia 1996, 1, 5–19. [Google Scholar] [CrossRef]
- Nguyen, Q.H.; Pervolarakis, N.; Blake, K.; Ma, D.; Davis, R.T.; James, N.; Phung, A.T.; Willey, E.; Kumar, R.; Jabart, E.; et al. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat. Commun. 2018, 9, 2028. [Google Scholar] [CrossRef] [PubMed]
- Jena, M.K.; Jaswal, S.; Kumar, S.; Mohanty, A.K. Molecular mechanism of mammary gland involution: An update. Dev. Biol. 2019, 445, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Paine, I.S.; Lewis, M.T. The Terminal End Bud: The Little Engine that Could. J. Mammary Gland Biol. Neoplasia 2017, 22, 93–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchcock, J.R.; Hughes, K.; Harris, O.B.; Watson, C.J. Dynamic architectural interplay between leucocytes and mammary epithelial cells. FEBS J. 2020, 287, 250–266. [Google Scholar] [CrossRef] [PubMed]
- Javed, A.; Lteif, A. Development of the human breast. Semin. Plast. Surg. 2013, 27, 5–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huebner, R.J.; Lechler, T.; Ewald, A.J. Developmental stratification of the mammary epithelium occurs through symmetry-breaking vertical divisions of apically positioned luminal cells. Development 2014, 141, 1085–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, I.K.H.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Ewald, A.J.; Brenot, A.; Duong, M.; Chan, B.S.; Werb, Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev. Cell 2008, 14, 570–581. [Google Scholar] [CrossRef] [Green Version]
- Unsworth, A.; Anderson, R.; Britt, K. Stromal fibroblasts and the immune microenvironment: Partners in mammary gland biology and pathology? J. Mammary Gland Biol. Neoplasia 2014, 19, 169–182. [Google Scholar] [CrossRef]
- Ingman, W.V.; Wyckoff, J.; Gouon-Evans, V.; Condeelis, J.; Pollard, J.W. Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2006, 235, 3222–3229. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, G.B. Tumour-stromal interactions. Role of the stroma in mammary development. Breast Cancer Res. BCR 2001, 3, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Moses, H.; Barcellos-Hoff, M.H. TGF-beta biology in mammary development and breast cancer. Cold Spring Harb. Perspect. Biol. 2011, 3, a003277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajikar, S.S.; Wang, C.C.; Borten, M.A.; Pereira, E.J.; Atkins, K.A.; Janes, K.A. Tumor-Suppressor Inactivation of GDF11 Occurs by Precursor Sequestration in Triple-Negative Breast Cancer. Dev. Cell 2017, 43, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Rossiter, H.; Barresi, C.; Ghannadan, M.; Gruber, F.; Mildner, M.; Födinger, D.; Tschachler, E. Inactivation of VEGF in mammary gland epithelium severely compromises mammary gland development and function. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 3994–4004. [Google Scholar] [CrossRef]
- Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. In situ identification of bipotent stem cells in the mammary gland. Nature 2014, 506, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Rocha, A.S.; Ousset, M.; Beck, B.; Bouvencourt, G.; Rock, J.; Sharma, N.; Dekoninck, S.; Blanpain, C. Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011, 479, 189–193. [Google Scholar] [CrossRef]
- Williams, J.M.; Daniel, C.W. Mammary ductal elongation: Differentiation of myoepithelium and basal lamina during branching morphogenesis. Dev. Biol. 1983, 97, 274–290. [Google Scholar] [CrossRef]
- Regan, J.; Sourisseau, T.; Soady, K.; Kendrick, H.; McCarthy, A.; Tang, C.; Brennan, K.; Linardopoulos, S.; White, D.E.; Smalley, M. Aurora A Kinase Regulates Mammary Epithelial Cell Fate by Determining Mitotic Spindle Orientation in a Notch-Dependent Manner. Cell Rep. 2013, 4, 110–123. [Google Scholar] [CrossRef]
- Sreekumar, A.; Toneff, M.J.; Toh, E.; Roarty, K.; Creighton, C.J.; Belka, G.K.; Lee, D.-K.; Xu, J.; Chodosh, L.A.; Richards, J.S.; et al. WNT-Mediated Regulation of FOXO1 Constitutes a Critical Axis Maintaining Pubertal Mammary Stem Cell Homeostasis. Dev. Cell 2017, 43, 436–448. [Google Scholar] [CrossRef]
- Scheele, C.L.G.J.; Hannezo, E.; Muraro, M.J.; Zomer, A.; Langedijk, N.S.M.; van Oudenaarden, A.; Simons, B.D.; van Rheenen, J. Identity and dynamics of mammary stem cells during branching morphogenesis. Nature 2017, 542, 313–317. [Google Scholar] [CrossRef]
- Huebner, R.J.; Ewald, A.J. Cellular foundations of mammary tubulogenesis. Semin. Cell Dev. Biol. 2014, 31, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.-L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chiang, H.C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef] [PubMed]
- Mahendralingam, M.J.; Kim, H.; McCloskey, C.W.; Aliar, K.; Casey, A.E.; Tharmapalan, P.; Pellacani, D.; Ignatchenko, V.; Garcia-Valero, M.; Palomero, L.; et al. Mammary epithelial cells have lineage-rooted metabolic identities. Nat. Metab. 2021, 3, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Kannan, N.; Nguyen, L.V.; Makarem, M.; Dong, Y.; Shih, K.; Eirew, P.; Raouf, A.; Emerman, J.T.; Eaves, C.J. Glutathione-dependent and -independent oxidative stress-control mechanisms distinguish normal human mammary epithelial cell subsets. Proc. Natl. Acad. Sci. USA 2014, 111, 7789–7794. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.M.; Rudolph, M.C.; McManaman, J.L.; Neville, M.C. Key stages in mammary gland development. Secretory activation in the mammary gland: It’s not just about milk protein synthesis! Breast Cancer Res. BCR 2007, 9, 204. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Hatzivassiliou, G.; Grigoriadou, C.; Romero, M.; Cavener, D.R.; Thompson, C.B.; Diehl, J.A. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 16314–16319. [Google Scholar] [CrossRef] [Green Version]
- Reginato, M.J.; Mills, K.R.; Paulus, J.K.; Lynch, D.K.; Sgroi, D.C.; Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 2003, 5, 733–740. [Google Scholar] [CrossRef]
- Wang, C.-C.; Jamal, L.; Janes, K.A. Normal morphogenesis of epithelial tissues and progression of epithelial tumors. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 51–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-C.; Bajikar, S.S.; Jamal, L.; Atkins, K.A.; Janes, K.A. A time- and matrix-dependent TGFBR3–JUND–KRT5 regulatory circuit in single breast epithelial cells and basal-like premalignancies. Nat. Cell Biol. 2014, 16, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-C.; Janes, K.A. Non-genetic heterogeneity caused by differential single-cell adhesion. Cell Cycle 2014, 13, 2149–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003, 30, 256–268. [Google Scholar] [CrossRef]
- Janes, K.A.; Wang, C.-C.; Holmberg, K.J.; Cabral, K.; Brugge, J.S. Identifying single-cell molecular programs by stochastic profiling. Nat. Methods 2010, 7, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Wen, Q.; Xie, Y.; Liu, J.; Kroemer, G.; et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 2018, 46, 441–455. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, M.; Tian, L.; Le, P.T.; DeMambro, V.E.; Becker, K.A.; Rosen, C.J.; Guntur, A.R. The mitophagy receptor Bcl-2–like protein 13 stimulates adipogenesis by regulating mitochondrial oxidative phosphorylation and apoptosis in mice. J. Biol. Chem. 2019, 294, 12683–12694. [Google Scholar] [CrossRef]
- Gwon, Y.; Maxwell, B.A.; Kolaitis, R.-M.; Zhang, P.; Kim, H.J.; Taylor, J.P. Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science 2021, 372, eabf6548. [Google Scholar] [CrossRef]
- Elko, E.A.; Manuel, A.M.; White, S.; Zito, E.; van der Vliet, A.; Anathy, V.; Janssen-Heininger, Y.M.W. Oxidation of peroxiredoxin-4 induces oligomerization and promotes interaction with proteins governing protein folding and endoplasmic reticulum stress. J. Biol. Chem. 2021, 296, 100665. [Google Scholar] [CrossRef]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Hyun, J.; Nah, J.; Han, J.; Kim, S.-H.; Park, J.; Oh, Y.; Gwon, Y.; Moon, S.; Jo, D.-G.; et al. SERP1 is an assembly regulator of γ-secretase in metabolic stress conditions. Sci. Signal. 2020, 13, eaax8949. [Google Scholar] [CrossRef] [PubMed]
- Gerald, D.; Berra, E.; Frapart, Y.M.; Chan, D.A.; Giaccia, A.J.; Mansuy, D.; Pouysségur, J.; Yaniv, M.; Mechta-Grigoriou, F. JunD Reduces Tumor Angiogenesis by Protecting Cells from Oxidative Stress. Cell 2004, 118, 781–794. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Mlynarczyk, C.; Fåhraeus, R. Endoplasmic reticulum stress sensitizes cells to DNA damage-induced apoptosis through p53-dependent suppression of p21CDKN1A. Nat. Commun. 2014, 5, 5067. [Google Scholar] [CrossRef] [PubMed]
- Samali, A.; Cai, J.; Zhivotovsky, B.; Jones, D.P.; Orrenius, S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of Jurkat cells. EMBO J. 1999, 18, 2040–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, X.; Broderick, R.; Bergoglio, V.; Zimmer, J.; Badie, S.; Niedzwiedz, W.; Hoffmann, J.-S.; Tarsounas, M. MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat. Commun. 2017, 8, 15983. [Google Scholar] [CrossRef] [Green Version]
- Tanioka, M.; Fan, C.; Parker, J.S.; Hoadley, K.A.; Hu, Z.; Li, Y.; Hyslop, T.M.; Pitcher, B.N.; Soloway, M.G.; Spears, P.A.; et al. Integrated Analysis of RNA and DNA from the Phase III Trial CALGB 40601 Identifies Predictors of Response to Trastuzumab-Based Neoadjuvant Chemotherapy in HER2-Positive Breast Cancer. Clin. Cancer Res. 2018, 24, 5292–5304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, E.J.; Burns, J.S.; Lee, C.Y.; Marohl, T.; Calderon, D.; Wang, L.; Atkins, K.A.; Wang, C.-C.; Janes, K.A. Sporadic activation of an oxidative stress–dependent NRF2-p53 signaling network in breast epithelial spheroids and premalignancies. Sci. Signal. 2020, 13, eaba4200. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Cho, P.; Selfors, L.M.; Kuiken, H.J.; Kaul, R.; Fujiwara, T.; Harris, I.S.; Zhang, T.; Gygi, S.P.; Brugge, J.S. 3D Culture Models with CRISPR Screens Reveal Hyperactive NRF2 as a Prerequisite for Spheroid Formation via Regulation of Proliferation and Ferroptosis. Mol. Cell 2020, 80, 828–844. [Google Scholar] [CrossRef]
- Davis, R.T.; Blake, K.; Ma, D.; Gabra, M.B.I.; Hernandez, G.A.; Phung, A.T.; Yang, Y.; Maurer, D.; Lefebvre, A.E.Y.T.; Alshetaiwi, H.; et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat. Cell Biol. 2020, 22, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Miyamoto, D.T.; Wittner, B.S.; Sullivan, J.P.; Aceto, N.; Jordan, N.V.; Yu, M.; Karabacak, N.M.; Comaills, V.; Morris, R.; et al. Expression of β-globin by cancer cells promotes cell survival during blood-borne dissemination. Nat. Commun. 2017, 8, 14344. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, R.; Zhu, W.; Chu, H.; Yu, H.; Wei, P.; Wu, X.; Zhu, H.; Gao, H.; Liang, J.; et al. UDP-glucose accelerates SNAI1 mRNA decay and impairs lung cancer metastasis. Nature 2019, 571, 127–131. [Google Scholar] [CrossRef]
- Elia, I.; Rossi, M.; Stegen, S.; Broekaert, D.; Doglioni, G.; van Gorsel, M.; Boon, R.; Escalona-Noguero, C.; Torrekens, S.; Verfaillie, C.; et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 2019, 568, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, G.B.; Ali, A.; Luengo, A.; Kodack, D.P.; Deik, A.; Abbott, K.L.; Bezwada, D.; Blanc, L.; Prideaux, B.; Jin, X.; et al. Fatty acid synthesis is required for breast cancer brain metastasis. Nat. Cancer 2021, 2, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-K.; Jeong, S.-H.; Jang, C.; Bae, H.; Kim, Y.H.; Park, I.; Kim, S.K.; Koh, G.Y. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 2019, 363, 644–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havas, K.M.; Milchevskaya, V.; Radic, K.; Alladin, A.; Kafkia, E.; Garcia, M.; Stolte, J.; Klaus, B.; Rotmensz, N.; Gibson, T.J.; et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J. Clin. Investig. 2017, 127, 2091–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Giraddi, R.R.; Chung, C.-Y.; Heinz, R.E.; Balcioglu, O.; Novotny, M.; Trejo, C.L.; Dravis, C.; Hagos, B.M.; Mehrabad, E.M.; Rodewald, L.W.; et al. Single-Cell Transcriptomes Distinguish Stem Cell State Changes and Lineage Specification Programs in Early Mammary Gland Development. Cell Rep. 2018, 24, 1653–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, 37, 168–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matoba, S.; Kang, J.-G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 Regulates Mitochondrial Respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damaghi, M.; West, J.; Robertson-Tessi, M.; Xu, L.; Ferrall-Fairbanks, M.C.; Stewart, P.A.; Persi, E.; Fridley, B.L.; Altrock, P.M.; Gatenby, R.A.; et al. The harsh microenvironment in early breast cancer selects for a Warburg phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2011342118. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Green, A.R.; Aleskandarany, M.A.; Agarwal, D.; Elsheikh, S.; Nolan, C.C.; Diez-Rodriguez, M.; Macmillan, R.D.; Ball, G.R.; Caldas, C.; Madhusudan, S.; et al. MYC functions are specific in biological subtypes of breast cancer and confers resistance to endocrine therapy in luminal tumours. Br. J. Cancer 2016, 114, 917–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; Talukdar, M.; Lu, Y.; Wang, X.; Hu, D.Z.; et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Investig. 2018, 128, 1283–1299. [Google Scholar] [CrossRef] [Green Version]
- Sicari, D.; Fantuz, M.; Bellazzo, A.; Valentino, E.; Apollonio, M.; Pontisso, I.; Di Cristino, F.; Dal Ferro, M.; Bicciato, S.; Del Sal, G.; et al. Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene 2019, 38, 6184–6195. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, F.; Brandt, D.T.; Schneikert, J.; Fuchs, J.; Grikscheit, K.; Wanzel, M.; Pavlakis, E.; Charles, J.P.; Timofeev, O.; Nist, A.; et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc. Natl. Acad. Sci. USA 2016, 113, E8433–E8442. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Ba-Alawi, W.; Deblois, G.; Cruickshank, J.; Duan, S.; Lima-Fernandes, E.; Haight, J.; Tonekaboni, S.A.M.; Fortier, A.-M.; Kuasne, H.; et al. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat. Commun. 2020, 11, 4205. [Google Scholar] [CrossRef] [PubMed]
- Dowling, C.M.; Hollinshead, K.E.R.; Di Grande, A.; Pritchard, J.; Zhang, H.; Dillon, E.T.; Haley, K.; Papadopoulos, E.; Mehta, A.K.; Bleach, R.; et al. Multiple screening approaches reveal HDAC6 as a novel regulator of glycolytic metabolism in triple-negative breast cancer. Sci. Adv. 2021, 7, eabc4897. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.J.; Hou, J.; Xu, B.; Cortez, M.; Potma, E.O.; Tromberg, B.J.; Razorenova, O.V. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2017, 114, E6556–E6565. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.-L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Gawrzak, S.; Rinaldi, L.; Gregorio, S.; Arenas, E.J.; Salvador, F.; Urosevic, J.; Figueras-Puig, C.; Rojo, F.; del Barco Barrantes, I.; Cejalvo, J.M.; et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER+ breast cancer. Nat. Cell Biol. 2018, 20, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Lukey, M.J.; Cluntun, A.A.; Katt, W.P.; Lin, M.-c.J.; Druso, J.E.; Ramachandran, S.; Erickson, J.W.; Le, H.H.; Wang, Z.-E.; Blank, B.; et al. Liver-Type Glutaminase GLS2 Is a Druggable Metabolic Node in Luminal-Subtype Breast Cancer. Cell Rep. 2019, 29, 76–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Liu, Z.; Desai, S.; Zhao, Y.; Liu, H.; Pannell, L.K.; Yi, H.; Wright, E.R.; Owen, L.B.; Dean-Colomb, W.; et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat. Commun. 2012, 3, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodack, D.P.; Askoxylakis, V.; Ferraro, G.B.; Fukumura, D.; Jain, R.K. Emerging Strategies for Treating Brain Metastases from Breast Cancer. Cancer Cell 2015, 27, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanojia, D.; Panek, W.K.; Cordero, A.; Fares, J.; Xiao, A.; Savchuk, S.; Kumar, K.; Xiao, T.; Pituch, K.C.; Miska, J.; et al. BET inhibition increases βIII-tubulin expression and sensitizes metastatic breast cancer in the brain to vinorelbine. Sci. Transl. Med. 2020, 12, eaax2879. [Google Scholar] [CrossRef]
- Cordero, A.; Kanojia, D.; Miska, J.; Panek, W.K.; Xiao, A.; Han, Y.; Bonamici, N.; Zhou, W.; Xiao, T.; Wu, M.; et al. FABP7 is a key metabolic regulator in HER2+ breast cancer brain metastasis. Oncogene 2019, 38, 6445–6460. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-C. Metabolic Stress Adaptations Underlie Mammary Gland Morphogenesis and Breast Cancer Progression. Cells 2021, 10, 2641. https://doi.org/10.3390/cells10102641
Wang C-C. Metabolic Stress Adaptations Underlie Mammary Gland Morphogenesis and Breast Cancer Progression. Cells. 2021; 10(10):2641. https://doi.org/10.3390/cells10102641
Chicago/Turabian StyleWang, Chun-Chao. 2021. "Metabolic Stress Adaptations Underlie Mammary Gland Morphogenesis and Breast Cancer Progression" Cells 10, no. 10: 2641. https://doi.org/10.3390/cells10102641
APA StyleWang, C.-C. (2021). Metabolic Stress Adaptations Underlie Mammary Gland Morphogenesis and Breast Cancer Progression. Cells, 10(10), 2641. https://doi.org/10.3390/cells10102641