
Enhanced Expression of microRNA-1273g-3p Contributes to Alzheimer’s Disease Pathogenesis by Regulating the Expression of Mitochondrial Genes

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Plasma and CSF Samples
2.2. Human Postmortem Brain Samples
2.3. Cell Culture
2.4. Plasmids, miRNA Mimic and Inhibitor and Transfection
2.5. Microarray
2.6. Quantitative Real-Time PCR (qPCR)
2.7. Biotinylated-miRNA Pull-Down Assay
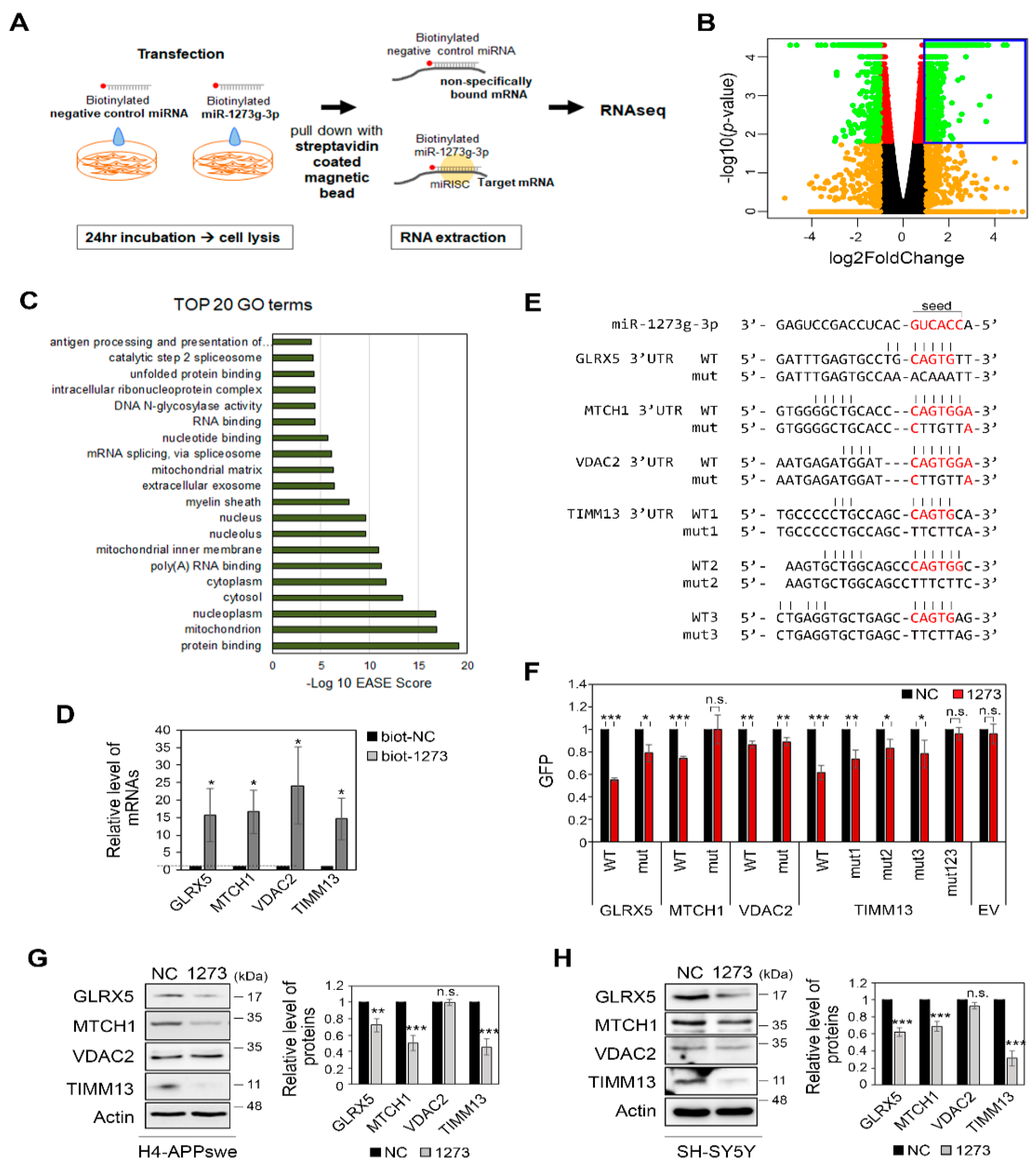
2.8. RNA Sequencing (RNAseq) and Gene Ontology (GO) Analysis
2.9. Bioinformatics Analysis
2.10. Measurements of Oxygen Consumption Rate (OCR)
2.11. Fluorescence Immunohistochemistry
2.12. Neurite Outgrowth Analysis
2.13. Statistical Analysis
3. Results
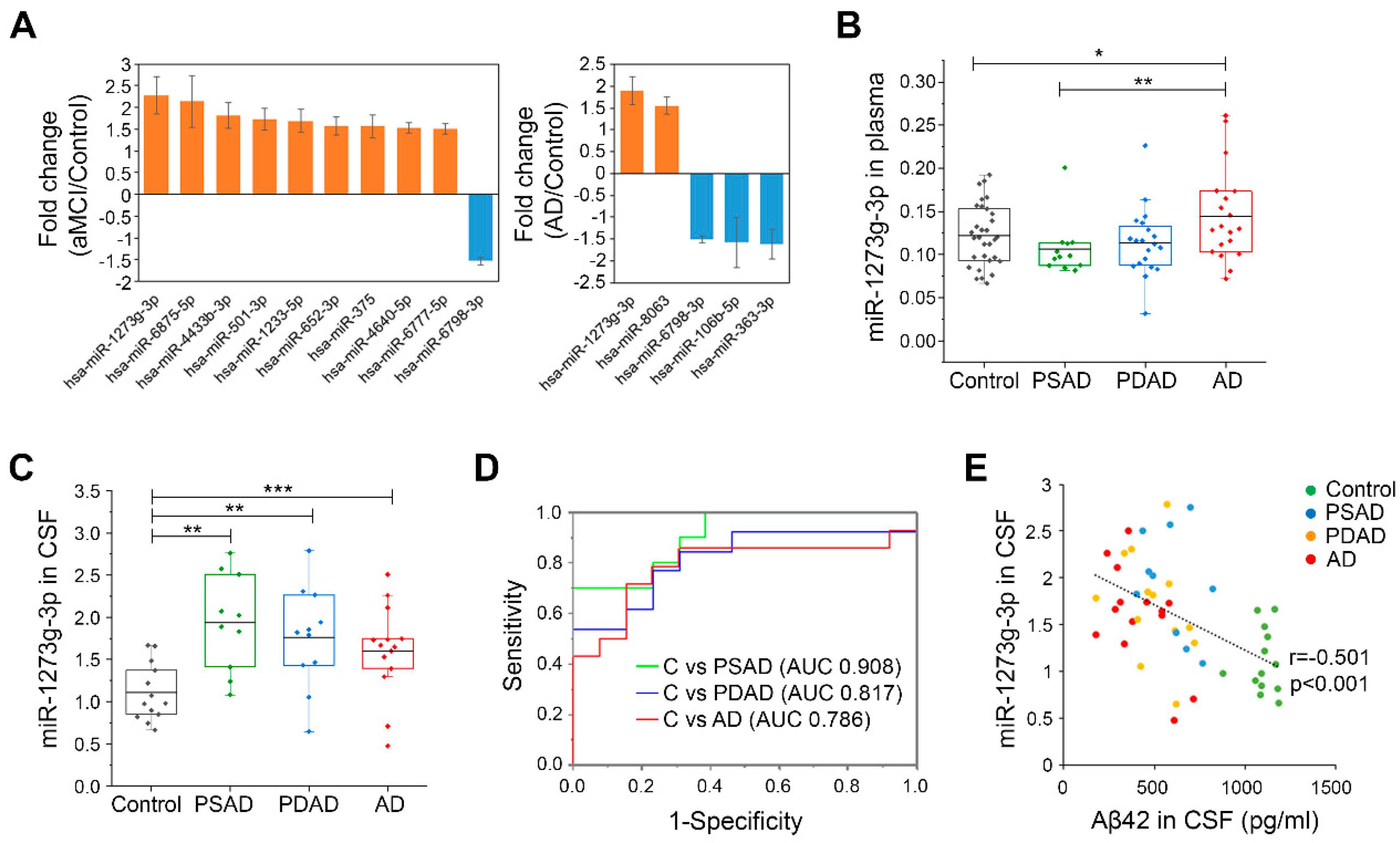
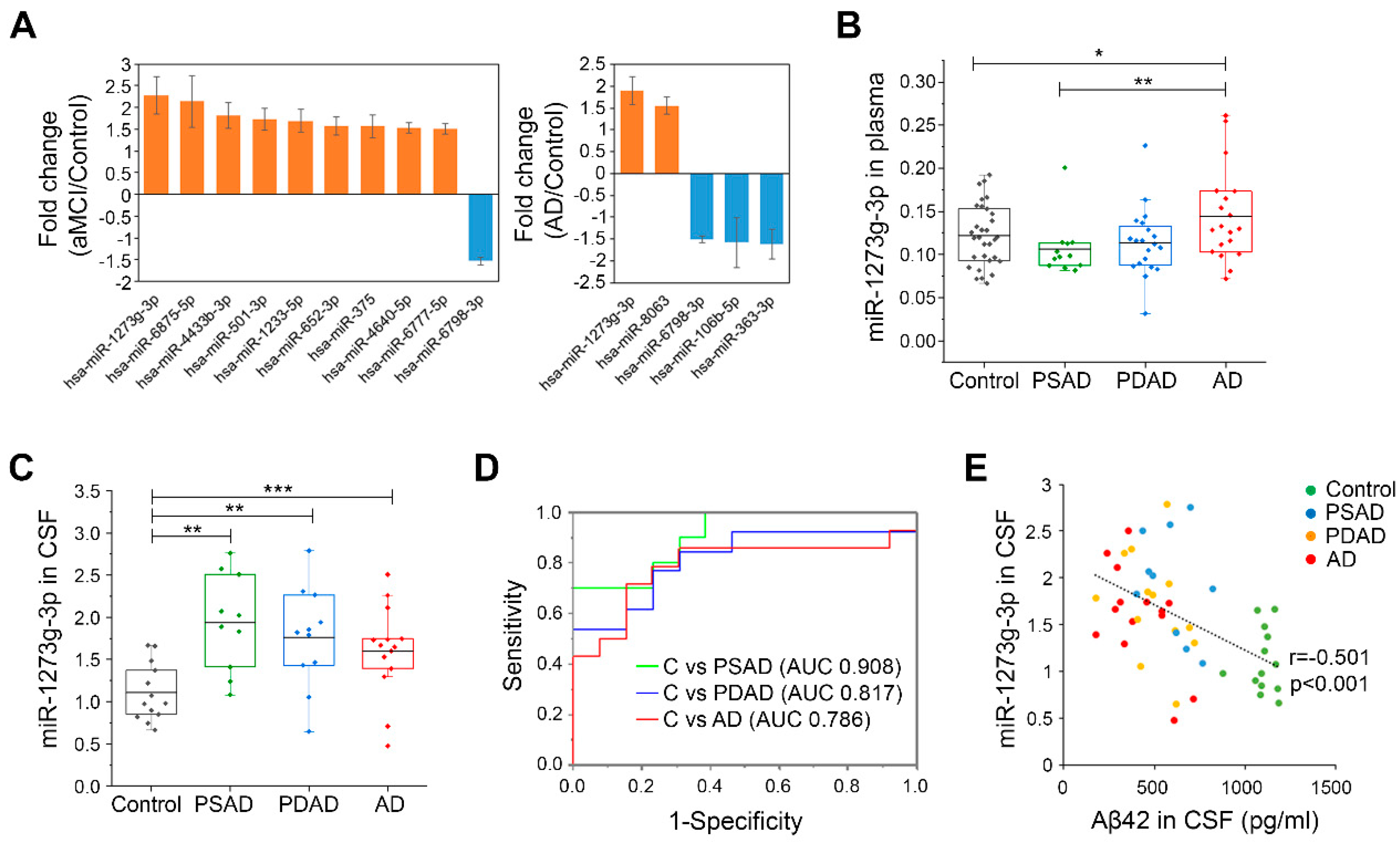
3.1. miR-1273g-3p Is Elevated in the Plasma and CSF of AD Patients
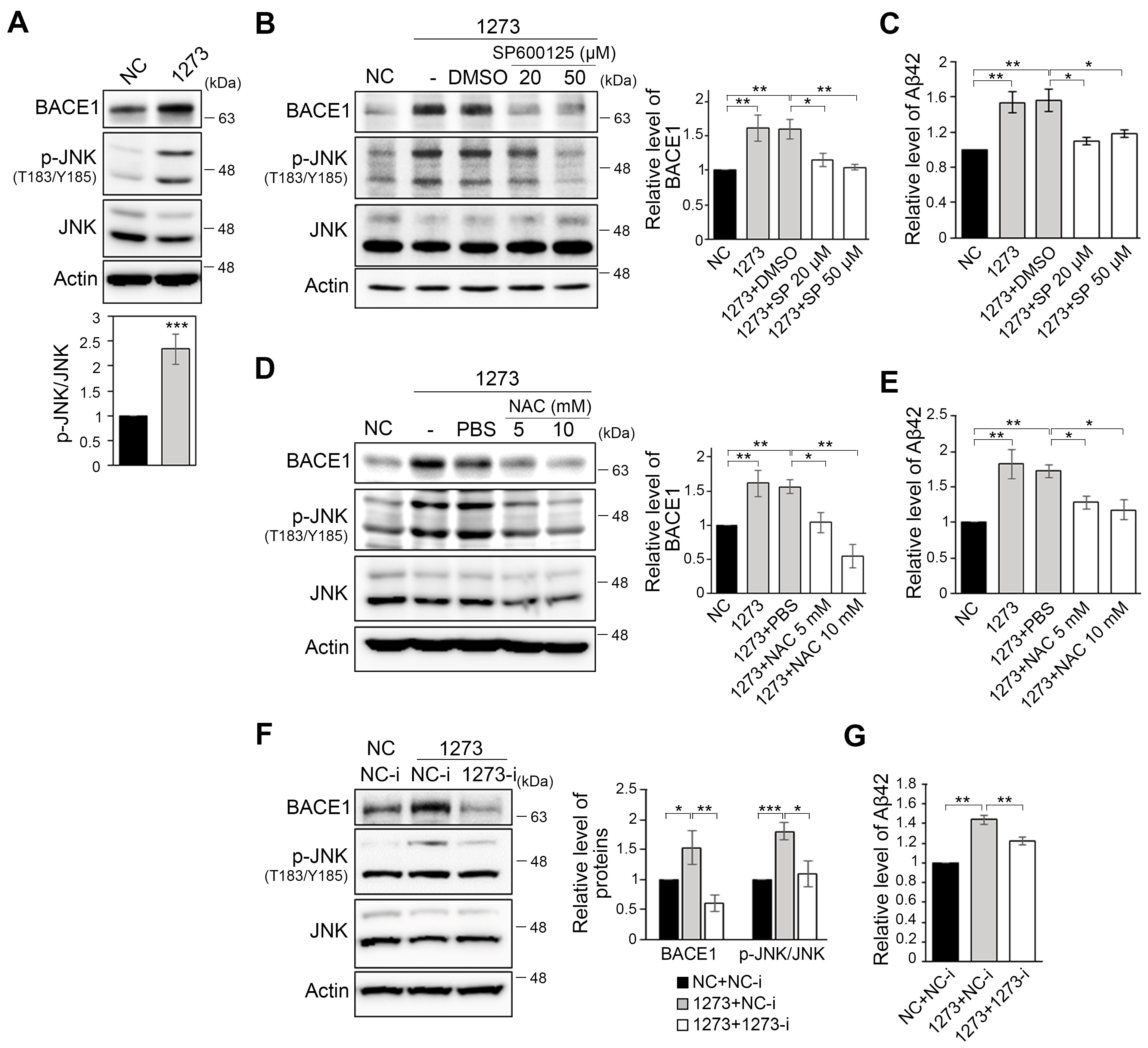
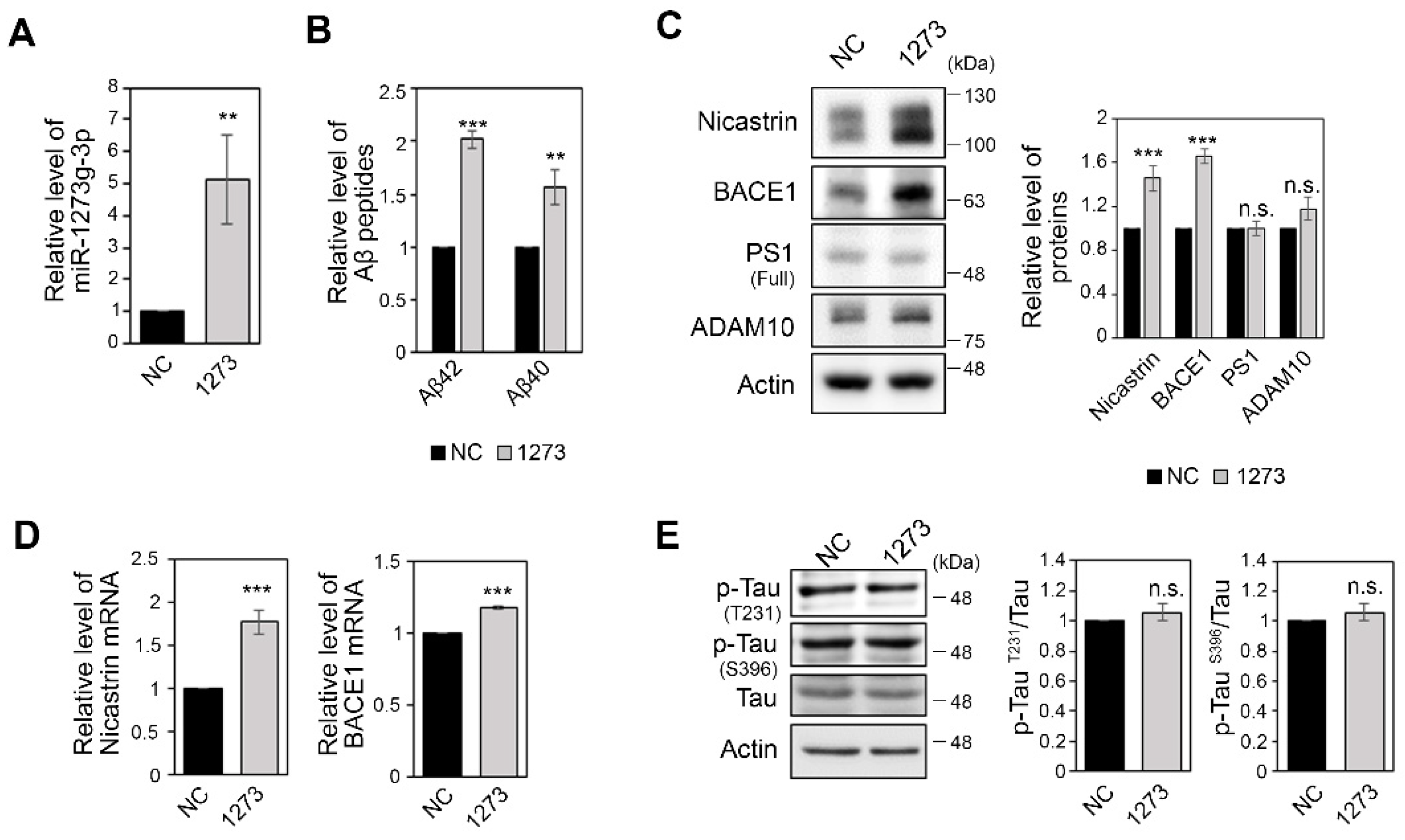
3.2. miR-1273g-3p Facilitates Aβ Production in an AD Model Cell Line
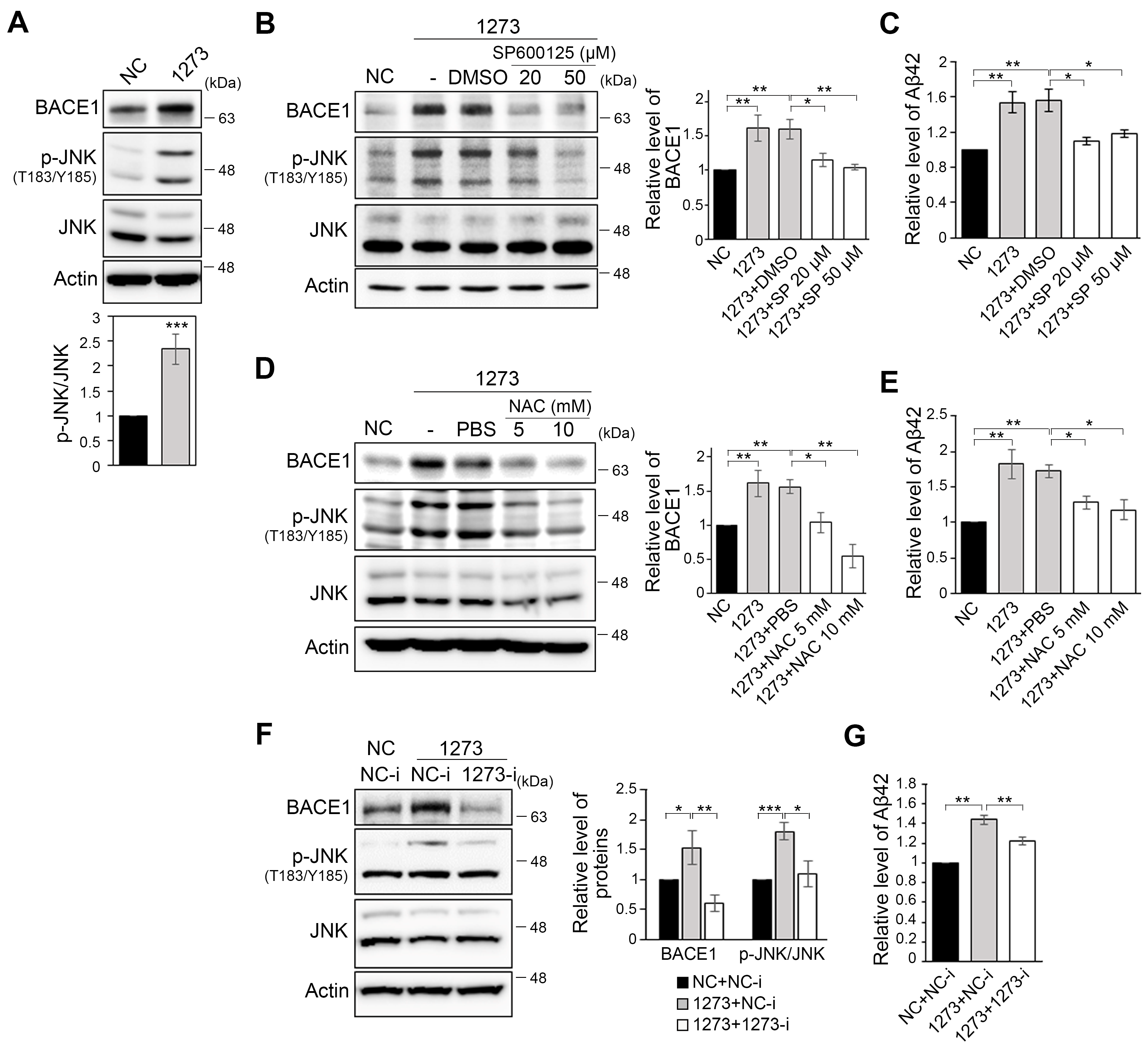
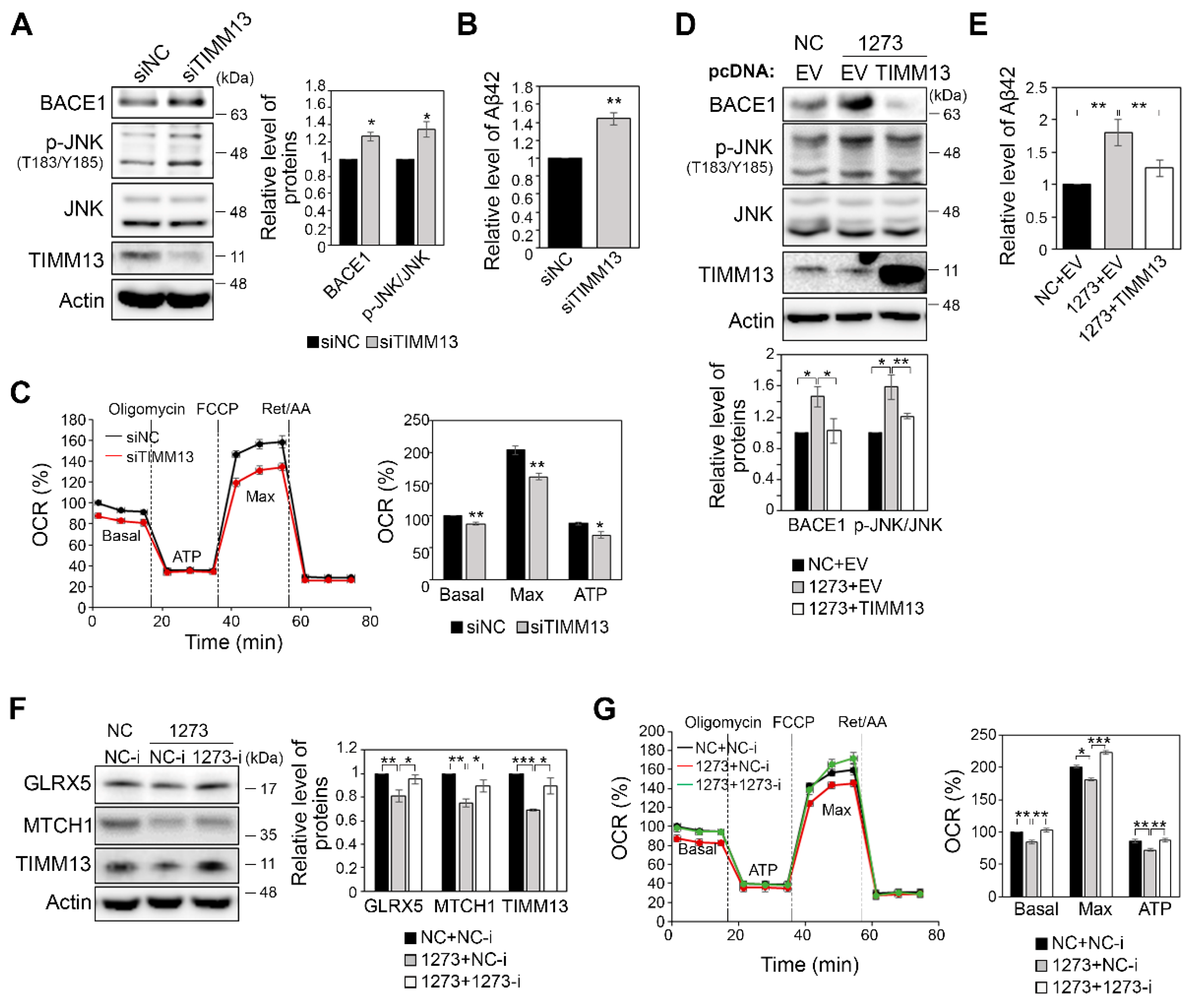
3.3. The miR-1273g-3p-Mediated Upregulation of BACE1 Is Caused by Oxidative Stress
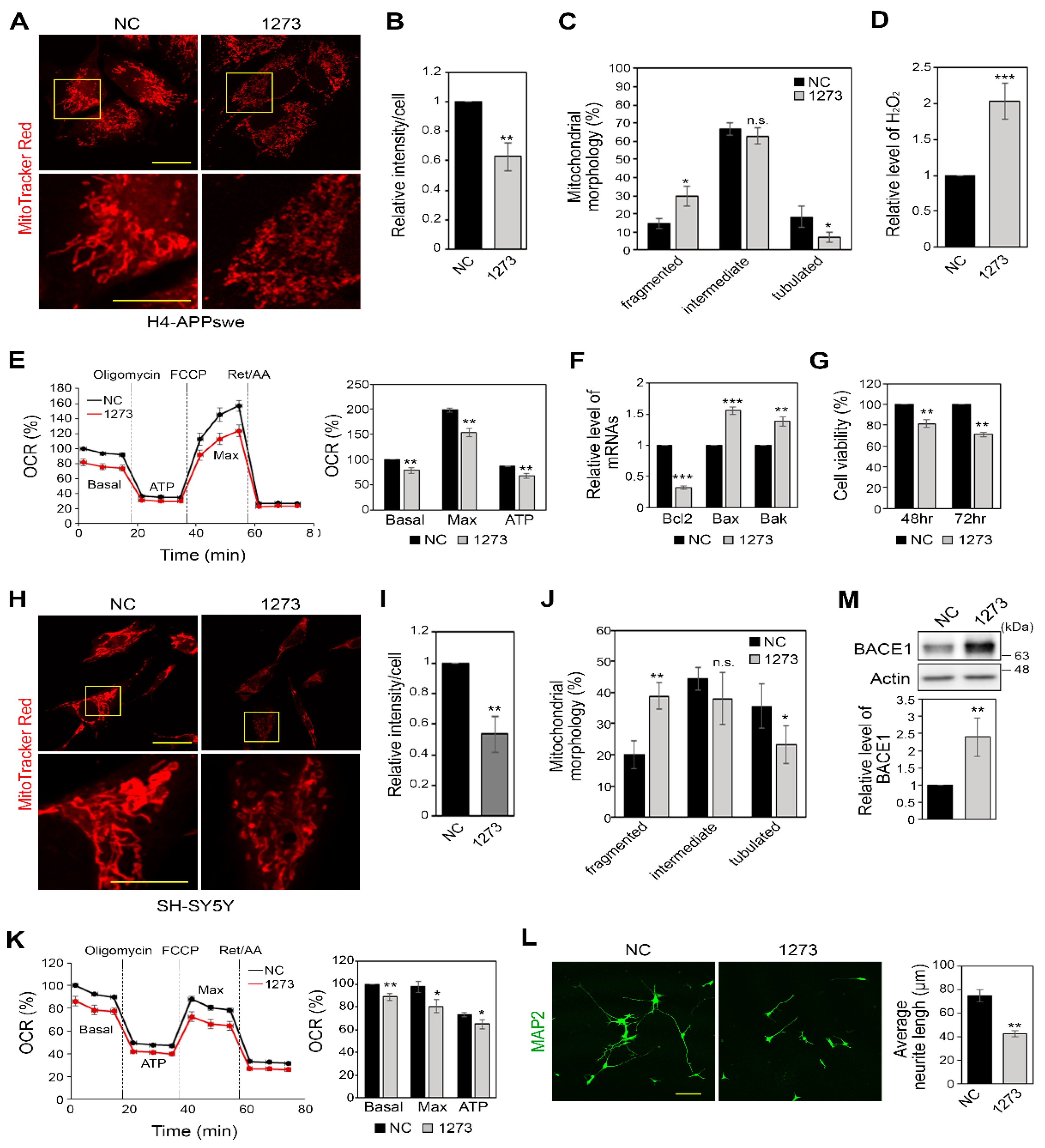
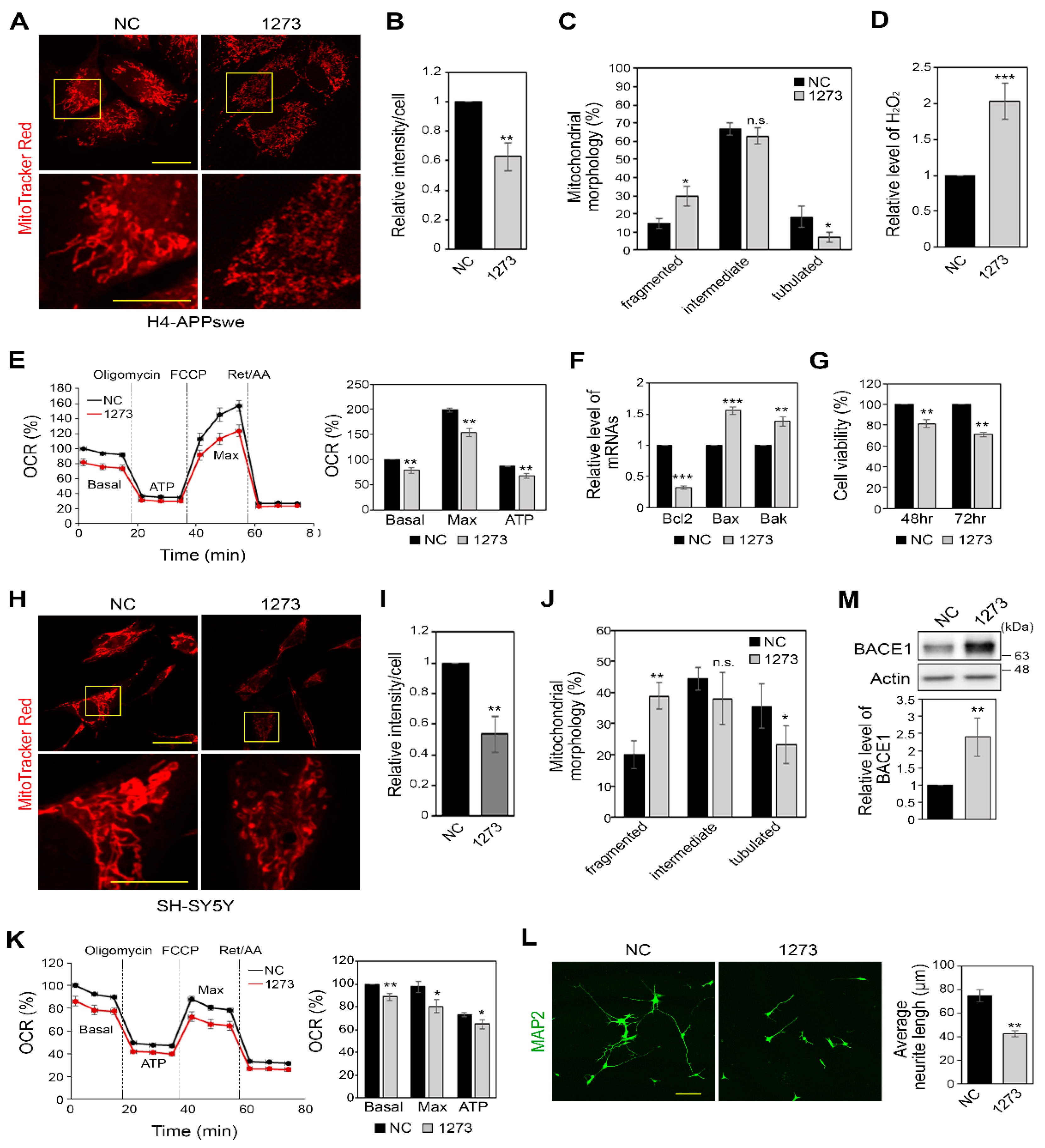
3.4. miR-1273g-3p Impairs Mitochondrial Function in H4-APPswe and SH-SY5Y Cells
3.5. miR-1273g-3p Interacts with and Downregulates the Expression Levels of Mitochondrial Genes in H4-APPswe Cells
3.6. Modulation of the Expression of miR-1273g-3p Target Genes Affects Mitochondrial Function and Aβ42 Production
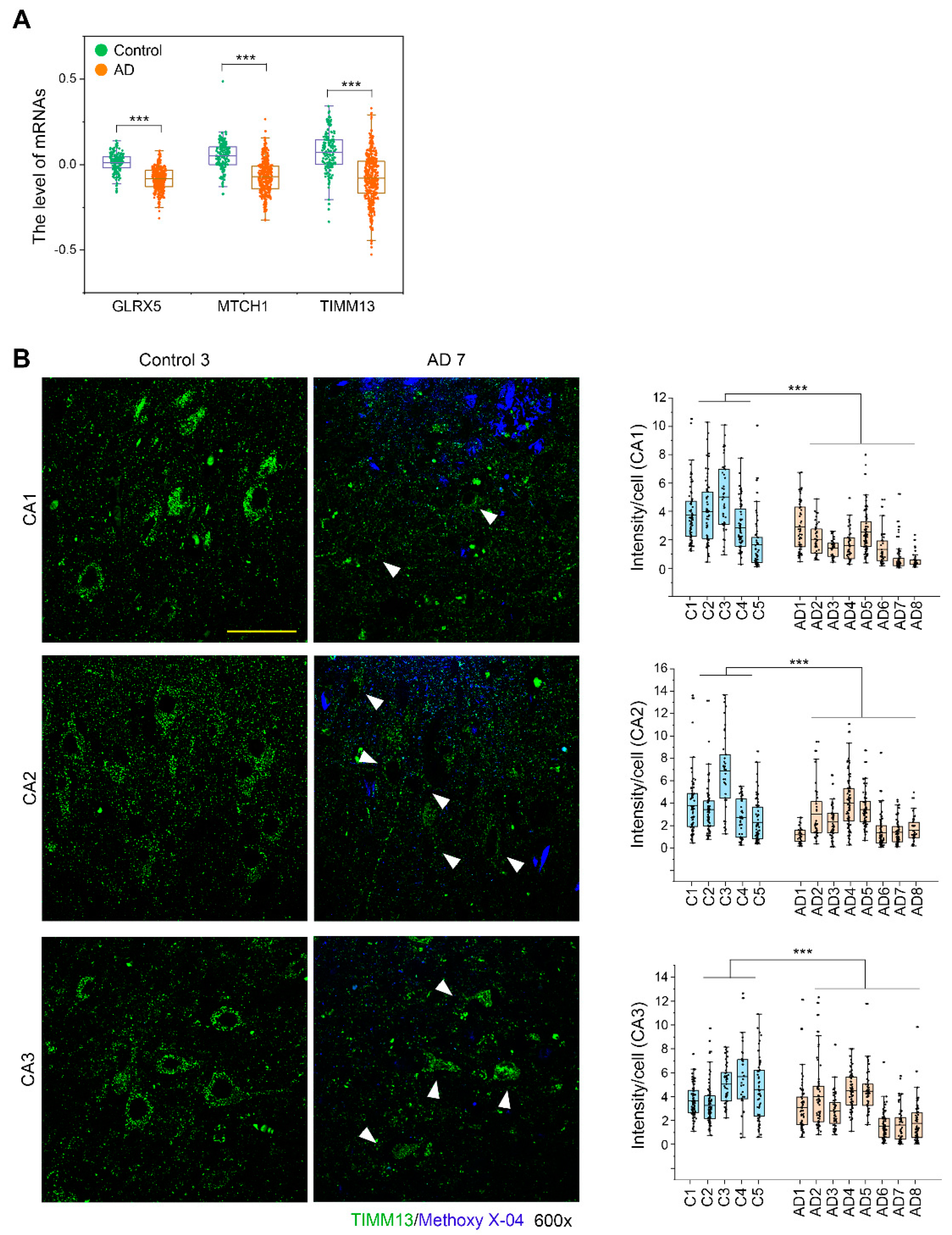
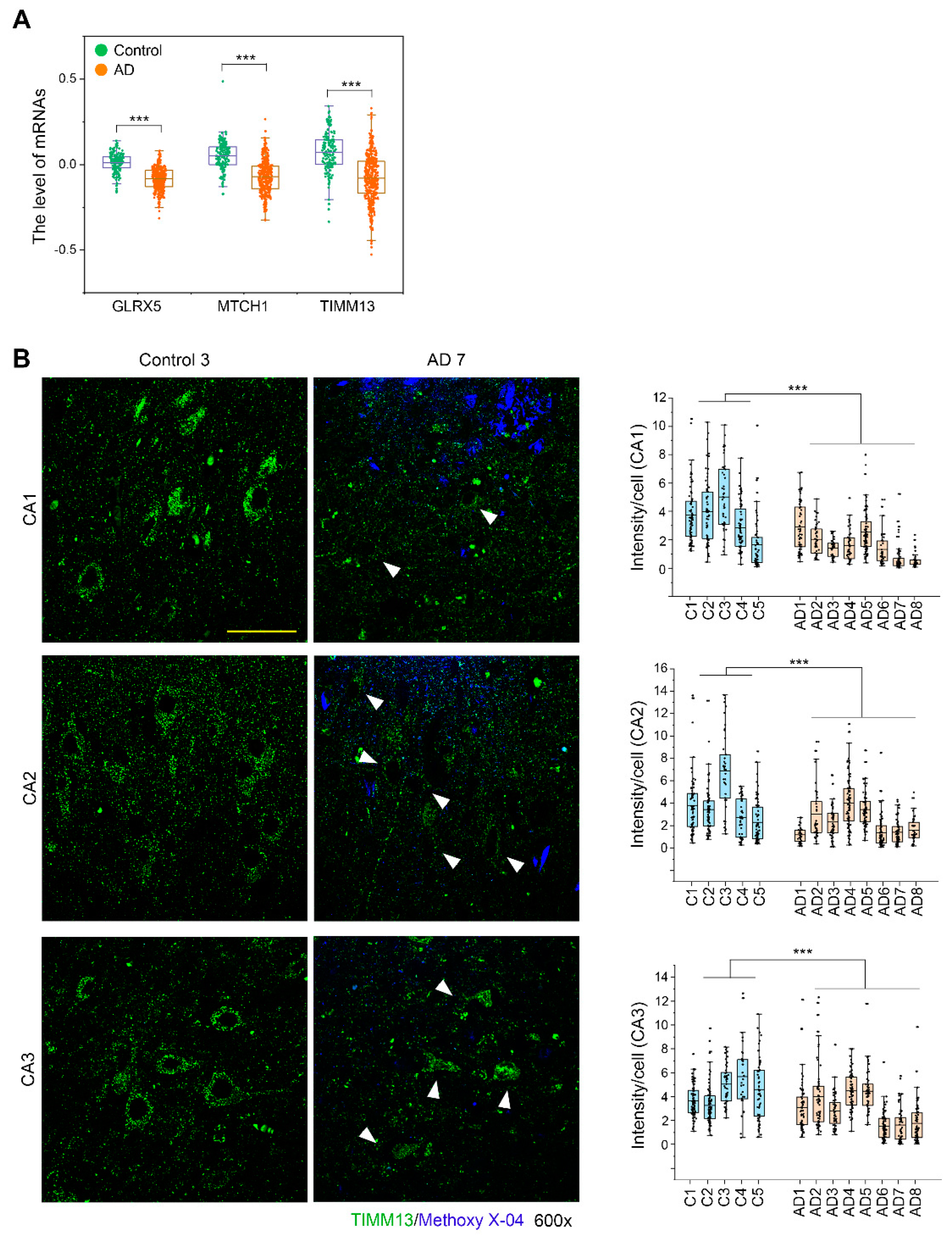
3.7. TIMM13 Is Downregulated in Hippocampi of Human AD Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jagust, W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Frere, S.; Slutsky, I. Alzheimer’s Disease: From Firing Instability to Homeostasis Network Collapse. Neuron 2018, 97, 32–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Benzinger, T.L.; Su, Y.; Christensen, J.; Friedrichsen, K.; Aldea, P.; McConathy, J.; Cairns, N.J.; Fagan, A.M.; Morris, J.C.; et al. Evaluation of Tau Imaging in Staging Alzheimer Disease and Revealing Interactions Between beta-Amyloid and Tauopathy. JAMA Neurol. 2016, 73, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in Neuroplasticity and Neurological Disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Blass, J.P.; SHEU, R.K.; Gibson, G.E. Inherent abnormalities in energy metabolism in Alzheimer disease: Interaction with cerebrovascular compromise. Ann. N. Y. Acad. Sci. 2000, 903, 204–221. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.M.; Cassarino, D.S.; Abramova, N.N.; Keeney, P.M.; Borland, M.K.; A Trimmer, P.; Krebs, C.T.; Bennett, J.C.; Parks, J.K.; Swerdlow, R.H.; et al. Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann. Neurol. 2000, 48, 148–155. [Google Scholar] [CrossRef]
- Trimmer, P.A.; Swerdlow, R.H.; Parks, J.K.; Keeneya, P.; Bennett, J.P., Jr.; Miller, S.W.; Davis, R.E.; Parker, W. Abnormal Mitochondrial Morphology in Sporadic Parkinson’s and Alzheimer’s Disease Cybrid Cell Lines. Exp. Neurol. 2000, 162, 37–50. [Google Scholar] [CrossRef]
- Swerdlow, R.; Parks, J.K.; Cassarino, D.S.; Maguire, D.J.; Maguire, R.S.; Bennett, J.P.; Davis, R.E.; Parker, W.D. Cybrids in Alzheimer’s disease: A cellular model of the disease? Neurology 1997, 49, 918–925. [Google Scholar] [CrossRef]
- Gabuzda, D.; Busciglio, J.; Chen, L.B.; Mabudaira, P.; Yankner, B.A. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 1994, 269, 13623–13628. [Google Scholar] [CrossRef]
- Leuner, K.; Schütt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 2012, 16, 1421–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouton-Liger, F.; Paquet, C.; Dumurgier, J.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2alpha pathway. Biochim. Biophys. Acta 2012, 1822, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, K.; Cai, H.; Luo, X.G.; Struble, R.G.; Clough, R.W.; Yan, X.X. Mitochondrial respiratory inhibition and oxidative stress elevate beta-secretase (BACE1) proteins and activity in vivo in the rat retina. Exp. Brain Res. 2007, 181, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Chami, L.; Checler, F. BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and beta-amyloid production in Alzheimer’s disease. Mol. Neurodegener. 2012, 7, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase ex-pression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, P.; Bossers, K.; Janky, R.S.; Salta, E.; Frigerio, C.S.; Barbash, S.; Rothmanet, R.; Sierksma, A.S.R.; Papadopoulou, A.S.; Achsel, T.; et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 1613–1634. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, J.; Chevillet, J.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.; Bennett, C.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [Green Version]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Lusardi, T.A.; Phillips, J.I.; Wiedrick, J.T.; Harrington, C.A.; Lind, B.; Lapidus, J.A.; Quinn, J.F.; Saugstad, J.A. MicroRNAs in Human Cerebrospinal Fluid as Biomarkers for Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 55, 1223–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anoop, A.; Singh, P.K.; Jacob, R.S.; Maji, S.K. CSF Biomarkers for Alzheimer’s Disease Diagnosis. Int. J. Alzheimer’s Dis. 2010, 2010, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siedlecki-Wullich, D.; Català-Solsona, J.; Fábregas, C.; Hernández, I.; Clarimon, J.; Lleó, A.; Boada, M.; Saura, C.A.; Rodríguez-Álvarez, J.; Miñano-Molina, A.J. Altered microRNAs related to synaptic function as potential plasma biomarkers for Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Burgos, K.; Malenica, I.; Metpally, R.; Courtright, A.; Rakela, B.; Beach, T.; Shill, H.; Adler, C.; Sabbagh, M.; Villa, S.; et al. Profiles of Extracellular miRNA in Cerebrospinal Fluid and Serum from Patients with Alzheimer’s and Parkinson’s Diseases Correlate with Disease Status and Features of Pathology. PLoS ONE 2014, 9, e94839. [Google Scholar] [CrossRef] [PubMed]
- Takousis, P.; Sadlon, A.; Schulz, J.; Wohlers, I.; Dobricic, V.; Middleton, L.; Lill, C.M.; Perneczky, R.; Bertram, L. Differential expression of microRNAs in Alzheimer’s disease brain, blood, and cerebrospinal fluid. Alzheimer’s Dement. 2019, 15, 1468–1477. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack Jr, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Martin, H.C.; Wani, S.; Steptoe, A.L.; Krishnan, K.; Nones, K.; Nourbakhsh, E.; Vlassov, A.; Grimmond, S.M.; Cloonan, N. Imperfect centered miRNA binding sites are common and can mediate repression of target mRNAs. Genome Biol. 2014, 15, R51. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, M.; Huynh, J.L.; Wang, K.; Yang, X.; Yoo, S.; McElwee, J.; Zhang, B.; Zhang, C.; Lamb, J.R.; Xie, T.; et al. Common dysregulation network in the human prefrontal cortex underlies two neurodegenerative diseases. Mol. Syst. Biol. 2014, 10, 743. [Google Scholar] [CrossRef] [PubMed]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. J. Vis. Exp. 2016, e53193. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, M.B.; Edelman, J.B.; Kao, S.C.-H.; Vallely, M.P.; Van Zandwijk, N.; Reid, G. The Impact of Hemolysis on Cell-Free microRNA Biomarkers. Front. Genet. 2013, 4, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.E.; Choi, K.Y.; Kim, B.C.; Choi, S.-M.; Song, M.-K.; Lee, J.J.; Kim, J.; Song, H.-C.; Kim, H.-W.; Ha, J.-M.; et al. Cerebrospinal Fluid Biomarkers for the Diagnosis of Prodromal Alzheimer’s Disease in Amnestic Mild Cognitive Impairment. Dement. Geriatr. Cogn. Disord. Extra 2019, 9, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-secretase BACE1 in Alzheimer’s disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Tamagno, E.; Guglielmotto, M.; Giliberto, L.; Vitali, A.; Borghi, R.; Autelli, R.; Danni, O.; Tabaton, M. JNK and ERK1/2 pathways have a dual opposite effect on the expression of BACE1. Neurobiol. Aging 2009, 30, 1563–1573. [Google Scholar] [CrossRef]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Hausenloy, D.J. Mitochondrial fusion and fission proteins: Novel therapeutic targets for combating cardiovascular disease. Br. J. Pharmacol. 2014, 171, 1890–1906. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- McDade, E.; Bateman, R.J. Stop Alzheimer’s before it starts. Nat. News 2017, 547, 153. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; EL Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jiao, B.; Shen, L. The Epigenetics of Alzheimer’s Disease: Factors and Therapeutic Implications. Front. Genet. 2018, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Denk, J.; Boelmans, K.; Siegismund, C.S.; Lassner, D.; Arlt, S.; Jahn, H. MicroRNA Profiling of CSF Reveals Potential Biomarkers to Detect Alzheimer’s Disease. PLoS ONE 2015, 10, e0126423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.Z.Y.; Thalamuthu, A.; Cheng, L.; Fowler, C.; Masters, C.L.; Sachdev, P.; Mather, K.A.; The Australian Imaging Biomarkers and Lifestyle Flagship Study of Ageing. Differential blood miRNA expression in brain amyloid imaging-defined Alzheimer’s disease and controls. Alzheimer’s Res. Ther. 2020, 12, 1–11. [Google Scholar] [CrossRef]
- Lutz, T.; Neupert, W.; Herrmann, J.M. Import of small Tim proteins into the mitochondrial intermembrane space. EMBO J. 2003, 22, 4400–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesch, K.; Hynds, P.J.; Varga, R.; Tranebjaerg, L.; Koehler, C.M. The calcium-binding aspartate/glutamate carriers, citrin and aralar1, are new substrates for the DDP1/TIMM8a-TIMM13 complex. Hum. Mol. Genet. 2004, 13, 2101–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neighbors, A.; Moss, T.; Holloway, L.; Yu, S.-H.; Annese, F.; Skinner, S.; Saneto, R.; Steet, R. Functional analysis of a novel mutation in the TIMM8A gene that causes deafness-dystonia-optic neuronopathy syndrome. Mol. Genet. Genom. Med. 2020, 8, e1121. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, A.; Potenza, F.; Schmid, A.W.; Ri-etsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Sommerkamp, P.; Cabezas-Wallscheid, N.; Trumpp, A. Alternative Polyadenylation in Stem Cell Self-Renewal and Differentiation. Trends Mol. Med. 2021, 27, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama-Nakamura, S.; Yoshikawa, H.; Homma, K.; Hayano, T.; Tsujimura-Takahashi, T.; Izumikawa, K.; Ishikawa, H.; Miyazawa, N.; Yanagida, M.; Miura, Y.; et al. Parvulin (Par14), a Peptidyl-Prolyl cis-trans Isomerase, Is a Novel rRNA Processing Factor That Evolved in the Metazoan Lineage. Mol. Cell. Proteom. 2009, 8, 1552–1565. [Google Scholar] [CrossRef] [Green Version]
- Cha, M.Y.; Han, S.H.; Son, S.M.; Hong, H.S.; Choi, Y.J.; Byun, J.; Mook-Jung, I. Mitochondria-specific accumulation of amyloid beta induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Vögtle, F.N.; Taskin, A.A.; Teixeira, P.F.; Ring, J.; Burkhart, J.M.; Burger, N.; Pinho, C.M.; Tadic, J.; Loreth, D.; et al. Amyloid-beta peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab. 2014, 20, 662–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supnet, C.; Bezprozvanny, I. Neuronal Calcium Signaling, Mitochondrial Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 20, S487–S498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tondo, G.; Iaccarino, L.; Caminiti, S.P.; Presotto, L.; Santangelo, R.; Iannaccone, S.; Magnani, G.; Perani, D. The combined effects of microglia activation and brain glucose hypometabolism in early-onset Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Jun, S.; Rellick, S.; Quintana, D.D.; Cavendish, J.Z.; Simpkins, J.W. Expression of microRNA-34a in Alzheimer’s disease brain targets genes linked to synaptic plasticity, energy metabolism, and resting state network activity. Brain Res. 2016, 1646, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhou, H.; Jiang, L.; Mao, Y.; Cui, X.; Xie, B.; Cui, D.; Wang, H.; Zhang, Q.; Xu, S. MiR-195 dependent roles of mitofusin2 in the mitochondrial dysfunction of hippocampal neurons in SAMP8 mice. Brain Res. 2016, 1652, 135–143. [Google Scholar] [CrossRef]
- Wang, Q.; Ge, X.; Zhang, J.; Chen, L. Effect of lncRNA WT1-AS regulating WT1 on oxidative stress injury and apoptosis of neurons in Alzheimer’s disease via inhibition of the miR-375/SIX4 axis. Aging 2020, 12, 23974–23995. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, Z.; Gao, J.; Yang, L.; Dang, E.; Fang, H.; Shao, S.; Zhang, S.; Xiao, C.; Yuan, X.; et al. Plasma exosomal miR-375-3p regulates mitochondria-dependent keratinocyte apoptosis by targeting XIAP in severe drug-induced skin reactions. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Dumortier, O.; Fabris, G.; Pisani, D.F.; Casamento, V.; Gautier, N.; Hinault, C.; Lebrun, P.; Duranton, C.; Tauc, M.; Dalle, S.; et al. microRNA-375 regulates glucose metabolism-related signaling for insulin secretion. J. Endocrinol. 2020, 244, 189–200. [Google Scholar] [CrossRef]
- Guo, J.; Sang, Y.; Yin, T.; Wang, B.; Yang, W.; Li, X.; Li, H.; Kang, Y. miR-1273g-3p participates in acute glucose fluctuation-induced autophagy, dysfunction, and proliferation attenuation in human umbilical vein endothelial cells. Am. J. Physiol. Metab. 2016, 310, E734–E743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Qian, X.; Zhu, M.; Li, A.; Fang, M.; Zhu, Y.; Zhang, J. miR-1273g-3p promotes proliferation, migration and invasion of LoVo cells via cannabinoid receptor 1 through activation of ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Mol. Med. Rep. 2018, 17, 4619–4626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, I.; Hebalkar, R.; Kar, S.; S, S.T.; Gutti, U.; Gutti, R.K. Systems biology approach to study the role of miRNA in promoter targeting during megakaryopoiesis. Exp. Cell Res. 2018, 366, 192–198. [Google Scholar] [CrossRef]
- Van der Vos, K.E.; Abels, E.R.; Zhang, X.; Lai, C.; Carrizosa, E.; Oakley, D.; Prabhakar, S.; Mardini, O.; Crommentuijn, M.H.W.; Skog, J.; et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro-Oncology 2015, 18, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | Diagnosis | n (M/F) | Age, y | p-Value | K-MMSE | p-Value | Amyloid PET |
|---|---|---|---|---|---|---|---|
| Microarray for miRNAs in plasma (Cohort 1) | Control | 36 (16/20) | 72.86 ± 4.72 | - | 27.31 ± 2.69 | - | - |
| aMCI | 24 (12/12) | 74.22 ± 4.51 | 0.115 | 25.94 ± 2.54 | 0.018 | - | |
| AD | 36 (20/16) | 73.58 ± 5.72 | 0.344 | 17.34 ± 8.17 | 2.35 × 10−9 | - | |
| qPCR for miRNAs in plasma (Cohort 2) | Control | 31 (18/13) | 72.85 ± 5.31 | - | 27.10 ± 1.83 | negative | |
| PSAD | 12 (6/6) | 73.99 ± 3.32 | 0.247 | 26.67 ± 3.55 | 0.28 | positive | |
| PDAD | 20 (10/10) | 74.38 ± 4.80 | 0.150 | 25.35 ± 3.69 | 1.45 × 10−2 | positive | |
| AD | 20 (8/12) | 72.54 ± 3.61 | 0.411 | 17.50 ± 4.83 | 8.67 × 10−14 | positive | |
| qPCR for miRNAs in CSF (Cohort 3) | Control | 13 (7/6) | 74.46 ± 4.66 | 26.62 ± 1.56 | negative | ||
| PSAD | 10 (5/5) | 73.75 ± 4.24 | 0.373 | 26.90 ± 3.28 | 0.393 | positive | |
| PDAD | 13 (7/6) | 74.08 ± 5.87 | 0.428 | 25.46 ± 4.03 | 0.173 | positive | |
| AD | 14 (7/7) | 70.39 ± 4.82 | 0.017 | 17.93 ± 4.91 | 1.13 × 10−6 | positive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.H.; Choi, K.Y.; Park, Y.; McLean, C.; Park, J.; Lee, J.H.; Lee, K.-H.; Kim, B.C.; Huh, Y.H.; Lee, K.H.; et al. Enhanced Expression of microRNA-1273g-3p Contributes to Alzheimer’s Disease Pathogenesis by Regulating the Expression of Mitochondrial Genes. Cells 2021, 10, 2697. https://doi.org/10.3390/cells10102697
Kim SH, Choi KY, Park Y, McLean C, Park J, Lee JH, Lee K-H, Kim BC, Huh YH, Lee KH, et al. Enhanced Expression of microRNA-1273g-3p Contributes to Alzheimer’s Disease Pathogenesis by Regulating the Expression of Mitochondrial Genes. Cells. 2021; 10(10):2697. https://doi.org/10.3390/cells10102697
Chicago/Turabian StyleKim, So Hee, Kyu Yeong Choi, Yega Park, Catriona McLean, Jiyu Park, Jung Hoon Lee, Kyung-Hwa Lee, Byeong C. Kim, Yun Hyun Huh, Kun Ho Lee, and et al. 2021. "Enhanced Expression of microRNA-1273g-3p Contributes to Alzheimer’s Disease Pathogenesis by Regulating the Expression of Mitochondrial Genes" Cells 10, no. 10: 2697. https://doi.org/10.3390/cells10102697