
Harnessing the Power of Induced Pluripotent Stem Cells and Gene Editing Technology: Therapeutic Implications in Hematological Malignancies


Abstract
1. Introduction
2. Disease Modeling Using Patient-Derived iPSCs
3. Disease Modeling Using Genetically Modified iPSCs
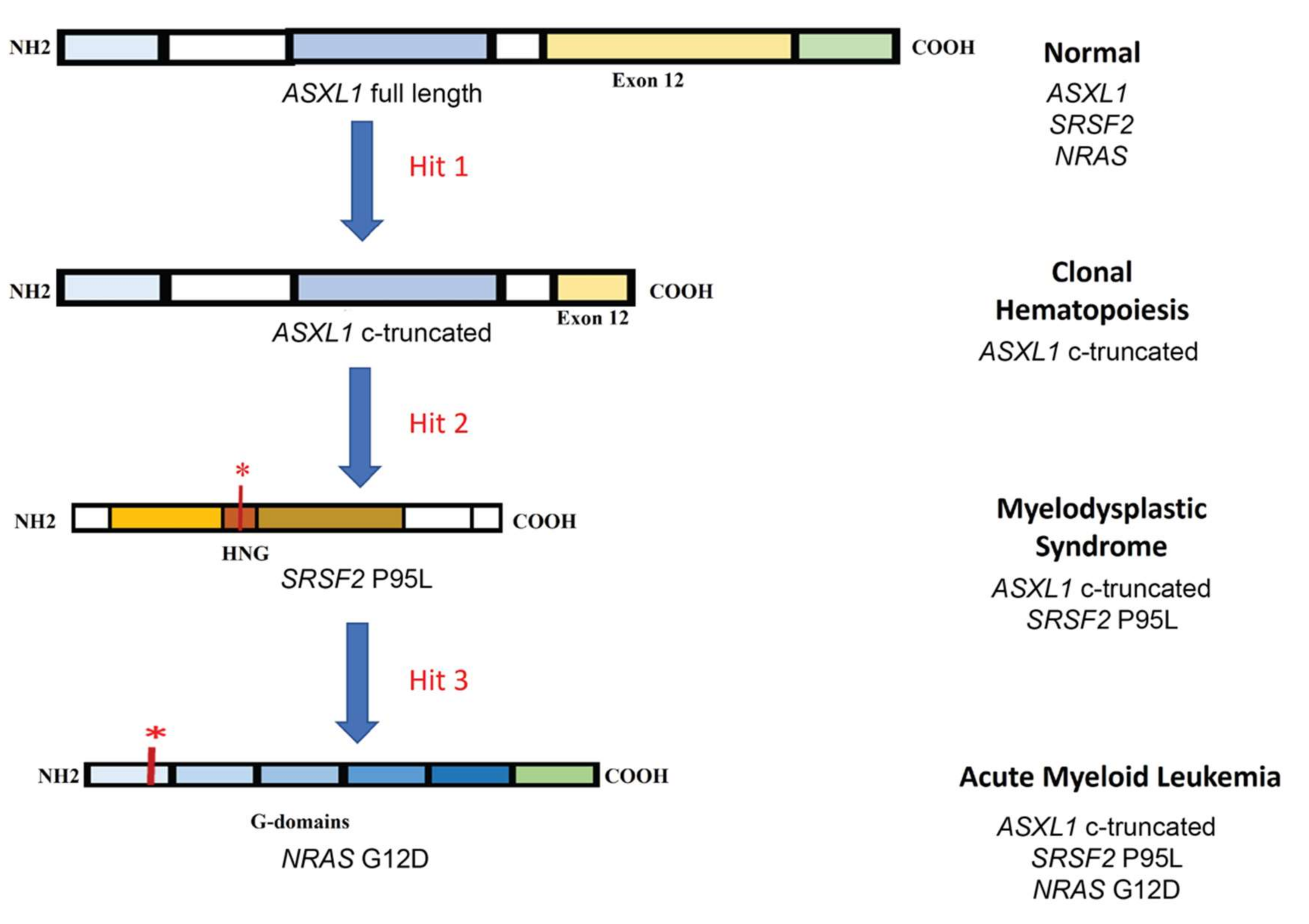
3.1. Clonal Evolution of AML: An Example of De Novo Leukemogenesis in Human iPSCs
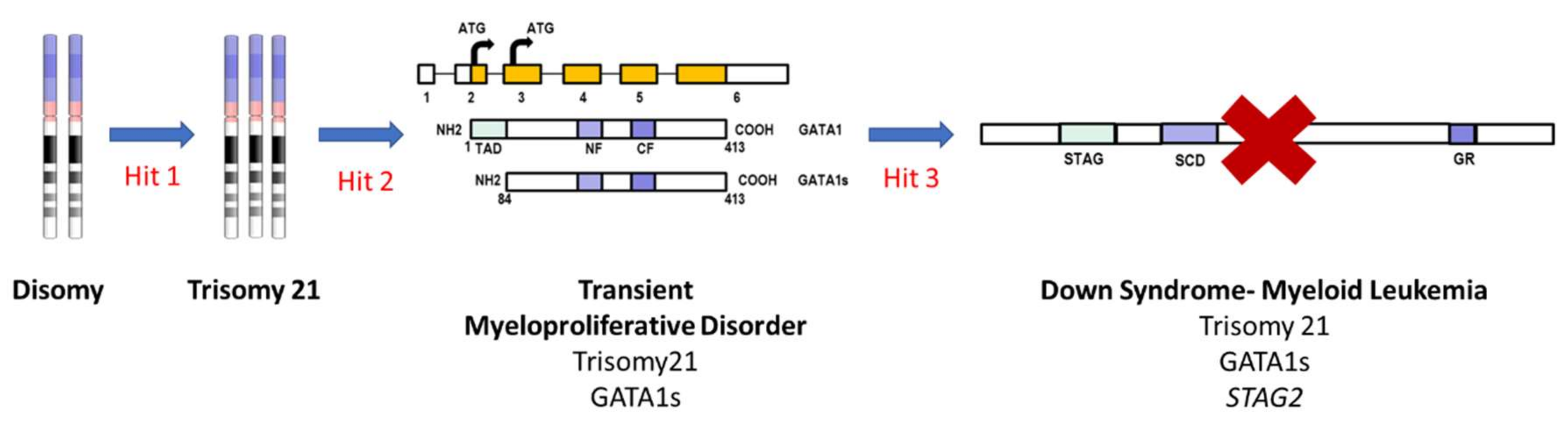
3.2. Down Syndrome-Myeloid Leukemia: An Example of iPSC-Based Sequential Disease Modeling
4. Identification of Therapeutic Targets using iPSCs—Clinical and Translational Implications
5. Hematopoietic Differentiation Approaches—2-Dimensional (2D) vs. 3-Dimensional (3D)
6. Perspectives and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALL | Acute Lymphocytic Leukemia |
| AML | Acute Myeloid Leukemia |
| B-ALL | B-cell Acute Lymphocytic Leukemia |
| cALL | Children Acute Lymphocytic Leukemia |
| CAR | Chimeric Antigen Receptor |
| CH | Clonal Hematopoiesis |
| CLL | Chronic Lymphocytic Leukemia |
| CML | Chronic Myelogenous Leukemia |
| CMML | Chronic Myelomonocytic leukemia |
| CN | Congenital Neutropenia |
| CTCL | Cutaneous T-cell Lymphoma |
| DLBCL | Diffuse Large B Cell Lymphoma |
| DS-ML | Down Syndrome-Myeloid Leukemia |
| ET | Essential Thrombocythemia |
| FPD/AML | Familial Platelet Disorder/Acute Myeloid Leukemia |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| HGBCL | High-grade B-cell Lymphoma |
| hnCD16 | High-affinity, Non-cleavable CD16 Fc receptor |
| HSC | Hematopoietic Stem Cells |
| HSPC | Hematopoietic Stem and Progenitor Cells |
| IL15RF | Interleukin 15 Receptor Fusion |
| iPSC | Induced Pluripotent Stem Cells |
| JMML | Juvenile Myelomonocytic leukemia |
| LSC | Leukemic Stem Cells |
| mAB | Monoclonal Antibody |
| MDS | Myelodysplastic Syndrome |
| MGUS | Monoclonal Gammopathy of Undetermined Significance |
| MM | Multiple Myeloma |
| MPN | Myeloproliferative Neoplasm |
| NHL | Non-Hodgkin Lymphoma |
| NK | Natural Killer Cells |
| NS/JMML | Noonan Syndrome/Juvenile Myelomonocytic Leukemia |
| NSG | NOD SCID IL2Rgnull |
| PTCL | Peripheral T-cell Lymphoma |
| PV | Polycythemia Vera |
| TKI | Tyrosine Kinase Inhibitors |
| TMD | Transient Myeloproliferative Disorder |
References
- Przybylski, G.K. Mouse models to study genes involved in hematological malignancies. Blood Sci. 2020, 2, 50–53. [Google Scholar] [CrossRef]
- Rossi, M.; Botta, C.; Arbitrio, M.; Grembiale, R.D.; Tagliaferri, P.; Tassone, P. Mouse models of multiple myeloma: Technologic platforms and perspectives. Oncotarget 2018, 9, 20119–20133. [Google Scholar] [CrossRef]
- Almosailleakh, M.; Schwaller, J. Murine Models of Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 453. [Google Scholar] [CrossRef]
- Law, L.W.; Taormina, V.; Boyle, P.J. Response of acute lymphocytic leukemias to the purine antagonist 6-mercaptopurine. Ann N. Y. Acad. Sci. 1954, 60, 244–250. [Google Scholar] [CrossRef]
- Friend, C. Cell-free transmission in adult Swiss mice of a disease having the character of a leukemia. J. Exp. Med. 1957, 105, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnapillai, A.; Kolb, E.A.; Dhanan, P.; Bojja, A.S.; Mason, R.W.; Corao, D.; Barwe, S.P. Generation of Pediatric Leukemia Xenograft Models in NSG-B2m Mice: Comparison with NOD/SCID Mice. Front. Oncol. 2016, 6, 162. [Google Scholar] [CrossRef] [PubMed]
- Omidvar, N.; Kogan, S.; Beurlet, S.; le Pogam, C.; Janin, A.; West, R.; Noguera, M.E.; Reboul, M.; Soulie, A.; Leboeuf, C.; et al. BCL-2 and mutant NRAS interact physically and functionally in a mouse model of progressive myelodysplasia. Cancer Res. 2007, 67, 11657–11667. [Google Scholar] [CrossRef] [PubMed]
- Uckelmann, H.J.; Kim, S.M.; Wong, E.M.; Hatton, C.; Giovinazzo, H.; Gadrey, J.Y.; Krivtsov, A.V.; Rücker, F.G.; Döhner, K.; McGeehan, G.M.; et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 2020, 367, 586–590. [Google Scholar] [CrossRef]
- Sontakke, P.; Carretta, M.; Jaques, J.; Brouwers-Vos, A.Z.; Lubbers-Aalders, L.; Yuan, H.; de Bruijn, J.D.; Martens, A.C.; Vellenga, E.; Groen, R.W.; et al. Modeling BCR-ABL and MLL-AF9 leukemia in a human bone marrow-like scaffold-based xenograft model. Leukemia 2016, 30, 2064–2073. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnapillai, A.; Kolb, E.A.; McCahan, S.M.; Barwe, S.P. Epigenetic drug combination induces remission in mouse xenograft models of pediatric acute myeloid leukemia. Leuk. Res. 2017, 58, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Carofino, B.L.; Ayanga, B.; Justice, M.J. A mouse model for inducible overexpression of Prdm14 results in rapid-onset and highly penetrant T-cell acute lymphoblastic leukemia (T-ALL). Dis. Model. Mech. 2013, 6, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Agnusdei, V.; Minuzzo, S.; Frasson, C.; Grassi, A.; Axelrod, F.; Satyal, S.; Gurney, A.; Hoey, T.; Seganfreddo, E.; Basso, G.; et al. Therapeutic antibody targeting of Notch1 in T-acute lymphoblastic leukemia xenografts. Leukemia 2014, 28, 278–288. [Google Scholar] [CrossRef]
- Quagliano, A.; Gopalakrishnapillai, A.; Kolb, E.A.; Barwe, S.P. CD81 knockout promotes chemosensitivity and disrupts in vivo homing and engraftment in acute lymphoblastic leukemia. Blood Adv. 2020, 4, 4393–4405. [Google Scholar] [CrossRef]
- Giotopoulos, G.; van der Weyden, L.; Osaki, H.; Rust, A.G.; Gallipoli, P.; Meduri, E.; Horton, S.J.; Chan, W.I.; Foster, D.; Prinjha, R.K.; et al. A novel mouse model identifies cooperating mutations and therapeutic targets critical for chronic myeloid leukemia progression. J. Exp. Med. 2015, 212, 1551–1569. [Google Scholar] [CrossRef]
- Peng, C.; Li, S. Chronic Myeloid Leukemia (CML) Mouse Model in Translational Research. Methods Mol. Biol. 2016, 1438, 225–243. [Google Scholar] [CrossRef]
- Herman, S.E.; Sun, X.; McAuley, E.M.; Hsieh, M.M.; Pittaluga, S.; Raffeld, M.; Liu, D.; Keyvanfar, K.; Chapman, C.M.; Chen, J.; et al. Modeling tumor-host interactions of chronic lymphocytic leukemia in xenografted mice to study tumor biology and evaluate targeted therapy. Leukemia 2013, 27, 2311–2321. [Google Scholar] [CrossRef] [PubMed]
- McClanahan, F.; Hanna, B.; Miller, S.; Clear, A.J.; Lichter, P.; Gribben, J.G.; Seiffert, M. PD-L1 checkpoint blockade prevents immune dysfunction and leukemia development in a mouse model of chronic lymphocytic leukemia. Blood 2015, 126, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Matthews, G.M.; Garbitt, V.M.; Palmer, S.E.; Shortt, J.; Lefebure, M.; Stewart, A.K.; Johnstone, R.W.; Bergsagel, P.L. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood 2012, 120, 376–385. [Google Scholar] [CrossRef]
- Hamouda, M.A.; Jacquel, A.; Robert, G.; Puissant, A.; Richez, V.; Cassel, R.; Fenouille, N.; Roulland, S.; Gilleron, J.; Griessinger, E.; et al. BCL-B (BCL2L10) is overexpressed in patients suffering from multiple myeloma (MM) and drives an MM-like disease in transgenic mice. J. Exp. Med. 2016, 213, 1705–1722. [Google Scholar] [CrossRef]
- Lykken, J.M.; Horikawa, M.; Minard-Colin, V.; Kamata, M.; Miyagaki, T.; Poe, J.C.; Tedder, T.F. Galectin-1 drives lymphoma CD20 immunotherapy resistance: Validation of a preclinical system to identify resistance mechanisms. Blood 2016, 127, 1886–1895. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; Tumang, J.R.; Sinha, A.; Currier, N.; Cardiff, R.D.; Rothstein, T.L.; Faller, D.V.; Denis, G.V. E mu-BRD2 transgenic mice develop B-cell lymphoma and leukemia. Blood 2004, 103, 1475–1484. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Egle, A.; Harris, A.W.; Bath, M.L.; O’Reilly, L.; Cory, S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood 2004, 103, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, J.C. Peripheral T cell lymphoma: New model + new insight. J. Exp. Med. 2010, 207, 911–913. [Google Scholar] [CrossRef]
- Mishra, A.; La Perle, K.; Kwiatkowski, S.; Sullivan, L.A.; Sams, G.H.; Johns, J.; Curphey, D.P.; Wen, J.; McConnell, K.; Qi, J.; et al. Mechanism, Consequences, and Therapeutic Targeting of Abnormal IL15 Signaling in Cutaneous T-cell Lymphoma. Cancer Discov. 2016, 6, 986–1005. [Google Scholar] [CrossRef]
- Alford, K.A.; Slender, A.; Vanes, L.; Li, Z.; Fisher, E.M.; Nizetic, D.; Orkin, S.H.; Roberts, I.; Tybulewicz, V.L. Perturbed hematopoiesis in the Tc1 mouse model of Down syndrome. Blood 2010, 115, 2928–2937. [Google Scholar] [CrossRef]
- Carmichael, C.L.; Majewski, I.J.; Alexander, W.S.; Metcalf, D.; Hilton, D.J.; Hewitt, C.A.; Scott, H.S. Hematopoietic defects in the Ts1Cje mouse model of Down syndrome. Blood 2009, 113, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Attarwala, H. TGN1412: From Discovery to Disaster. J. Young Pharm. 2010, 2, 332–336. [Google Scholar] [CrossRef]
- Cohen, Y.C.; Joffe, E.; Benyamini, N.; Dimopoulos, M.A.; Terpos, E.; Trestman, S.; Held-Kuznetsov, V.; Avivi, I.; Kastritis, E. Primary failure of bortezomib in newly diagnosed multiple myeloma--understanding the magnitude, predictors, and significance. Leuk Lymphoma 2016, 57, 1382–1388. [Google Scholar] [CrossRef]
- Wästerlid, T.; Harrysson, S.; Andersson, T.M.; Ekberg, S.; Enblad, G.; Andersson, P.O.; Jerkeman, M.; Eloranta, S.; Smedby, K.E. Outcome and determinants of failure to complete primary R-CHOP treatment for reasons other than non-response among patients with diffuse large B-cell lymphoma. Am. J. Hematol. 2020, 95, 740–748. [Google Scholar] [CrossRef]
- Zizioli, D.; Mione, M.; Varinelli, M.; Malagola, M.; Bernardi, S.; Alghisi, E.; Borsani, G.; Finazzi, D.; Monti, E.; Presta, M.; et al. Zebrafish disease models in hematology: Highlights on biological and translational impact. Biochim. Biophys. Acta. Mol. Basis. Dis. 2019, 1865, 620–633. [Google Scholar] [CrossRef]
- Baeten, J.T.; de Jong, J.L.O. Genetic Models of Leukemia in Zebrafish. Front Cell Dev. Biol. 2018, 6, 115. [Google Scholar] [CrossRef]
- Feng, H.; Langenau, D.M.; Madge, J.A.; Quinkertz, A.; Gutierrez, A.; Neuberg, D.S.; Kanki, J.P.; Look, A.T. Heat-shock induction of T-cell lymphoma/leukaemia in conditional Cre/lox-regulated transgenic zebrafish. Br. J. Haematol. 2007, 138, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Grebliunaite, R.; Feng, H.; Kozakewich, E.; Zhu, S.; Guo, F.; Payne, E.; Mansour, M.; Dahlberg, S.E.; Neuberg, D.S.; et al. Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J. Exp. Med. 2011, 208, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Borga, C.; Park, G.; Foster, C.; Burroughs-Garcia, J.; Marchesin, M.; Shah, R.; Hasan, A.; Ahmed, S.T.; Bresolin, S.; Batchelor, L.; et al. Simultaneous B and T cell acute lymphoblastic leukemias in zebrafish driven by transgenic MYC: Implications for oncogenesis and lymphopoiesis. Leukemia 2019, 33, 333–347. [Google Scholar] [CrossRef]
- Yeh, J.R.; Munson, K.M.; Chao, Y.L.; Peterson, Q.P.; Macrae, C.A.; Peterson, R.T. AML1-ETO reprograms hematopoietic cell fate by downregulating scl expression. Development 2008, 135, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.J.; Chen, F.Y.; Zhang, Y.; Cao, L.F.; Kuang, Y.; Zhong, M.; Wang, T.; Zhong, H. MYCN transgenic zebrafish model with the characterization of acute myeloid leukemia and altered hematopoiesis. PLoS ONE 2013, 8, e59070. [Google Scholar] [CrossRef] [PubMed]
- Pruvot, B.; Jacquel, A.; Droin, N.; Auberger, P.; Bouscary, D.; Tamburini, J.; Muller, M.; Fontenay, M.; Chluba, J.; Solary, E. Leukemic cell xenograft in zebrafish embryo for investigating drug efficacy. Haematologica 2011, 96, 612–616. [Google Scholar] [CrossRef]
- Zhang, B.; Shimada, Y.; Kuroyanagi, J.; Umemoto, N.; Nishimura, Y.; Tanaka, T. Quantitative phenotyping-based in vivo chemical screening in a zebrafish model of leukemia stem cell xenotransplantation. PLoS ONE 2014, 9, e85439. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, W.; Zhao, J.J.; Kwart, A.H.; Yang, C.; Ma, D.; Ren, X.; Tai, Y.T.; Anderson, K.C.; Handin, R.I.; et al. A clinically relevant in vivo zebrafish model of human multiple myeloma to study preclinical therapeutic efficacy. Blood 2016, 128, 249–252. [Google Scholar] [CrossRef]
- Quentmeier, H.; Pommerenke, C.; Dirks, W.G.; Eberth, S.; Koeppel, M.; MacLeod, R.A.F.; Nagel, S.; Steube, K.; Uphoff, C.C.; Drexler, H.G. The LL-100 panel: 100 cell lines for blood cancer studies. Sci. Rep. 2019, 9, 8218. [Google Scholar] [CrossRef]
- Sarin, V.; Yu, K.; Ferguson, I.D.; Gugliemini, O.; Nix, M.A.; Hann, B.; Sirota, M.; Wiita, A.P. Evaluating the efficacy of multiple myeloma cell lines as models for patient tumors via transcriptomic correlation analysis. Leukemia 2020, 34, 2754–2765. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Sfougataki, I.; Grafakos, I.; Varela, I.; Mitrakos, A.; Karagiannidou, A.; Tzannoudaki, M.; Poulou, M.; Mertzanian, A.; Roubelakis, G.M.; Stefanaki, K.; et al. Reprogramming of bone marrow derived mesenchymal stromal cells to human induced pluripotent stem cells from pediatric patients with hematological diseases using a commercial mRNA kit. Blood Cells Mol. Dis. 2019, 76, 32–39. [Google Scholar] [CrossRef]
- Bueno, C.; Sardina, J.L.; Di Stefano, B.; Romero-Moya, D.; Muñoz-López, A.; Ariza, L.; Chillón, M.C.; Balanzategui, A.; Castaño, J.; Herreros, A.; et al. Reprogramming human B cells into induced pluripotent stem cells and its enhancement by C/EBPα. Leukemia 2016, 30, 674–682. [Google Scholar] [CrossRef]
- Wattanapanitch, M. Recent Updates on Induced Pluripotent Stem Cells in Hematological Disorders. Stem Cells Int. 2019, 2019, 5171032. [Google Scholar] [CrossRef]
- Vo, L.T.; Daley, G.Q. De novo generation of HSCs from somatic and pluripotent stem cell sources. Blood 2015, 125, 2641–2648. [Google Scholar] [CrossRef]
- Kim, H.; Schaniel, C. Modeling Hematological Diseases and Cancer With Patient-Specific Induced Pluripotent Stem Cells. Front Immunol. 2018, 9, 2243. [Google Scholar] [CrossRef]
- Reilly, A.; Doulatov, S. Induced pluripotent stem cell models of myeloid malignancies and clonal evolution. Stem Cell Res. 2021, 52, 102195. [Google Scholar] [CrossRef]
- Donada, A.; Basso-Valentina, F.; Arkoun, B.; Monte-Mor, B.; Plo, I.; Raslova, H. Induced pluripotent stem cells and hematological malignancies: A powerful tool for disease modeling and drug development. Stem Cell Res. 2020, 49, 102060. [Google Scholar] [CrossRef]
- Lin, S.L.; Chang, D.C.; Chang-Lin, S.; Lin, C.H.; Wu, D.T.; Chen, D.T.; Ying, S.Y. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. Rna 2008, 14, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Gentles, A.J.; Chatterjee, S.; Lan, F.; Reinisch, A.; Corces, M.R.; Xavy, S.; Shen, J.; Haag, D.; Chanda, S.; et al. Human AML-iPSCs Reacquire Leukemic Properties after Differentiation and Model Clonal Variation of Disease. Cell Stem Cell 2017, 20, 329–344.e7. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Li, T.; Wang, X.; Bao, W.; Huang, J.; Ma, Y.; Li, S.; Wang, S.; Yang, Y.; et al. Generation of three iPSC lines from different types of pediatric acute leukemia patients. Stem Cell Res. 2021, 55, 102460. [Google Scholar] [CrossRef]
- Kotini, A.G.; Chang, C.J.; Boussaad, I.; Delrow, J.J.; Dolezal, E.K.; Nagulapally, A.B.; Perna, F.; Fishbein, G.A.; Klimek, V.M.; Hawkins, R.D.; et al. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, S.; Nishinaka-Arai, Y.; Kazuki, Y.; Oshimura, M.; Nakahata, T.; Niwa, A.; Saito, M.K. Pluripotent stem cell model of early hematopoiesis in Down syndrome reveals quantitative effects of short-form GATA1 protein on lineage specification. PLoS ONE 2021, 16, e0247595. [Google Scholar] [CrossRef]
- Byrska-Bishop, M.; VanDorn, D.; Campbell, A.E.; Betensky, M.; Arca, P.R.; Yao, Y.; Gadue, P.; Costa, F.F.; Nemiroff, R.L.; Blobel, G.A. Pluripotent stem cells reveal erythroid-specific activities of the GATA1 N-terminus. J. Clin. Investig. 2015, 125, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, M.; Kumano, K.; Taoka, K.; Arai, S.; Kataoka, K.; Ueda, K.; Kamikubo, Y.; Takayama, N.; Otsu, M.; Eto, K.; et al. Generation of induced pluripotent stem cells derived from primary and secondary myelofibrosis patient samples. Exp. Hematol. 2014, 42, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Pruszak, J.; Varadarajan, M.; Blomen, V.A.; Gokhale, S.; Camargo, F.D.; Wernig, M.; Jaenisch, R.; Brummelkamp, T.R. Generation of iPSCs from cultured human malignant cells. Blood 2010, 115, 4039–4042. [Google Scholar] [CrossRef] [PubMed]
- Telliam, G.; Féraud, O.; Griscelli, F.; Opolon, P.; Divers, D.; Bennaceur-Griscelli, A.; Turhan, A.G. Generation of an induced pluripotent stem cell line from a patient with chronic myeloid leukemia (CML) resistant to targeted therapies. Stem Cell Res. 2016, 17, 235–237. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Muñoz-López, A.; Romero-Moya, D.; Prieto, C.; Ramos-Mejía, V.; Agraz-Doblas, A.; Varela, I.; Buschbeck, M.; Palau, A.; Carvajal-Vergara, X.; Giorgetti, A.; et al. Development Refractoriness of MLL-Rearranged Human B Cell Acute Leukemias to Reprogramming into Pluripotency. Stem Cell Rep. 2016, 7, 602–618. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, J.H.; Kang, K.W.; Lee, B.H.; Kim, B.S. Generation of normal induced pluripotent stem cell line KUMCi002-A from bone marrow CD34+ cells of patient with multiple myeloma disease having 13q deletion and IGH translocation. Stem Cell Res. 2020, 49, 102030. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Kunimoto, H.; Watanabe, N.; Fukuchi, Y.; Yuasa, S.; Yamazaki, S.; Nishimura, T.; Sadahira, K.; Fukuda, K.; Okano, H.; et al. Impaired hematopoietic differentiation of RUNX1-mutated induced pluripotent stem cells derived from FPD/AML patients. Leukemia 2014, 28, 2344–2354. [Google Scholar] [CrossRef]
- Antony-Debré, I.; Manchev, V.T.; Balayn, N.; Bluteau, D.; Tomowiak, C.; Legrand, C.; Langlois, T.; Bawa, O.; Tosca, L.; Tachdjian, G.; et al. Level of RUNX1 activity is critical for leukemic predisposition but not for thrombocytopenia. Blood 2015, 125, 930–940. [Google Scholar] [CrossRef]
- Chang, T.Y.; Dvorak, C.C.; Loh, M.L. Bedside to bench in juvenile myelomonocytic leukemia: Insights into leukemogenesis from a rare pediatric leukemia. Blood 2014, 124, 2487–2497. [Google Scholar] [CrossRef]
- Gandre-Babbe, S.; Paluru, P.; Aribeana, C.; Chou, S.T.; Bresolin, S.; Lu, L.; Sullivan, S.K.; Tasian, S.K.; Weng, J.; Favre, H.; et al. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood 2013, 121, 4925–4929. [Google Scholar] [CrossRef]
- Tasian, S.K.; Casas, J.A.; Posocco, D.; Gandre-Babbe, S.; Gagne, A.L.; Liang, G.; Loh, M.L.; Weiss, M.J.; French, D.L.; Chou, S.T. Mutation-specific signaling profiles and kinase inhibitor sensitivities of juvenile myelomonocytic leukemia revealed by induced pluripotent stem cells. Leukemia 2019, 33, 181–190. [Google Scholar] [CrossRef]
- Ye, Z.; Liu, C.F.; Lanikova, L.; Dowey, S.N.; He, C.; Huang, X.; Brodsky, R.A.; Spivak, J.L.; Prchal, J.T.; Cheng, L. Differential sensitivity to JAK inhibitory drugs by isogenic human erythroblasts and hematopoietic progenitors generated from patient-specific induced pluripotent stem cells. Stem Cells 2014, 32, 269–278. [Google Scholar] [CrossRef]
- Hsu, J.; Reilly, A.; Hayes, B.J.; Clough, C.A.; Konnick, E.Q.; Torok-Storb, B.; Gulsuner, S.; Wu, D.; Becker, P.S.; Keel, S.B.; et al. Reprogramming identifies functionally distinct stages of clonal evolution in myelodysplastic syndromes. Blood 2019, 134, 186–198. [Google Scholar] [CrossRef]
- Beke, A.; Laplane, L.; Riviere, J.; Yang, Q.; Torres-Martin, M.; Dayris, T.; Rameau, P.; Saada, V.; Bilhou-Nabera, C.; Hurtado, A.; et al. Multilayer intraclonal heterogeneity in chronic myelomonocytic leukemia. Haematologica 2020, 105, 112–123. [Google Scholar] [CrossRef]
- Kotini, A.G.; Chang, C.J.; Chow, A.; Yuan, H.; Ho, T.C.; Wang, T.; Vora, S.; Solovyov, A.; Husser, C.; Olszewska, M.; et al. Stage-Specific Human Induced Pluripotent Stem Cells Map the Progression of Myeloid Transformation to Transplantable Leukemia. Cell Stem Cell 2017, 20, 315–328.e7. [Google Scholar] [CrossRef]
- Wesely, J.; Kotini, A.G.; Izzo, F.; Luo, H.; Yuan, H.; Sun, J.; Georgomanoli, M.; Zviran, A.; Deslauriers, A.G.; Dusaj, N.; et al. Acute Myeloid Leukemia iPSCs Reveal a Role for RUNX1 in the Maintenance of Human Leukemia Stem Cells. Cell Rep. 2020, 31, 107688. [Google Scholar] [CrossRef] [PubMed]
- Mulero-Navarro, S.; Sevilla, A.; Roman, A.C.; Lee, D.F.; D’Souza, S.L.; Pardo, S.; Riess, I.; Su, J.; Cohen, N.; Schaniel, C.; et al. Myeloid Dysregulation in a Human Induced Pluripotent Stem Cell Model of PTPN11-Associated Juvenile Myelomonocytic Leukemia. Cell Rep. 2015, 13, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Göhring, G.; Heuser, M.; Ganser, A.; Schambach, A.; Morgan, M.A. Letter to the Editor: Production of Mature Healthy Hematopoietic Cells from Induced Pluripotent Stem Cells Derived from an AML Diagnostic Sample Containing the t(8;21) Translocation. Stem Cells 2016, 34, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, A.E.; King, N.E.; Matsui, H.; Jepsen, K.; Panopoulos, A.D. Two iPSC lines generated from the bone marrow of a relapsed/refractory AML patient display normal karyotypes and myeloid differentiation potential. Stem. Cell Res. 2019, 41, 101587. [Google Scholar] [CrossRef] [PubMed]
- Al-Attar, S.; Westra, E.R.; van der Oost, J.; Brouns, S.J. Clustered regularly interspaced short palindromic repeats (CRISPRs): The hallmark of an ingenious antiviral defense mechanism in prokaryotes. Biol. Chem. 2011, 392, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Pine, A.R.; Kotini, A.G.; Yuan, H.; Zamparo, L.; Starczynowski, D.T.; Leslie, C.; Papapetrou, E.P. Sequential CRISPR gene editing in human iPSCs charts the clonal evolution of myeloid leukemia and identifies early disease targets. Cell Stem. Cell 2021, 28, 1074–1089.e7. [Google Scholar] [CrossRef]
- Gerritsen, M.; Yi, G.; Tijchon, E.; Kuster, J.; Schuringa, J.J.; Martens, J.H.A.; Vellenga, E. RUNX1 mutations enhance self-renewal and block granulocytic differentiation in human in vitro models and primary AMLs. Blood Adv. 2019, 3, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Tijchon, E.; Yi, G.; Mandoli, A.; Smits, J.G.A.; Ferrari, F.; Heuts, B.M.H.; Wijnen, F.; Kim, B.; Janssen-Megens, E.M.; Schuringa, J.J.; et al. The acute myeloid leukemia associated AML1-ETO fusion protein alters the transcriptome and cellular progression in a single-oncogene expressing in vitro induced pluripotent stem cell based granulocyte differentiation model. PLoS ONE 2019, 14, e0226435. [Google Scholar] [CrossRef]
- Nasri, M.; Ritter, M.; Mir, P.; Dannenmann, B.; Aghaallaei, N.; Amend, D.; Makaryan, V.; Xu, Y.; Fletcher, B.; Bernhard, R.; et al. CRISPR/Cas9-mediated ELANE knockout enables neutrophilic maturation of primary hematopoietic stem and progenitor cells and induced pluripotent stem cells of severe congenital neutropenia patients. Haematologica 2020, 105, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Dannenmann, B.; Klimiankou, M.; Oswald, B.; Solovyeva, A.; Mardan, J.; Nasri, M.; Ritter, M.; Zahabi, A.; Arreba-Tutusaus, P.; Mir, P.; et al. iPSC modeling of stage-specific leukemogenesis reveals BAALC as a key oncogene in severe congenital neutropenia. Cell Stem. Cell 2021, 28, 906–922.e6. [Google Scholar] [CrossRef] [PubMed]
- Nilsri, N.; Jangprasert, P.; Pawinwongchai, J.; Israsena, N.; Rojnuckarin, P. Distinct effects of V617F and exon12-mutated JAK2 expressions on erythropoiesis in a human induced pluripotent stem cell (iPSC)-based model. Sci. Rep. 2021, 11, 5255. [Google Scholar] [CrossRef] [PubMed]
- Böiers, C.; Richardson, S.E.; Laycock, E.; Zriwil, A.; Turati, V.A.; Brown, J.; Wray, J.P.; Wang, D.; James, C.; Herrero, J.; et al. A Human IPS Model Implicates Embryonic B-Myeloid Fate Restriction as Developmental Susceptibility to B Acute Lymphoblastic Leukemia-Associated ETV6-RUNX1. Dev. Cell 2018, 44, 362–377.e7. [Google Scholar] [CrossRef]
- Barwe, S.P.; Sidhu, I.; Kolb, E.A.; Gopalakrishnapillai, A. Modeling Transient Abnormal Myelopoiesis Using Induced Pluripotent Stem Cells and CRISPR/Cas9 Technology. Mol. Methods Clin. Dev. 2020, 19, 201–209. [Google Scholar] [CrossRef]
- Banno, K.; Omori, S.; Hirata, K.; Nawa, N.; Nakagawa, N.; Nishimura, K.; Ohtaka, M.; Nakanishi, M.; Sakuma, T.; Yamamoto, T. Systematic cellular disease models reveal synergistic interaction of trisomy 21 and GATA1 mutations in hematopoietic abnormalities. Cell Rep. 2016, 15, 1228–1241. [Google Scholar] [CrossRef]
- Sidhu, I.; Barwe, S.P.; Kiick, K.E.; Kolb, E.A.; Gopalakrishnapillai, A. A 3-D Hydrogel Based System for Hematopoietic Differentiation and its Use in Modeling Down Syndrome Associated Transient Myeloproliferative Disorder. Biomater. Sci. 2021. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Tallman, M.S. Emerging therapeutic drugs for AML. Blood 2016, 127, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Malinge, S.; Bliss-Moreau, M.; Kirsammer, G.; Diebold, L.; Chlon, T.; Gurbuxani, S.; Crispino, J.D. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J. Clin. Investig. 2012, 122, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, I.Q.; Splendore, A.; Emerenciano, M.; Figueiredo, A.; Ferrari, I.; Pombo-de-Oliveira, M.S. GATA1 mutations in acute leukemia in children with Down syndrome. Cancer Genet. Cytogenet 2006, 166, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Hollanda, L.M.; Lima, C.S.; Cunha, A.F.; Albuquerque, D.M.; Vassallo, J.; Ozelo, M.C.; Joazeiro, P.P.; Saad, S.T.; Costa, F.F. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat. Genet. 2006, 38, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Gruber, T.A.; Downing, J.R. The biology of pediatric acute megakaryoblastic leukemia. Blood 2015, 126, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, S.I.; Santoni, F.; Vannier, A.; Falconnet, E.; Giarin, E.; Basso, G.; Hoischen, A.; Veltman, J.A.; Groet, J.; Nizetic, D.; et al. Exome sequencing identifies putative drivers of progression of transient myeloproliferative disorder to AMKL in infants with Down syndrome. Blood 2013, 122, 554–561. [Google Scholar] [CrossRef]
- Yoshida, K.; Toki, T.; Okuno, Y.; Kanezaki, R.; Shiraishi, Y.; Sato-Otsubo, A.; Sanada, M.; Park, M.J.; Terui, K.; Suzuki, H.; et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat. Genet. 2013, 45, 1293–1299. [Google Scholar] [CrossRef]
- O’Hare, T.; Zabriskie, M.S.; Eiring, A.M.; Deininger, M.W. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat. Rev. Cancer 2012, 12, 513–526. [Google Scholar] [CrossRef]
- Mahon, F.X.; Réa, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Imagawa, J.; Tanaka, H.; Okada, M.; Nakamae, H.; Hino, M.; Murai, K.; Ishida, Y.; Kumagai, T.; Sato, S.; Ohashi, K.; et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): A multicentre phase 2 trial. Lancet Haematol. 2015, 2, e528–e535. [Google Scholar] [CrossRef]
- Miyauchi, M.; Koya, J.; Arai, S.; Yamazaki, S.; Honda, A.; Kataoka, K.; Yoshimi, A.; Taoka, K.; Kumano, K.; Kurokawa, M. ADAM8 Is an Antigen of Tyrosine Kinase Inhibitor-Resistant Chronic Myeloid Leukemia Cells Identified by Patient-Derived Induced Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Taoka, K.; Arai, S.; Kataoka, K.; Hosoi, M.; Miyauchi, M.; Yamazaki, S.; Honda, A.; Aixinjueluo, W.; Kobayashi, T.; Kumano, K.; et al. Using patient-derived iPSCs to develop humanized mouse models for chronic myelomonocytic leukemia and therapeutic drug identification, including liposomal clodronate. Sci. Rep. 2018, 8, 15855. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Kotini, A.G.; Olszewska, M.; Georgomanoli, M.; Teruya-Feldstein, J.; Sperber, H.; Sanchez, R.; DeVita, R.; Martins, T.J.; Abdel-Wahab, O.; et al. Dissecting the Contributions of Cooperating Gene Mutations to Cancer Phenotypes and Drug Responses with Patient-Derived iPSCs. Stem. Cell Rep. 2018, 10, 1610–1624. [Google Scholar] [CrossRef] [PubMed]
- Skokowa, J.; Dale, D.C.; Touw, I.P.; Zeidler, C.; Welte, K. Severe congenital neutropenias. Nat. Rev. Dis. Primers 2017, 3, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rhyasen, G.W.; Bolanos, L.; Fang, J.; Jerez, A.; Wunderlich, M.; Rigolino, C.; Mathews, L.; Ferrer, M.; Southall, N.; Guha, R.; et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell 2013, 24, 90–104. [Google Scholar] [CrossRef]
- Smith, M.A.; Choudhary, G.S.; Pellagatti, A.; Choi, K.; Bolanos, L.C.; Bhagat, T.D.; Gordon-Mitchell, S.; Von Ahrens, D.; Pradhan, K.; Steeples, V.; et al. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nat. Cell. Biol. 2019, 21, 640–650. [Google Scholar] [CrossRef]
- Nianias, A.; Themeli, M. Induced Pluripotent Stem Cell (iPSC)-Derived Lymphocytes for Adoptive Cell Immunotherapy: Recent Advances and Challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef]
- Ueda, N.; Uemura, Y.; Zhang, R.; Kitayama, S.; Iriguchi, S.; Kawai, Y.; Yasui, Y.; Tatsumi, M.; Ueda, T.; Liu, T.Y.; et al. Generation of TCR-Expressing Innate Lymphoid-like Helper Cells that Induce Cytotoxic T Cell-Mediated Anti-leukemic Cell Response. Stem Cell Rep. 2018, 10, 1935–1946. [Google Scholar] [CrossRef]
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat. Commun. 2021, 12, 430. [Google Scholar] [CrossRef]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 153. [Google Scholar] [CrossRef]
- Zhu, H.; Blum, R.H.; Bjordahl, R.; Gaidarova, S.; Rogers, P.; Lee, T.T.; Abujarour, R.; Bonello, G.B.; Wu, J.; Tsai, P.-F. Pluripotent stem cell–derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020, 135, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, J.P.; Bjordahl, R.; Mahmood, S.; Reiser, J.; Gaidarova, S.; Blum, R.; Cichocki, F.; Chu, H.-y.; Bonello, G.; Lee, T. FT576: Multi-specific off-the-shelf CAR-NK cell therapy engineered for enhanced persistence, avoidance of self-fratricide and optimized mab combination therapy to prevent antigenic escape and elicit a deep and durable response in multiple myeloma. Blood 2020, 136, 4–5. [Google Scholar] [CrossRef]
- Lengerke, C.; Grauer, M.; Niebuhr, N.I.; Riedt, T.; Kanz, L.; Park, I.-H.; Daley, G.Q. Hematopoietic development from human induced pluripotent stem cells. Ann. N. Y. Acad. Sci. 2009, 1176, 219. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhan, H.; Mali, P.; Dowey, S.; Williams, D.M.; Jang, Y.-Y.; Dang, C.V.; Spivak, J.L.; Moliterno, A.R.; Cheng, L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood J. Am. Soc. Hematol. 2009, 114, 5473–5480. [Google Scholar] [CrossRef]
- Choi, K.D.; Yu, J.; Smuga-Otto, K.; Salvagiotto, G.; Rehrauer, W.; Vodyanik, M.; Thomson, J.; Slukvin, I. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells 2009, 27, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Salvagiotto, G.; Burton, S.; Daigh, C.A.; Rajesh, D.; Slukvin, I.I.; Seay, N.J. A defined, feeder-free, serum-free system to generate in vitro hematopoietic progenitors and differentiated blood cells from hESCs and hiPSCs. PLoS ONE 2011, 6, e17829. [Google Scholar] [CrossRef]
- Marí-Buyé, N.; Semino, C.E. Differentiation of mouse embryonic stem cells in self-assembling peptide scaffolds. Methods Mol. Biol. 2011, 690, 217–237. [Google Scholar] [CrossRef]
- Xu, Y.; Shan, W.; Li, X.; Wang, B.; Liu, S.; Wang, Y.; Long, Y.; Tie, R.; Wang, L.; Cai, S.; et al. A synthetic three-dimensional niche system facilitates generation of functional hematopoietic cells from human-induced pluripotent stem cells. J. Hematol. Oncol. 2016, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef]
- Tsou, Y.-H.; Khoneisser, J.; Huang, P.-C.; Xu, X. Hydrogel as a bioactive material to regulate stem cell fate. Bioact. Mater. 2016, 1, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.E.; Shah, D.A.; Rogers, C.; Hall, S.; Weston, N.; Parmenter, C.D.; McNally, D.; Denning, C.; Shakesheff, K.M. Combined hydrogels that switch human pluripotent stem cells from self-renewal to differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 5580–5585. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Martin, L.M.; Bosco, D.B.; Bundy, J.L.; Nowakowski, R.S.; Sang, Q.X.; Li, Y. Differential effects of acellular embryonic matrices on pluripotent stem cell expansion and neural differentiation. Biomaterials 2015, 73, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.K.; Chowdhury, M.F.; Iyer, R.K.; Stanford, W.L.; Radisic, M. Engineering surfaces for site-specific vascular differentiation of mouse embryonic stem cells. Acta. Biomater. 2010, 6, 1904–1916. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Shih, Y.V.; Hwang, Y.; Wen, C.; Rao, V.; Seo, T.; Varghese, S. Mineralized gelatin methacrylate-based matrices induce osteogenic differentiation of human induced pluripotent stem cells. Acta. Biomater. 2014, 10, 4961–4970. [Google Scholar] [CrossRef]
- Lam, J.; Lowry, W.E.; Carmichael, S.T.; Segura, T. Delivery of iPS-NPCs to the stroke cavity within a hyaluronic acid matrix promotes the differentiation of transplanted cells. Adv. Funct. Mater. 2014, 24, 7053–7062. [Google Scholar] [CrossRef]
- Tarunina, M.; Humbert, C.; Ali, S.; Choo, Y.; Chuang, A.T.T.; Saw, D. Methods for Producing Cells of the Hematopoietic Lineage Using Hydrogel Encapsulation. U.S. Patent Application No. 16/669650, 16 April 2020. [Google Scholar]



{kind=link}
{kind=link}
| Disease | Model | Outcome | Reference |
|---|---|---|---|
| Acute myeloid leukemia (AML) | Conditional transgenic targeting NRAS and BCL-2 | MDS/AML transformation | [7] |
| Npm1c/Dnmt3a mutant knock-in mice | Epigenetic therapy in preleukemic stage | [8] | |
| Humanized NSG xenograft expressing BCR–ABL1 and MLL-AF9 | Efficient engraftment in humanized niche | [9] | |
| Cell line-derived xenograft model | Epigenetic therapy in pediatric AML | [10] | |
| Acute lymphoblastic leukemia (ALL) | Cre-recombinase-inducible mouse model for PRDM14 | Rapid onset T-ALL model | [11] |
| Humanized NSG xenograft expressing MLL-AF9 | Efficient engraftment in humanized niche, efficacy of the I-BET151 inhibitor | [9] | |
| T-ALL xenograft model | Targeted monoclonal antibody against NOTCH1 | [12] | |
| CD81 knockout cell line xenograft | Role of CD81 in homing and engraftment | [13] | |
| Chronic myeloid leukemia (CML) | Transposon-based insertional mutagenesis | Identification of mechanisms of blast crisis | [14] |
| Conditional gene knock-out strains | Identification of tumor repressor PTEN in BCR-ABL background | [15] | |
| Chronic lymphocytic leukemia (CLL) | NSG xenograft mice | Effect of BTK inhibitor ibrutinib | [16] |
| Serial transplantation in TCL-1 transgenic mice | Efficacy of programmed cell death (PD-1) immune checkpoint inhibitors | [17] | |
| Multiple myeloma (MM) | Vk*MYC transgenic mice | Identification of novel drugs | [18] |
| BCL2L10 transgenic mice | Recapitulation of MM phenotype for validation of new therapies | [19] | |
| B-cell lymphoma | Conditional transgenic for MYC and RAS | Preclinical testing for CD20 | [20,21] |
| Follicular lymphoma | Transgenic linked to Vav regulatory sequence | Development of germinal center hyperplasia followed by follicular lymphoma | [22] |
| Peripheral T-cell lymphoma (PTCL) | Inducible transgenic for ITK-SYK | Efficacy of Syk inhibitors | [23] |
| Cutaneous T-cell lymphoma (CTCL) | Transgenic for IL15 | Efficacy of HDAC inhibitors | [24] |
| Disease | Model | Outcome | Reference |
|---|---|---|---|
| Acute myeloid leukemia (AML) | SRSF2-ASXL1-NRAS triple mutant | Mechanism of clonal evolution and identification of early target genes | [80] |
| RUNXI S291fs300X mutant | Blocked granulocytic differentiation via CEBPA downregulation | [81] | |
| RUNX1-RUNX1T1 fusion | Blocked granulocytic differentiation via altering the acetylome during differentiation | [82] | |
| Congenital neutropenia (CN)/AML | ELANE mutant knock-out | Revert the maturation arrest | [83] |
| CSF3R or RUNX1 mutant | MK2a phosphorylation targeting | [84] | |
| Polycythemia vera (PV) | JAK2 V617F mutant | Erythrocytosis and thrombocytosis; interferon alpha and arsenic trioxide | [85] |
| JAK2 exon 12 N542-E543del mutant | Erythrocytosis; interferon alpha and arsenic trioxide therapy | [85] | |
| Acute lymphoblastic leukemia (ALL)—pediatric | ETV6-RUNX1 | Initiation model during fetal development | [86] |
| Transient myeloproliferative disorder/Down syndrome myeloid leukemia—pediatric | Trisomy 21 + GATA1 mutant | Initiation and progression model | [87,88,89] |
| Therapy | Features | Disease | Clinical Trial Identifier |
|---|---|---|---|
| FT516 | NK cells expressing hnCD16 | AML | NCT04023071 |
| NK cells expressing hnCD16 + mAB (rituximab or obinutuzumab) | B-lymphoma | NCT04023071 | |
| FT596 | NK cells expressing hnCD16, IL15RF + mAB (rituximab) | NHL, DLBCL, HGBCL | NCT04555811 |
| NK cells expressing hnCD16, IL15RF +/− mAB (rituximab or obinutuzumab) | CLL, B-lymphoma | NCT04245722 | |
| iCAR NK Cells | Anti-CD19 | B-lymphoma | NCT03824951 |
| FT819 | A novel 1XX CAR targeting CD19 inserted into the T-cell receptor alpha constant (TRAC) locus and edited for elimination of T-cell receptor (TCR) expression | CLL, B-lymphoma, B-ALL | NCT04629729 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidhu, I.; Barwe, S.P.; Pillai, R.K.; Gopalakrishnapillai, A. Harnessing the Power of Induced Pluripotent Stem Cells and Gene Editing Technology: Therapeutic Implications in Hematological Malignancies. Cells 2021, 10, 2698. https://doi.org/10.3390/cells10102698
Sidhu I, Barwe SP, Pillai RK, Gopalakrishnapillai A. Harnessing the Power of Induced Pluripotent Stem Cells and Gene Editing Technology: Therapeutic Implications in Hematological Malignancies. Cells. 2021; 10(10):2698. https://doi.org/10.3390/cells10102698
Chicago/Turabian StyleSidhu, Ishnoor, Sonali P. Barwe, Raju K. Pillai, and Anilkumar Gopalakrishnapillai. 2021. "Harnessing the Power of Induced Pluripotent Stem Cells and Gene Editing Technology: Therapeutic Implications in Hematological Malignancies" Cells 10, no. 10: 2698. https://doi.org/10.3390/cells10102698
APA StyleSidhu, I., Barwe, S. P., Pillai, R. K., & Gopalakrishnapillai, A. (2021). Harnessing the Power of Induced Pluripotent Stem Cells and Gene Editing Technology: Therapeutic Implications in Hematological Malignancies. Cells, 10(10), 2698. https://doi.org/10.3390/cells10102698