
Quantifying Renin-Angiotensin-System Alterations in COVID-19

 ,
,  ,
, 
Abstract
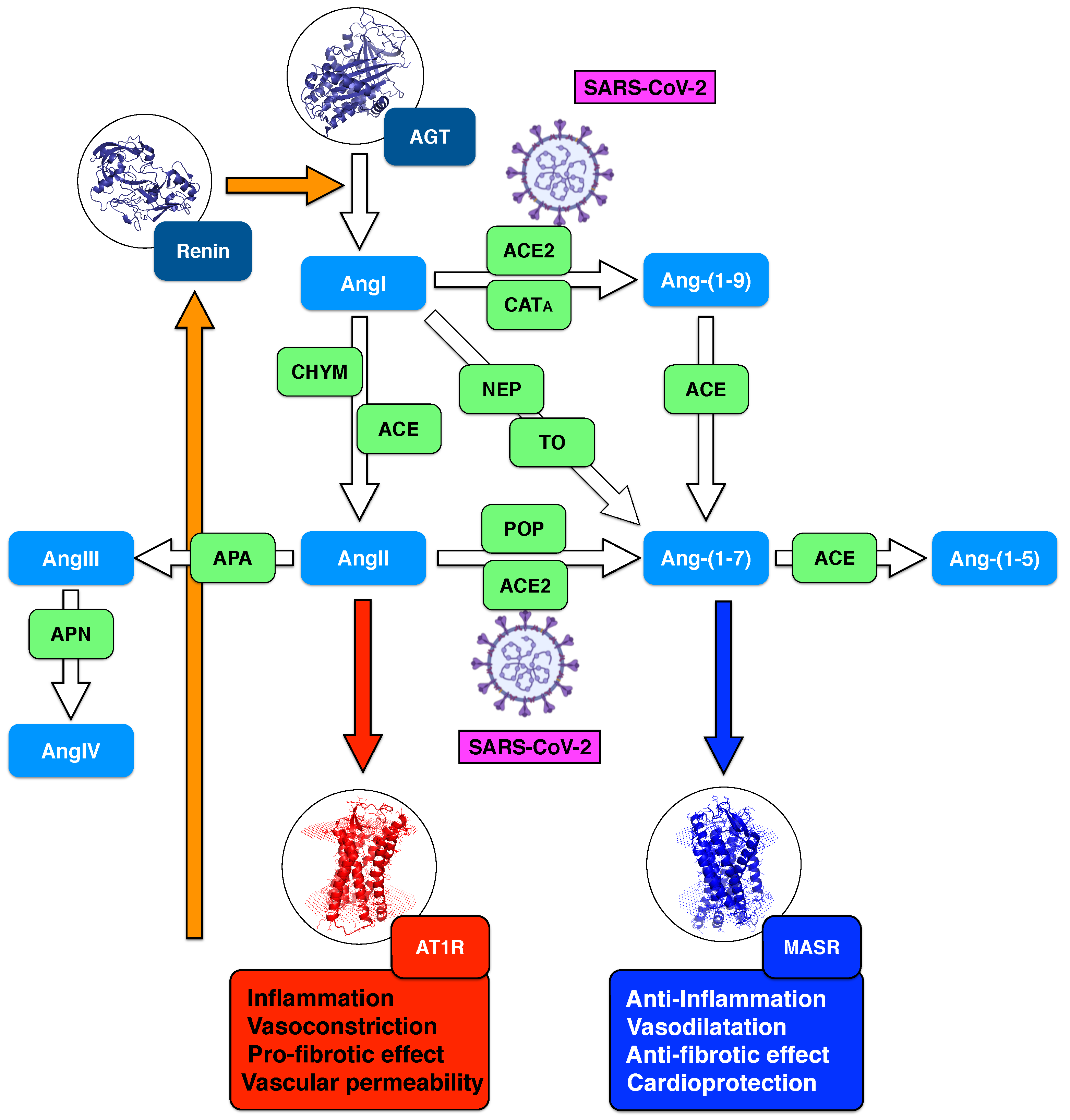
:1. Introduction: Normal RAS Regulation
- angiotensin 1-9 (Ang-(1-9)) nonapeptide via angiotensin-converting enzyme 2 (ACE2) or cathepsin A (CAT);
- angiotensin-II (AngII) octapeptide via angiotensin-converting enzyme (ACE) or chymase (CHY);
- angiotensin-(1-7) (Ang-(1-7)) heptapeptide via neprilysin (NEP) or thimet oligopeptidase (TO).
2. RAS Dysregulation in the First SARS-CoV Infections
3. RAS Dysregulation in Current SARS-CoV-2 Infections: New Data and Hypotheses
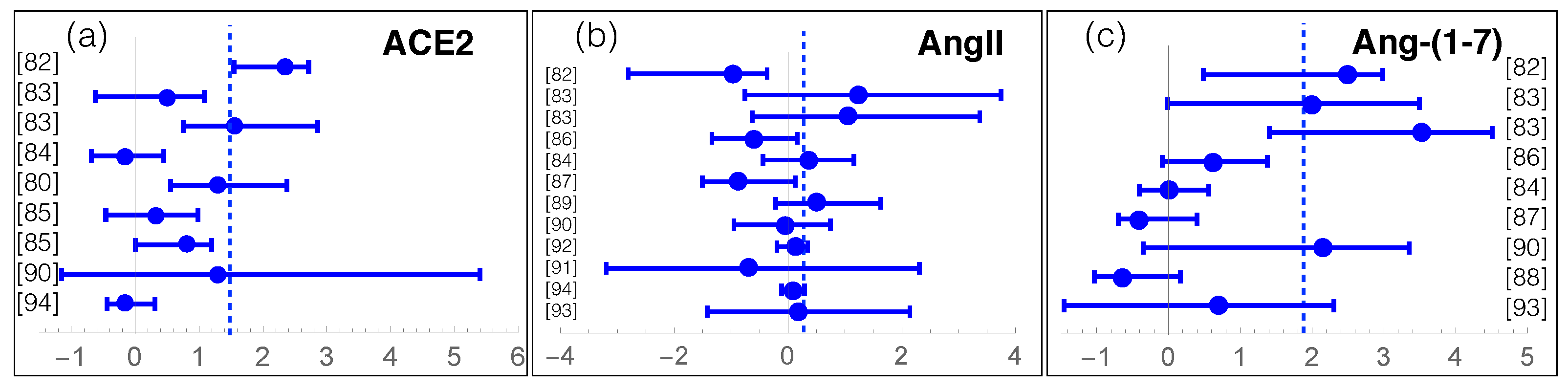
4. Meta-Analysis on RAS Components in COVID-19
5. RAS-Targeting Drugs in COVID-19
6. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Raizada, M.K.; Phillips, M.I.; Sumners, C. Cellular and Molecular Biology of the Renin-Angiotensin System; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Casarini, D.E.; Arita, D.Y.; Cunha, T.S.; Colucci, J.A. New Aspects of the Renin Angiotensin System in Cardiovascular and Renal Diseases; Bentham: Sharjah, United Arab Emirates, 2016. [Google Scholar]
- Wu, C.H.; Mohammadmoradi, S.; Chen, J.Z.; Sawada, H.; Daugherty, A.; Lu, H.S. Renin-angiotensin system and cardiovascular functions. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e108–e116. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, C.M. Role of angiotensin II in cardiovascular disease—Therapeutic implications of more than a century of research. J. Renin-Angiotensin-Aldosterone Syst. 2006, 7, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.S.D.; Liao, W.; Zhou, S.; Mei, D.; Wong, W.S.F. Targeting the renin–angiotensin system as novel therapeutic strategy for pulmonary diseases. Curr. Opin. Pharmacol. 2018, 40, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Rüster, C.; Wolf, G. Renin-angiotensin-aldosterone system and progression of renal disease. J. Am. Soc. Nephrol. 2006, 17, 2985–2991. [Google Scholar] [CrossRef] [PubMed]
- e Silva, A.C.S.; Miranda, A.S.; Rocha, N.P.; Teixeira, A.L. Renin angiotensin system in liver diseases: Friend or foe? World J. Gastroenterol. 2017, 23, 3396. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Simoes e Silva, A.; Silveira, K.; Ferreira, A.; Teixeira, M. ACE2, angiotensin-(1-7) and M as receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/angiotensin-(1-7)/MAS axis of the renin-angiotensin system: Focus on angiotensin-(1-7). Physiol. Rev. 2017. [Google Scholar] [CrossRef] [Green Version]
- Iwai, M.; Horiuchi, M. Devil and angel in the renin–angiotensin system: ACE–angiotensin II–AT 1 receptor axis vs. ACE2–angiotensin-(1-7)–Mas receptor axis. Hypertens. Res. 2009, 32, 533–536. [Google Scholar] [CrossRef] [Green Version]
- Zaman, M.A.; Oparil, S.; Calhoun, D.A. Drugs targeting the renin–angiotensin–aldosterone system. Nat. Rev. Drug Discov. 2002, 1, 621–636. [Google Scholar] [CrossRef]
- Vian, J.; Pereira, C.; Chavarria, V.; Köhler, C.; Stubbs, B.; Quevedo, J.; Kim, S.W.; Carvalho, A.F.; Berk, M.; Fernandes, B.S. The renin–angiotensin system: A possible new target for depression. BMC Med. 2017, 15, 144. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: Celebrating the 20th anniversary of the discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Matavelli, L.C.; Siragy, H.M. AT2 receptor activities and pathophysiological implications. J. Cardiovasc. Pharmacol. 2015, 65, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juillerat-Jeanneret, L. The Other Angiotensin II Receptor: AT2R as a Therapeutic Target. J. Med. Chem. 2020, 63, 1978–1995. [Google Scholar] [CrossRef]
- Yugandhar, V.G.; Clark, M.A. Angiotensin III: A physiological relevant peptide of the renin angiotensin system. Peptides 2013, 46, 26–32. [Google Scholar] [CrossRef]
- Hrenak, J.; Paulis, L.; Simko, F. Angiotensin A/Alamandine/MrgD axis: Another clue to understanding cardiovascular pathophysiology. Int. J. Mol. Sci. 2016, 17, 1098. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Zhang, J.; Zhuo, J.L. The vasoprotective axes of the renin-angiotensin system: Physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney diseases. Pharmacol. Res. 2017, 125, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Nehme, A.; Zouein, F.A.; Deris Zayeri, Z.; Zibara, K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8. [Google Scholar] [CrossRef]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascolo, A.; Scavone, C.; Rafaniello, C.; Ferrajolo, C.; Racagni, G.; Berrino, L.; Paolisso, G.; Rossi, F.; Capuano, A. Renin-angiotensin system and Coronavirus disease 2019: A narrative review. Front. Cardiovasc. Med. 2020, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Hanff, T.C.; Harhay, M.O.; Brown, T.S.; Cohen, J.B.; Mohareb, A.M. Is there an association between COVID-19 mortality and the renin-angiotensin system? A call for epidemiologic investigations. Clin. Infect. Dis. 2020, 71, 870–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingraham, N.E.; Barakat, A.G.; Reilkoff, R.; Bezdicek, T.; Schacker, T.; Chipman, J.G.; Tignanelli, C.J.; Puskarich, M.A. Understanding the renin–angiotensin–aldosterone–SARS-CoV axis: A comprehensive review. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): A review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiris, J.; Guan, Y.; Yuen, K. Severe acute respiratory syndrome. Nat. Med. 2004, 10, S88–S97. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Yuan, S.; Kok, K.H.; To, K.K.W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.Y.; Poon, R.W.S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Gurwitz, D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev. Res. 2020, 81, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
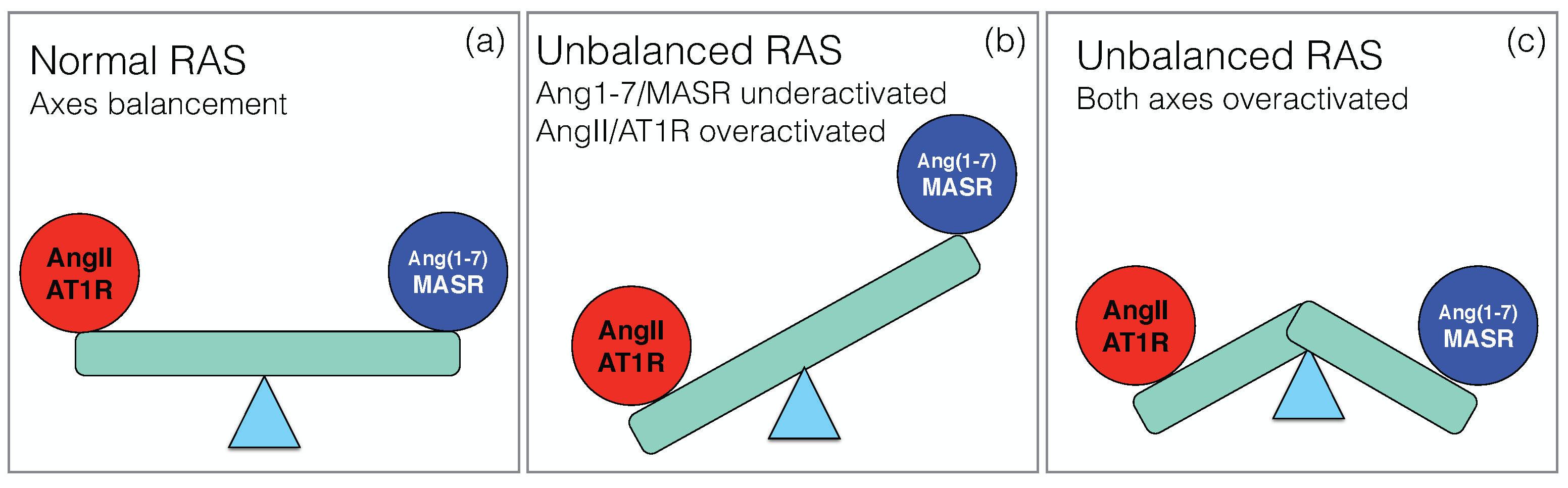
- Lanza, K.; Perez, L.G.; Costa, L.B.; Cordeiro, T.M.; Palmeira, V.A.; Ribeiro, V.T.; Simões e Silva, A.C. Covid-19: The renin–angiotensin system imbalance hypothesis. Clin. Sci. 2020, 134, 1259–1264. [Google Scholar] [CrossRef]
- Dean, A.Q.; Bozza, W.P.; Twomey, J.D.; Luo, S.; Nalli, A.; Zhang, B. The fight against COVID-19: Striking a balance in the renin–angiotensin system. Drug Discov. Today 2021. [Google Scholar] [CrossRef]
- Chung, M.K.; Karnik, S.; Saef, J.; Bergmann, C.; Barnard, J.; Lederman, M.M.; Tilton, J.; Cheng, F.; Harding, C.V.; Young, J.B.; et al. SARS-CoV-2 and ACE2: The biology and clinical data settling the ARB and ACEI controversy. EBioMedicine 2020, 58, 102907. [Google Scholar] [CrossRef]
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell. Cardiol. 2014, 66, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Satou, R.; Penrose, H.; Navar, L.G. Inflammation as a regulator of the renin-angiotensin system and blood pressure. Curr. Hypertens. Rep. 2018, 20, 100. [Google Scholar] [CrossRef]
- Santos, R.A.; e Silva, A.C.S.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [Green Version]
- Arnold, R.H. COVID-19—Does this disease kill due to imbalance of the renin angiotensin system (RAS) caused by genetic and gender differences in the response to viral ACE 2 attack? Heart Lung Circ. 2020, 29, 964–972. [Google Scholar] [CrossRef]
- Wiese, O.; Allwood, B.; Zemlin, A. COVID-19 and the renin-angiotensin system (RAS): A spark that sets the forest alight? Med. Hypotheses 2020, 144, 110231. [Google Scholar] [CrossRef]
- Seltzer, S. Linking ACE2 and angiotensin II to pulmonary immunovascular dysregulation in SARS-CoV-2 infection. Int. J. Infect. Dis. 2020, 101, 42–45. [Google Scholar] [CrossRef]
- Bastolla, U.; Chambers, P.; Abia, D.; García-Bermejo, M.L.; Fresno, M. Is Covid-19 severity associated with ACE2 degradation? arXiv 2021, arXiv:q-bio.TO/2102.13210. [Google Scholar]
- Pucci, F.; Bogaerts, P.; Rooman, M. Modeling the Molecular Impact of SARS-CoV-2 Infection on the Renin-Angiotensin System. Viruses 2020, 12, 1367. [Google Scholar] [CrossRef] [PubMed]
- Rysz, S.; Al-Saadi, J.; Sjöström, A.; Farm, M.; Jalde, F.C.; Plattén, M.; Eriksson, H.; Klein, M.; Vargas-Paris, R.; Nyrén, S.; et al. COVID-19 pathophysiology may be driven by an imbalance in the renin-angiotensin-aldosterone system. Nat. Commun. 2021, 12, 2417. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, F.; Manla, Y.; Atallah, B.; Starling, R.C. Heart failure and COVID-19. Heart Fail. Rev. 2021, 26, 1–10. [Google Scholar] [CrossRef]
- Migliaccio, M.G.; Di Mauro, M.; Ricciolino, R.; Spiniello, G.; Carfora, V.; Verde, N.; Mottola, F.F.; Coppola, N. Renal Involvement in COVID-19: A Review of the Literature. Infect. Drug Resist. 2021, 14, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Saguner, A.M.; An, J.; Ning, Y.; Yan, Y.; Li, G. Dysfunctional coagulation in COVID-19: From cell to bedside. Adv. Therapy 2020, 37, 3033–3039. [Google Scholar] [CrossRef] [PubMed]
- Mortus, J.R.; Manek, S.E.; Brubaker, L.S.; Loor, M.; Cruz, M.A.; Trautner, B.W.; Rosengart, T.K. Thromboelastographic results and hypercoagulability syndrome in patients with coronavirus disease 2019 who are critically ill. JAMA Netw. Open 2020, 3, e2011192. [Google Scholar] [CrossRef]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Zamai, L. Upregulation of the Renin–Angiotensin System Pathways and SARS-CoV-2 Infection: The Rationale for the Administration of Zinc-Chelating Agents in COVID-19 Patients. Cells 2021, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- Zamai, L. The Yin and Yang of ACE/ACE2 Pathways: The Rationale for the Use of Renin-Angiotensin System Inhibitors in COVID-19 Patients. Cells 2020, 9, 1704. [Google Scholar] [CrossRef] [PubMed]
- Zipeto, D.; Palmeira, J.d.F.; Argañaraz, G.A.; Argañaraz, E.R. ACE2/ADAM17/TMPRSS2 interplay may be the main risk factor for COVID-19. Front. Immunol. 2020, 11, 2642. [Google Scholar] [CrossRef]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- Montanari, M.; Canonico, B.; Nordi, E.; Vandini, D.; Barocci, S.; Benedetti, S.; Carlotti, E.; Zamai, L. Which ones, when and why should renin-angiotensin system inhibitors work against COVID-19? Adv. Biol. Regul. 2021, 81, 100820. [Google Scholar] [CrossRef]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-α-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-α production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef] [Green Version]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Sun, P.D. High affinity binding of SARS-CoV-2 spike protein enhances ACE2 carboxypeptidase activity. J. Biol. Chem. 2020, 295, 18579–18588. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.L.; Teng, J.L.L.; Jia, L.; Zhang, C.; Huang, C.; Cai, J.P.; Zhou, R.; Chan, K.H.; Zhao, H.; Zhu, L.; et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell 2021, 184, 2212–2228. [Google Scholar] [CrossRef] [PubMed]
- Zoufaly, A.; Poglitsch, M.; Aberle, J.H.; Hoepler, W.; Seitz, T.; Traugott, M.; Grieb, A.; Pawelka, E.; Laferl, H.; Wenisch, C.; et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 2020, 8, 1154–1158. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Leisman, D.E.; Mehta, A.; Li, Y.; Kays, K.R.; Li, J.Z.; Filbin, M.R.; Goldberg, M.B. Vasopressin infusion in COVID-19 critical illness is not associated with impaired viral clearance: A pilot study. BJA Br. J. Anaesth. 2021, 127, e146–e148. [Google Scholar] [CrossRef] [PubMed]
- Narula, S.; Yusuf, S.; Chong, M.; Ramasundarahettige, C.; Rangarajan, S.; Bangdiwala, S.I.; van Eikels, M.; Leineweber, K.; Wu, A.; Pigeyre, M.; et al. Plasma ACE2 and risk of death or cardiometabolic diseases: A case-cohort analysis. Lancet 2020, 396, 968–976. [Google Scholar] [CrossRef]
- Epelman, S.; Shrestha, K.; Troughton, R.W.; Francis, G.S.; Sen, S.; Klein, A.L.; Tang, W.W. Soluble angiotensin-converting enzyme 2 in human heart failure: Relation with myocardial function and clinical outcomes. J. Card. Fail. 2009, 15, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Ramchand, J.; Patel, S.K.; Srivastava, P.M.; Farouque, O.; Burrell, L.M. Elevated plasma angiotensin converting enzyme 2 activity is an independent predictor of major adverse cardiac events in patients with obstructive coronary artery disease. PLoS ONE 2018, 13, e0198144. [Google Scholar] [CrossRef]
- Úri, K.; Fagyas, M.; Kertész, A.; Borbély, A.; Jenei, C.; Bene, O.; Csanádi, Z.; Paulus, W.J.; Édes, I.; Papp, Z.; et al. Circulating ACE2 activity correlates with cardiovascular disease development. J. Renin-Angiotensin-Aldosterone Syst. 2016, 17, 1470320316668435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velkoska, E.; Dean, R.G.; Griggs, K.; Burchill, L.; Burrell, L.M. Angiotensin-(1-7) infusion is associated with increased blood pressure and adverse cardiac remodelling in rats with subtotal nephrectomy. Clin. Sci. 2011, 120, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Schweda, F.; Blumberg, F.C.; Schweda, A.; Kammerl, M.; Holmer, S.R.; Riegger, G.A.; Pfeifer, M.; Krämer, B.K. Effects of chronic hypoxia on renal renin gene expression in rats. Nephrol. Dial. Transplant. 2000, 15, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Zwaveling, S.; van Wijk, R.G.; Karim, F. Pulmonary edema in COVID-19: Explained by bradykinin? J. Allergy Clin. Immunol. 2020, 146, 1454–1455. [Google Scholar] [CrossRef]
- van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. Elife 2020, 9, e57555. [Google Scholar] [CrossRef]
- Mehrabadi, M.E.; Hemmati, R.; Tashakor, A.; Homaei, A.; Yousefzadeh, M.; Hemmati, K.; Hosseinkhani, S. Induced dysregulation of ACE2 by SARS-CoV-2 plays a key role in COVID-19 severity. Biomed. Pharmacother. 2021, 137, 111363. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Cai, T.; Fan, L.; Lou, K.; Hua, X.; Huang, Z.; Gao, G. The potential role of serum angiotensin-converting enzyme in coronavirus disease 2019. BMC Infect. Dis. 2020, 20, 883. [Google Scholar] [CrossRef] [PubMed]
- Lundström, A.; Ziegler, L.; Havervall, S.; Rudberg, A.S.; von Meijenfeldt, F.; Lisman, T.; Mackman, N.; Sandén, P.; Thålin, C. Soluble angiotensin-converting enzyme 2 is transiently elevated in COVID-19 and correlates with specific inflammatory and endothelial markers. J. Med. Virol. 2021, 93, 5908–5916. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.K.; Juno, J.A.; Lee, W.S.; Wragg, K.M.; Hogarth, P.M.; Kent, S.J.; Burrell, L.M. Plasma ACE2 activity is persistently elevated following SARS-CoV-2 infection: Implications for COVID-19 pathogenesis and consequences. Eur. Respir. J. 2021, 57, 2003730. [Google Scholar] [CrossRef]
- van Lier, D.; Kox, M.; Santos, K.; van der Hoeven, H.; Pillay, J.; Pickkers, P. Increased blood Angiotensin Converting Enzyme 2 activity in critically ill COVID-19 patients. ERJ Open Res. 2021, 7. [Google Scholar] [CrossRef]
- Reindl-Schwaighofer, R.; Hödlmoser, S.; Eskandary, F.; Poglitsch, M.; Bonderman, D.; Strassl, R.; Aberle, J.H.; Oberbauer, R.; Zoufaly, A.; Hecking, M. ACE2 elevation in severe COVID-19. Am. J. Respirat. Crit. Care Med. 2021, 203, 1191–1196. [Google Scholar] [CrossRef]
- Osman, I.O.; Melenotte, C.; Brouqui, P.; Million, M.; Lagier, J.C.; Parola, P.; Stein, A.; La Scola, B.; Meddeb, L.; Mege, J.L.; et al. Expression of ACE2, Soluble ACE2, Angiotensin I, Angiotensin II and Angiotensin-(1-7) Is Modulated in COVID-19 Patients. Front. Immunol. 2021, 12, 2350. [Google Scholar] [CrossRef]
- Kragstrup, T.W.; Singh, H.S.; Grundberg, I.; Nielsen, A.L.L.; Rivellese, F.; Mehta, A.; Goldberg, M.B.; Filbin, M.R.; Qvist, P.; Bibby, B.M. Plasma ACE2 predicts outcome of COVID-19 in hospitalized patients. PLoS ONE 2021, 16, e0252799. [Google Scholar] [CrossRef]
- Martins, A.L.V.; da Silva, F.A.; Bolais-Ramos, L.; de Oliveira, G.C.; Ribeiro, R.C.; Pereira, D.A.A.; Annoni, F.; Diniz, M.M.L.; Silva, T.G.F.; Zivianni, B.; et al. Increased circulating levels of angiotensin-(1-7) in severely ill COVID-19 patients. ERJ Open Res. 2021. [Google Scholar] [CrossRef]
- Eleuteri, D.; Montini, L.; Cutuli, S.L.; Rossi, C.; Alcaro, F.; Antonelli, M. Renin–angiotensin system dysregulation in critically ill patients with acute respiratory distress syndrome due to COVID-19: A preliminary report. Crit. Care 2021, 25, 95. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.M.; Benoit, J.L.; Berger, B.A.; Pulvino, C.; Lavie, C.J.; Lippi, G.; Benoit, S.W. Coronavirus disease 2019 is associated with low circulating plasma levels of angiotensin 1 and angiotensin 1, 7. J. Med.Virol. 2021, 93, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hu, R.; Zhang, C.; Ren, W.; Yu, A.; Zhou, X. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit. Care 2020, 24, 290. [Google Scholar] [CrossRef]
- Burns, K.; Cheng, M.; Lee, T.; McGeer, A.; Sweet, D.; Tran, K.; Lee, T.; Murthy, S.; Boyd, J.; Singer, J.; et al. Sustained Dysregulation of the Plasma Renin-angiotensin System in Acute COVID-19. Res. Square 2021. [Google Scholar] [CrossRef]
- Ozkan, S.; Cakmak, F.; Konukoglu, D.; Biberoglu, S.; Ipekci, A.; Akdeniz, Y.S.; Bolayirli, I.M.; Balkan, I.I.; Dumanli, G.Y.; Ikizceli, I. Efficacy of Serum Angiotensin II Levels in Prognosis of Patients with Coronavirus Disease 2019. Crit. Care Med. 2021, 49, e613–e623. [Google Scholar] [CrossRef]
- Liu, N.; Hong, Y.; Chen, R.G.; Zhu, H.M. High rate of increased level of plasma Angiotensin II and its gender difference in COVID-19: An analysis of 55 hospitalized patients with COVID-19 in a single hospital, WuHan, China. medRxiv 2020. [Google Scholar] [CrossRef]
- Files, D.C.; Gibbs, K.W.; Schaich, C.L.; Collins, S.P.; Gwathmey, T.M.; Casey, J.D.; Self, W.H.; Chappell, M.C. A Pilot Study to Assess the Circulating Renin-Angiotensin-System in COVID-19 Acute Respiratory Failure. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2021, 321, L213–L218. [Google Scholar] [CrossRef]
- Rieder, M.; Wirth, L.; Pollmeier, L.; Jeserich, M.; Goller, I.; Baldus, N.; Schmid, B.; Busch, H.J.; Hofmann, M.; Kern, W.; et al. Serum ACE2, Angiotensin II, and Aldosterone Levels Are Unchanged in Patients with COVID-19. Am. J. Hypertens. 2021, 34, 278–281. [Google Scholar] [CrossRef]
- Serfozo, P.; Wysocki, J.; Gulua, G.; Schulze, A.; Ye, M.; Liu, P.; Jin, J.; Bader, M.; Myöhänen, T.; García-Horsman, J.A.; et al. Ang II (angiotensin II) conversion to angiotensin-(1-7) in the circulation is POP (prolyloligopeptidase)-dependent and ACE2 (angiotensin-converting enzyme 2)-independent. Hypertension 2020, 75, 173–182. [Google Scholar] [CrossRef]
- Nagy, B., Jr.; Fejes, Z.; Szentkereszty, Z.; Süto, R.; Várkonyi, I.; Ajzner, É.; Kappelmayer, J.; Papp, Z.; Tóth, A.; Fagyas, M. A dramatic rise in serum ACE2 activity in a critically ill COVID-19 patient. Int. J. Infect. Dis. 2021, 103, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Chappell, M.C.; Pirro, N.T.; South, A.M.; Gwathmey, T.M. Concerns on the Specificity of Commercial ELISAs for the Measurement of Angiotensin (1–7) and Angiotensin II in Human Plasma. Hypertension 2021, 77, e29–e31. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, H.R.; Adhikari, S.; Pulgarin, C.; Troxel, A.B.; Iturrate, E.; Johnson, S.B.; Hausvater, A.; Newman, J.D.; Berger, J.S.; Bangalore, S.; et al. Renin–angiotensin–aldosterone system inhibitors and risk of Covid-19. N. Engl. J. Med. 2020, 382, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Mancia, G.; Rea, F.; Ludergnani, M.; Apolone, G.; Corrao, G. Renin–angiotensin–aldosterone system blockers and the risk of Covid-19. N. Engl. J. Med. 2020, 382, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Baral, R.; Tsampasian, V.; Debski, M.; Moran, B.; Garg, P.; Clark, A.; Vassiliou, V.S. Association between Renin-Angiotensin-Aldosterone System Inhibitors and Clinical Outcomes in Patients with COVID-19: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4, e213594. [Google Scholar] [CrossRef]
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 3594. [Google Scholar] [CrossRef]
- Gu, H.; Xie, Z.; Li, T.; Zhang, S.; Lai, C.; Zhu, P.; Wang, K.; Han, L.; Duan, Y.; Zhao, Z.; et al. Angiotensin-converting enzyme 2 inhibits lung injury induced by respiratory syncytial virus. Sci. Rep. 2016, 6, 19840. [Google Scholar] [CrossRef]
- Lei, C.; Qian, K.; Li, T.; Zhang, S.; Fu, W.; Ding, M.; Hu, S. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat. Commun. 2020, 11, 2070. [Google Scholar] [CrossRef] [Green Version]
- Monteil, V.; Dyczynski, M.; Lauschke, V.M.; Kwon, H.; Wirnsberger, G.; Youhanna, S.; Zhang, H.; Slutsky, A.S.; Hurtado Del Pozo, C.; Horn, M.; et al. Human soluble ACE2 improves the effect of remdesivir in SARS-CoV-2 infection. EMBO Mol. Med. 2021, 13, e13426. [Google Scholar] [CrossRef]
- Haschke, M.; Schuster, M.; Poglitsch, M.; Loibner, H.; Salzberg, M.; Bruggisser, M.; Penninger, J.; Krähenbühl, S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin. Pharmacokinet. 2013, 52, 783–792. [Google Scholar] [CrossRef]
- Khan, A.; Benthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care 2017, 21, 234. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Cui, Y.; Zhu, Y. Recombinant human ACE2: Potential therapeutics of SARS-CoV-2 infection and its complication. Acta Pharmacol. Sin. 2020, 41, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Lezutekong, J.N.; Chen, X.; Oudit, G.Y. Recombinant human ACE2 and the angiotensin 1-7 axis as potential new therapies for heart failure. Can. J. Cardiol. 2017, 33, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wysocki, J.; Souma, T.; Ye, M.; Ramirez, V.; Zhou, B.; Wilsbacher, L.D.; Quaggin, S.E.; Batlle, D.; Jin, J. Novel ACE2-Fc chimeric fusion provides long-lasting hypertension control and organ protection in mouse models of systemic renin angiotensin system activation. Kidney Int. 2018, 94, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Talotta, R.; Roberston, E.S. Perspectives: Potential therapeutic approach with inhalation of ACE2-derived peptides for SARS-CoV-2 infection. Am. J. Clin. Exp. Immunol. 2020, 9, 73–80. [Google Scholar] [PubMed]
- Han, Y.; Král, P. Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Pomplun, S.; Loftis, A.R.; Loas, A.; Pentelute, B.L. The first-in-class peptide binder to the SARS-CoV-2 spike protein. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Zambelli, V.; Bellani, G.; Borsa, R.; Pozzi, F.; Grassi, A.; Scanziani, M.; Castiglioni, V.; Masson, S.; Decio, A.; Laffey, J.G.; et al. Angiotensin-(1-7) improves oxygenation, while reducing cellular infiltrate and fibrosis in experimental acute respiratory distress syndrome. Intensive Care Med. Exp. 2015, 3, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wösten-van Asperen, R.M.; Lutter, R.; Specht, P.A.; Moll, G.N.; van Woensel, J.B.; van der Loos, C.M.; van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J. Pathol. 2011, 225, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Issa, H.; Eid, A.H.; Berry, B.; Takhviji, V.; Khosravi, A.; Mantash, S.; Nehme, R.; Hallal, R.; Karaki, H.; Dhayni, K.; et al. Combination of Angiotensin (1-7) Agonists and Convalescent Plasma as a New Strategy to Overcome Angiotensin Converting Enzyme 2 (ACE2) Inhibition for the Treatment of COVID-19. Front. Med. 2021, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Savergnini, S.Q.; Beiman, M.; Lautner, R.Q.; de Paula-Carvalho, V.; Allahdadi, K.; Pessoa, D.C.; Costa-Fraga, F.P.; Fraga-Silva, R.A.; Cojocaru, G.; Cohen, Y.; et al. Vascular relaxation, antihypertensive effect, and cardioprotection of a novel peptide agonist of the MAS receptor. Hypertension 2010, 56, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Povlsen, A.L.; Grimm, D.; Wehland, M.; Infanger, M.; Krüger, M. The vasoactive Mas receptor in essential hypertension. J. Clin. Med. 2020, 9, 267. [Google Scholar] [CrossRef] [Green Version]
- Namsolleck, P.; Moll, G.N. Does activation of the protective Renin-Angiotensin System have therapeutic potential in COVID-19? Mol. Med. 2020, 26, 80. [Google Scholar] [CrossRef]
- Jiang, F.; Yang, J.; Zhang, Y.; Dong, M.; Wang, S.; Zhang, Q.; Liu, F.F.; Zhang, K.; Zhang, C. Angiotensin-converting enzyme 2 and angiotensin 1–7: Novel therapeutic targets. Nat. Rev. Cardiol. 2014, 11, 413–426. [Google Scholar] [CrossRef]
- Palau, V.; Riera, M.; Soler, M.J. ADAM17 inhibition may exert a protective effect on COVID-19. Nephrol. Dial. Transplant. 2020, 35, 1071–1072. [Google Scholar] [CrossRef]
- Yuan, S.; Wang, R.; Chan, J.F.W.; Zhang, A.J.; Cheng, T.; Chik, K.K.H.; Ye, Z.W.; Wang, S.; Lee, A.C.Y.; Jin, L.; et al. Metallodrug ranitidine bismuth citrate suppresses SARS-CoV-2 replication and relieves virus-associated pneumonia in Syrian hamsters. Nat. Microbiol. 2020, 5, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Lartey, N.L.; Valle-Reyes, S.; Vargas-Robles, H.; Jiménez-Camacho, K.E.; Guerrero-Fonseca, I.M.; Castellanos-Martínez, R.; Montoya-García, A.; García-Cordero, J.; Cedillo-Barrón, L.; Nava, P.; et al. ADAM17 inhibition prevents neutrophilia and lung injury in a mouse model of Covid-19. bioRxiv 2021. [Google Scholar] [CrossRef]
- Deshotels, M.R.; Xia, H.; Sriramula, S.; Lazartigues, E.; Filipeanu, C.M. Angiotensin-II mediates ACE2 Internalization and Degradation through an Angiotensin-II type I receptor-dependent mechanism. Hypertension 2014, 64, 1368–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, U.O.; Kintscher, U. ACE2 and SARS-CoV-2: Tissue or Plasma, Good or Bad? Am. J. Hypertens. 2021, 34, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Ocaranza, M.P.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| RAS Component | COVID-19 Effect | p-Value | N |
|---|---|---|---|
| ACE | −10% | - | 124 |
| ACE2 | +158% | < | 1024 |
| AngI | −22% | - | 263 |
| AngII | +74% | ∼ | 840 |
| Ang-(1-7) | +1100% | < | 602 |
| Ang-(1-5) | −28% | - | 53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pucci, F.; Annoni, F.; dos Santos, R.A.S.; Taccone, F.S.; Rooman, M. Quantifying Renin-Angiotensin-System Alterations in COVID-19. Cells 2021, 10, 2755. https://doi.org/10.3390/cells10102755
Pucci F, Annoni F, dos Santos RAS, Taccone FS, Rooman M. Quantifying Renin-Angiotensin-System Alterations in COVID-19. Cells. 2021; 10(10):2755. https://doi.org/10.3390/cells10102755
Chicago/Turabian StylePucci, Fabrizio, Filippo Annoni, Robson Augusto Souza dos Santos, Fabio Silvio Taccone, and Marianne Rooman. 2021. "Quantifying Renin-Angiotensin-System Alterations in COVID-19" Cells 10, no. 10: 2755. https://doi.org/10.3390/cells10102755
APA StylePucci, F., Annoni, F., dos Santos, R. A. S., Taccone, F. S., & Rooman, M. (2021). Quantifying Renin-Angiotensin-System Alterations in COVID-19. Cells, 10(10), 2755. https://doi.org/10.3390/cells10102755