
Integrated CGH/WES Analyses Advance Understanding of Aggressive Neuroblastoma Evolution: A Case Study


 ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Information, Tumor Sample Collection, and Primary Cells Maintenance
2.2. Histological and Immunohistochemical Analyses
2.3. DNA Extraction, Library Construction, and WES
2.4. WES Analysis
2.5. aCGH Analysis
2.6. Pharmacogenetics Study
2.7. Statistical and Clonal Analysis
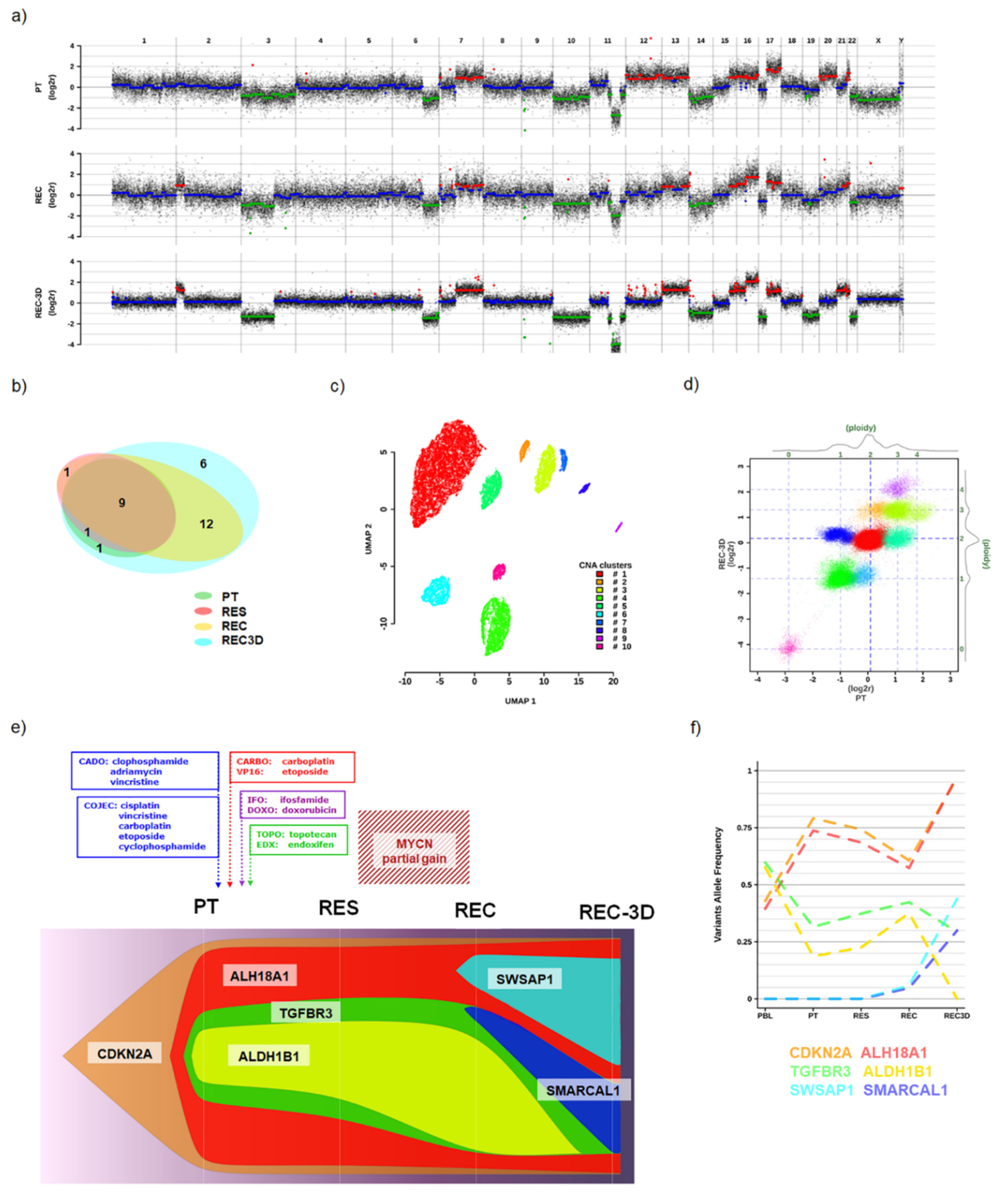
3. Results
3.1. Case Description
3.1.1. The 3D In Vitro Study
3.1.2. Genomic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Luksch, R.; Castellani, M.R.; Collini, P.; De Bernardi, B.; Conte, M.; Gambini, C.; Gandola, L.; Garaventa, A.; Biasoni, D.; Podda, M.; et al. Neuroblastoma (Peripheral neuroblastic tumours). Crit. Rev. Oncol. Hematol. 2016, 107, 163–181. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Daage, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonini, G.P. Growth, progression and chromosome instability of Neuroblastoma: A new scenario of tumorigenesis? BMC Cancer 2017, 17, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, E.; Desai, A. The Evolution of Risk Classification for Neuroblastoma. Children 2019, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Janoueix-Lerosey, I.; Schleiermacher, G.; Michels, E.; Mosseri, V.; Ribeiro, A.; Lequin, D.; Vermeulen, J.; Couturier, J.; Peuchmaur, M.; Valent, A.; et al. Overall Genomic Pattern Is a Predictor of Outcome in Neuroblastoma. J. Clin. Oncol. 2009, 27, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The International Neuroblastoma Risk Group (INRG) Staging System: An INRG Task Force Report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambros, I.M.; Benard, J.; Boavida, M.; Bown, N.; Caron, H.; Combaret, V.; Couturier, J.; Darnfors, C.; Delattre, O.; Freeman-Edward, J.; et al. Quality Assessment of Genetic Markers Used for Therapy Stratification. J. Clin. Oncol. 2003, 21, 2077–2084. [Google Scholar] [CrossRef]
- Scaruffi, P.; Coco, S.; Cifuentes, F.; Albino, D.; Nair, M.; Defferrari, R.; Mazzocco, K.; Tonini, G.P. Identification and characterization of DNA imbalances in neuroblastoma by high-resolution oligonucleotide array comparative genomic hybridization. Cancer Genet. Cytogenet. 2007, 177, 20–29. [Google Scholar] [CrossRef]
- Maris, J.M.; Weiss, M.J.; Guo, C.; Gerbing, R.B.; Stram, D.O.; White, P.S.; Hogarty, M.D.; Sulman, E.P.; Thompson, P.M.; Lukens, J.N.; et al. Loss of Heterozygosity at 1p36 Independently Predicts for Disease Progression But Not Decreased Overall Survival Probability in Neuroblastoma Patients: A Children’s Cancer Group Study. J. Clin. Oncol. 2000, 18, 1888–1899. [Google Scholar] [CrossRef]
- Park, S.-J.; Park, C.-J.; Kim, S.; Jang, S.; Chi, H.-S.; Kim, M.J.; Im, H.-J.; Seo, J.-J. Detection of Bone Marrow Metastases of Neuroblastoma With Immunohistochemical Staining of CD56, Chromogranin A, and Synaptophysin. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 348–352. [Google Scholar] [CrossRef]
- Zhong, Z.-Y.; Shi, B.-J.; Zhou, H.; Wang, W.-B. CD133 expression and MYCN amplification induce chemoresistance and reduce average survival time in pediatric neuroblastoma. J. Int. Med. Res. 2018, 46, 1209–1220. [Google Scholar] [CrossRef]
- Tadeo, I.; Berbegall, A.P.; Castel, V.; García-Miguel, P.; Callaghan, R.; Påhlman, S.; Navarro, S.; Noguera, R. Extracellular matrix composition defines an ultra-high-risk group of neuroblastoma within the high-risk patient cohort. Br. J. Cancer 2016, 115, 480–489. [Google Scholar] [CrossRef] [Green Version]
- London, W.B.; Castel, V.; Monclair, T.; Ambros, P.F.; Pearson, A.D.J.; Cohn, S.L.; Berthold, F.; Nakagawara, A.; Ladenstein, R.L.; Iehara, T.; et al. Clinical and Biologic Features Predictive of Survival After Relapse of Neuroblastoma: A Report From the International Neuroblastoma Risk Group Project. J. Clin. Oncol. 2011, 29, 3286–3292. [Google Scholar] [CrossRef] [Green Version]
- Olivera, G.; Sendra, L.; Herrero, M.J.; Berlanga, P.; Gargallo, P.; Yáñez, Y.; Urtasun, A.; Font de Mora, J.; Castel, V.; Cañete, A.; et al. Pharmacogenetics implementation in the clinics: Information and guidelines for germline variants. Cancer Drug Resist. 2019, 2, 53–68. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.; Chen, H.; Lo, P.-K. In Vitro Tumorsphere Formation Assays. BIO-PROTOCOL 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosby, K.; Simendinger, J.; Grange, C.; Ferrante, M.; Bernier, T.; Standen, C. Immunohistochemistry Protocol for Paraffin-embedded Tissue Sections. JoVE 2014. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attiyeh, E.F.; London, W.B.; Mossé, Y.P.; Wang, Q.; Winter, C.; Khazi, D.; McGrady, P.W.; Seeger, R.C.; Look, A.T.; Shimada, H.; et al. Chromosome 1p and 11q Deletions and Outcome in Neuroblastoma. N. Engl. J. Med. 2005, 353, 2243–2253. [Google Scholar] [CrossRef]
- Garaventa, A.; Poetschger, U.; Valteau-Couanet, D.; Castel, V.; Elliott, M.; Ash, S.; Chan, G.C.-F.; Laureys, G.; Beck Popovic, M.; Vettenranta, K.; et al. The randomised induction for high-risk neuroblastoma comparing COJEC and N5-MSKCC regimens: Early results from the HR-NBL1.5/SIOPEN trial. J. Clin. Oncol. 2018, 36, 10507. [Google Scholar] [CrossRef]
- Burgos-Panadero, R.; Noguera, I.; Cañete, A.; Navarro, S.; Noguera, R. Vitronectin as a molecular player of the tumor microenvironment in neuroblastoma. BMC Cancer 2019, 19, 479. [Google Scholar] [CrossRef]
- Guo, Y.-F.; Duan, J.-J.; Wang, J.; Li, L.; Wang, D.; Liu, X.-Z.; Yang, J.; Zhang, H.-R.; Lv, J.; Yang, Y.-J.; et al. Inhibition of the ALDH18A1-MYCN positive feedback loop attenuates MYCN -amplified neuroblastoma growth. Sci. Transl. Med. 2020, 12, eaax8694. [Google Scholar] [CrossRef]
- Carr-Wilkinson, J.; O’Toole, K.; Wood, K.M.; Challen, C.C.; Baker, A.G.; Board, J.R.; Evans, L.; Cole, M.; Cheung, N.-K.V.; Boos, J.; et al. High Frequency of p53/MDM2/p14ARF Pathway Abnormalities in Relapsed Neuroblastoma. Clin. Cancer Res. 2010, 16, 1108–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iolascon, A.; Giordani, L.; Borriello, A.; Carbone, R.; Izzo, A.; Tonini, G.P.; Gambini, C.; Ragione, F. Della Reduced expression of transforming growth factor-beta receptor type III in high stage neuroblastomas. Br. J. Cancer 2000, 82, 1171–1176. [Google Scholar] [CrossRef]
- Longo, L.; Tonini, G.P.; Ceccherini, I.; Perri, P. Oligogenic inheritance in neuroblastoma. Cancer Lett. 2005, 228, 65–69. [Google Scholar] [CrossRef]
- Esposito, M.R.; Aveic, S.; Seydel, A.; Tonini, G.P. Neuroblastoma treatment in the post-genomic era. J. Biomed. Sci. 2017, 24, 14. [Google Scholar] [CrossRef] [Green Version]
- Manica, M.; Kim, H.R.; Mathis, R.; Chouvarine, P.; Rutishauser, D.; De Vargas Roditi, L.; Szalai, B.; Wagner, U.; Oehl, K.; Saba, K.; et al. Inferring clonal composition from multiple tumor biopsies. NPJ Syst. Biol. Appl. 2020, 6, 27. [Google Scholar] [CrossRef]
- Kjeldsen, E. Oligo-based high-resolution aCGH analysis enhances routine cytogenetic diagnostics in haematological malignancies. Cancer Genom. Proteom. 2015, 12, 301–337. [Google Scholar]
- Depuydt, P.; Boeva, V.; Hocking, T.D.; Cannoodt, R.; Ambros, I.M.; Ambros, P.F.; Asgharzadeh, S.; Attiyeh, E.F.; Combaret, V.; Defferrari, R.; et al. Genomic Amplifications and Distal 6q Loss: Novel Markers for Poor Survival in High-risk Neuroblastoma Patients. JNCI J. Natl. Cancer Inst. 2018, 110, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Olivera, G.G.; Yáñez, Y.; Gargallo, P.; Sendra, L.; Aliño, S.F.; Segura, V.; Sanz, M.Á.; Cañete, A.; Castel, V.; Font De Mora, J.; et al. MTHFR and VDR Polymorphisms Improve the Prognostic Value of MYCN Status on Overall Survival in Neuroblastoma Patients. Int. J. Mol. Sci. 2020, 21, 2714. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Genotypes | SNP Array | WES | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Drug | Gene | SNP | NO Risk | Risk | PBL | PBL | PT | RES | REC | REC-3D |
| Azathioprine | TPMT | rs1800462 | CC | CG,GG | CC | CC | CC | CC | CC | CC |
| TPMT | rs1800584 | CC | CT,TT | CC | CC | CC | CC | CC | CC | |
| TPMT | rs1142345 | TT | TC,CC | TT | TT | TT | TT | TT | TT | |
| TPMT | rs1800460 | CC | CT,TT | CC | CC | CC | CC | CC | CC | |
| NUDT15 | rs116855232 | CC | CT,TT | CC | CC | CC | CC | CC | CC | |
| Carboplatin * | ERCC1 | rs11615 | GG | AG,AA | AA | AA | AA | AA | AA | AA |
| ERCC1 | rs3212986 | AA | AC,CC | CC | ||||||
| GSTP1 | rs1695 | GG | AG,AA | AA | AA | AA | AA | AA | AA | |
| MTHFR | rs1801133 | AA | AG,GG | AA | AA | AA | AA | AA | AA | |
| NQO1 | rs1800566 | GG | AG,AA | AG | ||||||
| XRCC1 | rs25487 | CC | CT,TT | CC | CC | |||||
| Cyclophosphamide * | GSTP1 | rs1695 | AA,AG | GG | AA | AA | AA | AA | AA | AA |
| SOD2 | rs4880 | AA | AG,GG | GG | GG | GG | a-GG | GG | ||
| TP53 | rs1042522 | CC | CG,GG | CC | CC-g | CC-g | CC-g | CC-g | CC-g | |
| Cisplatin * | ERCC1 | rs11615 | GG | AG,AA | AA | AA | AA | AA | AA | AA |
| ERCC1 | rs3212986 | AA | AC,CC | CC | ||||||
| GSTP1 | rs1695 | GG | AG,AA | AA | AA | AA | AA | AA | AA | |
| MTHFR | rs1801133 | AA | AG,GG | AA | AA | AA | AA | AA | AA | |
| NQO1 | rs1800566 | GG | AG,GG | AG | ||||||
| TP53 | rs1042522 | CC | CG,GG | CC | CC-g | CC-g | CC-g | CC-g | CC-g | |
| TPMT | rs1800462 | CC | CG,GG | CC | CC | CC | CC | CC | CC | |
| TPMT | rs1800584 | CC | CT,TT | CC | CC | CC | CC | CC | CC | |
| TPMT | rs1142345 | TT | TC,CC | TT | TT | TT | TT | TT | TT | |
| TPMT | rs1800460 | CC | CT,TT | CC | CC | CC | CC | CC | CC | |
| XRCC1 | rs25487 | CC | CT,TT | CC | ||||||
| XPC | rs2228001 | TT | GT,GG | TT | ||||||
| Doxorubicin * | NQO1 | rs1800566 | GG | AG,GG | AG | |||||
| Etoposide * | DYNC2H1 | rs716274 | AA | AG,GG | AG | |||||
| Opioids | ABCB1 | rs1045642 | AA,AG | GG | AG | AG | g-AG | g-AG | AG | g-AG |
| Irinotecan | C8orf34 | rs1517114 | GG | CG,CC | GG | |||||
| SEMA3C | rs7779029 | TT | CT,CC | TT | ||||||
| UGT1A1 | rs4148323 | GG | GA,AA | GG | GG | GG | GG | GG | GG | |
| Mercaptopurine | TPMT | rs1800462 | CC | CG,GG | CC | CC | CC | CC | CC | CC |
| TPMT | rs1800584 | CC | CT,TT | CC | CC | CC | CC | CC | CC | |
| TPMT | rs1142345 | TT | TC,CC | TT | TT | TT | TT | TT | TT | |
| TPMT | rs1800460 | CC | CT,TT | CC | CC | CC | CC | CC | CC | |
| NUDT15 | rs116855232 | CC | CT,TT | CC | CC | CC | CC | CC | CC | |
| Methotrexate | ABCB1 | rs1045642 | GG | AG,AA | AG | AG | g-AG | g-AG | AG | g-AG |
| SLCO1B1 | rs11045879 | CC | CT,TT | CT | ||||||
| MTHFR | rs1801133 | GG | AA,AG | AA | AA | AA | AA | AA | ||
| MTRR | rs1801394 | AA | AG,GG | GG | GG | a-GG | GG | GG | a-GG | |
| ATIC | rs4673993 | CC,CT | TT | CT | ||||||
| Ondansetron | ABCB1 | rs1045642 | AA | AG,GG | AG | AG | g-AG | g-AG | AG | g-AG |
| Thioguanine | TPMT | rs1800462 | CC | CG,GG | CC | CC | CC | CC | CC | CC |
| TPMT | rs1800584 | CC | CT,TT | CC | CC | CC | CC | CC | CC | |
| TPMT | rs1142345 | TT | TC,CC | TT | TT | TT | TT | TT | TT | |
| TPMT | rs1800460 | CC | CT,TT | CC | CC | CC | CC | CC | CC | |
| NUDT15 | rs116855232 | CC | CT,TT | CC | CC | CC | CC | CC | CC | |
| Vincristine * | CEP72 | rs924607 | CC,CT | TT | CC | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corallo, D.; Zanon, C.; Pantile, M.; Tonini, G.P.; Zin, A.; Francescato, S.; Rossi, B.; Trevisson, E.; Pinato, C.; Monferrer, E.; et al. Integrated CGH/WES Analyses Advance Understanding of Aggressive Neuroblastoma Evolution: A Case Study. Cells 2021, 10, 2695. https://doi.org/10.3390/cells10102695
Corallo D, Zanon C, Pantile M, Tonini GP, Zin A, Francescato S, Rossi B, Trevisson E, Pinato C, Monferrer E, et al. Integrated CGH/WES Analyses Advance Understanding of Aggressive Neuroblastoma Evolution: A Case Study. Cells. 2021; 10(10):2695. https://doi.org/10.3390/cells10102695
Chicago/Turabian StyleCorallo, Diana, Carlo Zanon, Marcella Pantile, Gian Paolo Tonini, Angelica Zin, Samuela Francescato, Bartolomeo Rossi, Eva Trevisson, Claudia Pinato, Ezequiel Monferrer, and et al. 2021. "Integrated CGH/WES Analyses Advance Understanding of Aggressive Neuroblastoma Evolution: A Case Study" Cells 10, no. 10: 2695. https://doi.org/10.3390/cells10102695
APA StyleCorallo, D., Zanon, C., Pantile, M., Tonini, G. P., Zin, A., Francescato, S., Rossi, B., Trevisson, E., Pinato, C., Monferrer, E., Noguera, R., Aliño, S. F., Herrero, M. J., Biffi, A., Viscardi, E., & Aveic, S. (2021). Integrated CGH/WES Analyses Advance Understanding of Aggressive Neuroblastoma Evolution: A Case Study. Cells, 10(10), 2695. https://doi.org/10.3390/cells10102695