
Sildenafil-Mediated Neuroprotection from Adult to Neonatal Brain Injury: Evidence, Mechanisms, and Future Translation

Abstract
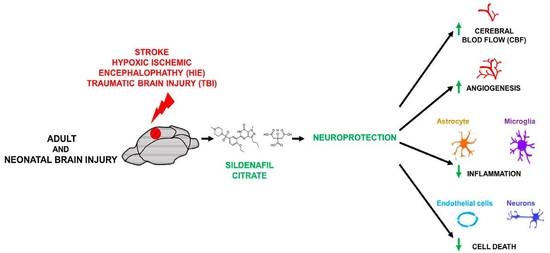
:
1. Introduction
2. Materials and Methods
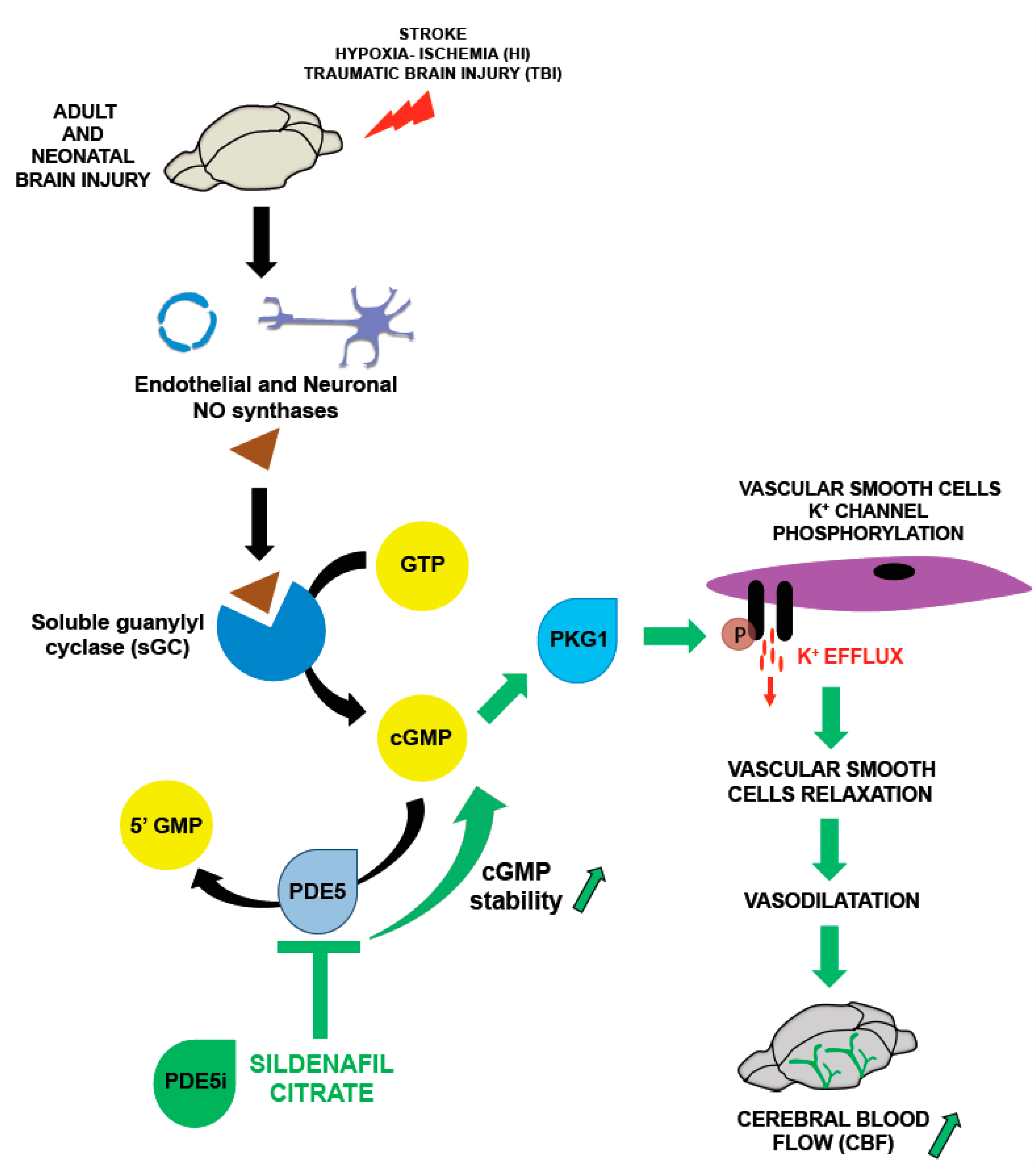
3. Nitric Oxide Pathway and Sildenafil: Key Modulators of Cerebral Blood Flow (CBF)
3.1. Nitric Oxide and CBF
3.2. Phosphodiesterases (PDEs) and CBF: A Key Role for Sildenafil
4. Other Effects of Sildenafil in the Brain
4.1. Angiogenesis
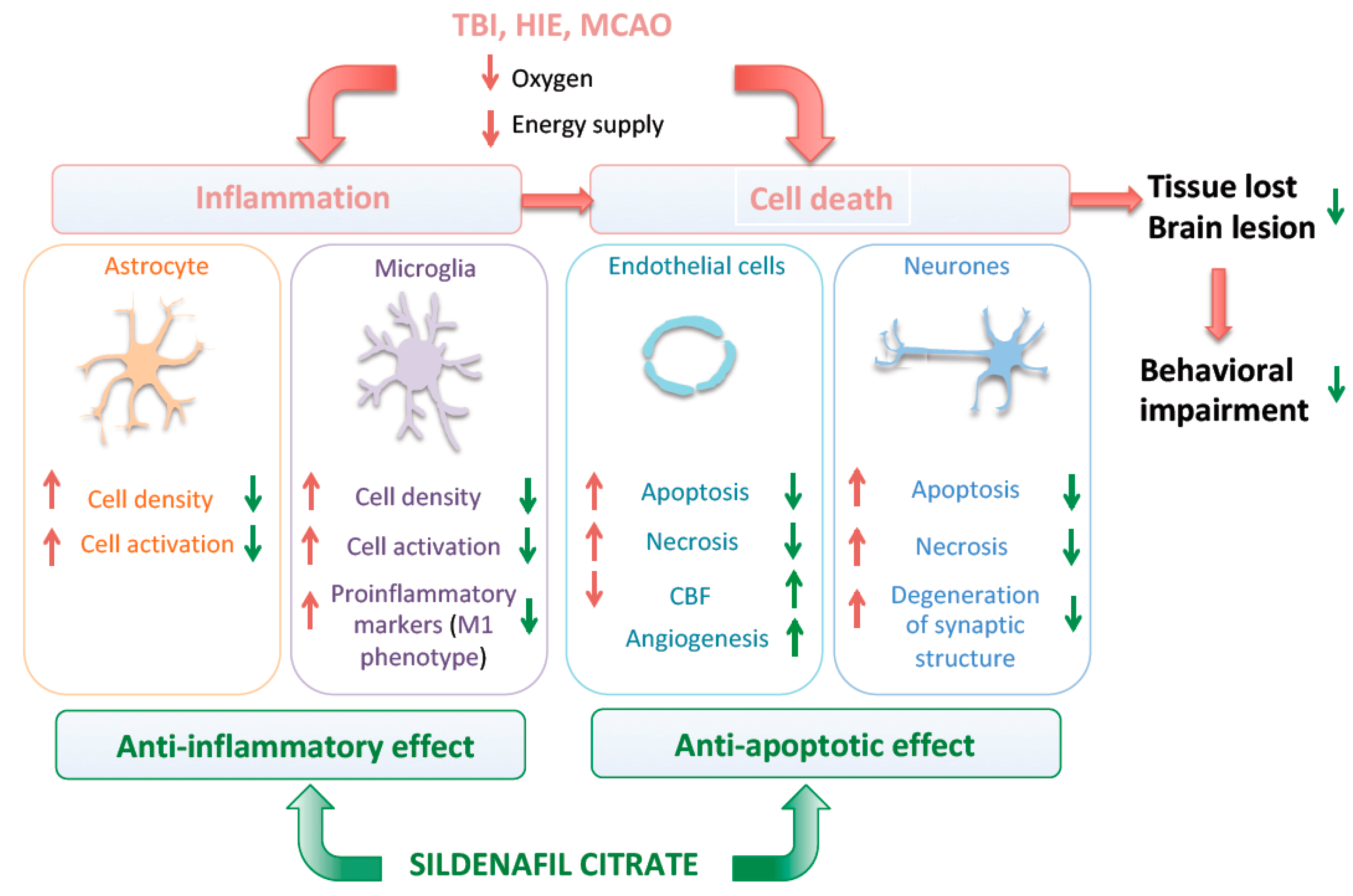
4.2. Neuro-Inflammation
4.3. Cell Death
4.4. Neurogenesis
5. Sildenafil Exposure and Behavioral Outcomes in Response to Brain Injury
6. Sex Difference in Brain Injury Outcome
7. Other PDE5 Inhibitors as Potential Neuroprotectors in Preclinical Models
8. Main Human Brain Injuries in Adults and Neonates as Potential Targets for Sildenafil
8.1. Adult Brain Injury
8.2. Neonatal Brain Injury
9. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- James, S.L.; Theadom, A.; Ellenbogen, R.G.; Bannick, M.S.; Montjoy-Venning, W.; Lucchesi, L.R.; Abbasi, N.; Abdulkader, R.; Abraha, H.N.; Adsuar, J.C.; et al. Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 56–87. [Google Scholar] [CrossRef] [Green Version]
- Clive, B.; Vincer, M.; Ahmad, T.; Khan, N.; Afifi, J.; El-Naggar, W. Epidemiology of neonatal stroke: A population-based study. Paediatr. Child Health 2019, 25, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Wafa, H.A.; Wolfe, C.D.; Emmett, E.; Roth, G.A.; Johnson, C.O.; Wang, Y. Burden of Stroke in Europe. Stroke 2020, 51, 2418–2427. [Google Scholar] [CrossRef]
- Toda, N.; Ayajiki, K.; Okamura, T. Cerebral Blood Flow Regulation by Nitric Oxide: Recent Advances. Pharmacol. Rev. 2009, 61, 62–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, C.J.; Blood, A.B.; White, C.R.; Pearce, W.J.; Power, G.G. Role of Nitric Oxide in Hypoxic Cerebral Vasodilatation in the Ovine Fetus. J. Physiol. 2003, 549, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Hortobágyi, L.; Kis, B.; Hrabák, A.; Horvath, B.; Huszty, G.; Schweer, H.; Benyó, B.; Sándor, P.; Busija, D.W.; Benyó, Z. Adaptation of the hypothalamic blood flow to chronic nitric oxide deficiency is independent of vasodilator prostanoids. Brain Res. 2007, 1131, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoccoli, G.; Grant, D.A.; Wild, J.; Walker, A.M. Nitric oxide inhibition abolishes sleep-wake differences in cerebral circulation. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2598–H2606. [Google Scholar] [CrossRef]
- Dayoub, H.; Rodionov, R.N.; Lynch, C.; Cooke, J.P.; Arning, E.; Bottiglieri, T.; Lentz, S.R.; Faraci, F.M. Overexpression of Dimethylarginine Dimethylaminohydrolase Inhibits Asymmetric Dimethylarginine–Induced Endothelial Dysfunction in the Cerebral Circulation. Stroke 2008, 39, 180–184. [Google Scholar] [CrossRef] [Green Version]
- Coumans, A.; Garnier, Y.; Supçun, S.; Jensen, A.; Hasaart, T.H.; Berger, R. The role of nitric oxide on fetal cardiovascular control during normoxia and acute hypoxia in 0.75 gestation sheep. J. Soc. Gynecol. Investig. 2003, 10, 275–282. [Google Scholar] [CrossRef]
- Hlatky, R.; Lui, H.; Cherian, L.; Goodman, J.C.; O’Brien, W.E.; Contant, C.F.; Robertson, C.S. The Role of Endothelial Nitric Oxide Synthase in the Cerebral Hemodynamics after Controlled Cortical Impact Injury in Mice. J. Neurotrauma 2003, 20, 995–1006. [Google Scholar] [CrossRef]
- Barnes, M.; Brisbois, E.J. Clinical use of inhaled nitric oxide: Local and systemic applications. Free. Radic. Biol. Med. 2020, 152, 422–431. [Google Scholar] [CrossRef]
- Terpolilli, N.A.; Kim, S.-W.; Thal, S.C.; Kataoka, H.; Zeisig, V.; Nitzsche, B.; Klaesner, B.; Zhu, C.; Schwarzmaier, S.; Meissner, L.; et al. Inhalation of Nitric Oxide Prevents Ischemic Brain Damage in Experimental Stroke by Selective Dilatation of Collateral Arterioles. Circ. Res. 2012, 110, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Sun, Y.; Gao, J.; Wang, X.; Plesnila, N.; Blomgren, K. Inhaled Nitric Oxide Protects Males But not Females from Neonatal Mouse Hypoxia-Ischemia Brain Injury. Transl. Stroke Res. 2012, 4, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Charriaut-Marlangue, C.; Bonnin, P.; Gharib, A.; Leger, P.-L.; Villapol, S.; Pocard, M.; Gressens, P.; Renolleau, S.; Baud, O. Inhaled Nitric Oxide Reduces Brain Damage by Collateral Recruitment in a Neonatal Stroke Model. Stroke 2012, 43, 3078–3084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate–nitrite–nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 2008, 7, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends Neurosci. 1991, 14, 60–67. [Google Scholar] [CrossRef]
- Hollas, M.A.; Ben Aissa, M.; Lee, S.H.; Gordon-Blake, J.M.; Thatcher, G.R.J. Pharmacological manipulation of cGMP and NO/cGMP in CNS drug discovery. Nitric Oxide 2019, 82, 59–74. [Google Scholar] [CrossRef]
- Ghalayini, I.F. Nitric oxide-cyclic GMP pathway with some emphasis on cavernosal contractility. Int. J. Impot. Res. 2004, 16, 459–469. [Google Scholar] [CrossRef] [Green Version]
- Greene, S.J.; Gheorghiade, M.; Borlaug, B.A.; Pieske, B.; Vaduganathan, M.; Burnett, J.C.; Roessig, L.; Stasch, J.; Solomon, S.D.; Paulus, W.J.; et al. The cGMP Signaling Pathway as a Therapeutic Target in Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2013, 2, e000536. [Google Scholar] [CrossRef] [Green Version]
- Menniti, F.; Faraci, W.S.; Schmidt, C.J. Phosphodiesterases in the CNS: Targets for drug development. Nat. Rev. Drug Discov. 2006, 5, 660–670. [Google Scholar] [CrossRef]
- Picón-Pagès, P.; Garcia-Buendia, J.; Muñoz, F.J. Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef]
- Liu, H.; Manganiello, V.; Waleh, N.; Clyman, I.R. Expression, Activity, and Function of Phosphodiesterases in the Mature and Immature Ductus Arteriosus. Pediatr. Res. 2008, 64, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Kruuse, C.; Khurana, T.S.; Rybalkin, S.D.; Birk, S.; Engel, U.; Edvinsson, L.; Olesen, J. Phosphodiesterase 5 and effects of sildenafil on cerebral arteries of man and guinea pig. Eur. J. Pharmacol. 2005, 521, 105–114. [Google Scholar] [CrossRef]
- Osterloh, I.H. The discovery and development of Viagra® (sildenafil citrate). In Sildenafil; Birkhäuser: Basel, Switzerland, 2004; pp. 1–13. ISBN 978-3-0348-7945-3. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Osterloh, I.H.; Grimminger, F. Sildenafil: From angina to erectile dysfunction to pulmonary hypertension and beyond. Nat. Rev. Drug Discov. 2006, 5, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, V.G.; Ugarte, A.; García-Barroso, C.; Cuadrado-Tejedor, M.; Szczupak, B.; Reyes, I.G.-D.; Lanciego, J.; Garcia-Osta, A.; Llop, J.; Oyarzabal, J.; et al. Pharmacokinetic investigation of sildenafil using positron emission tomography and determination of its effect on cerebrospinal fluid cGMP levels. J. Neurochem. 2015, 136, 403–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salloum, F.; Abbate, A.; Das, A.; Houser, J.-E.; Mudrick, C.A.; Qureshi, I.Z.; Hoke, N.N.; Roy, S.K.; Brown, W.R.; Prabhakar, S.; et al. Sildenafil (Viagra) attenuates ischemic cardiomyopathy and improves left ventricular function in mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 294, H1398–H1406. [Google Scholar] [CrossRef] [PubMed]
- Salloum, F.; Ockaili, R.; Wittkamp, M.; Marwaha, V.; Kukreja, R. Vardenafil: A novel type 5 phosphodiesterase inhibitor reduces myocardial infarct size following ischemia/reperfusion injury via opening of mitochondrial KATP channels in rabbits. J. Mol. Cell. Cardiol. 2006, 40, 405–411. [Google Scholar] [CrossRef]
- Schwartz, B.G.; Levine, L.A.; Comstock, G.; Stecher, V.J.; Kloner, R.A. Cardiac Uses of Phosphodiesterase-5 Inhibitors. J. Am. Coll. Cardiol. 2012, 59, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Ding, G.; Jiang, Q.; Li, L.; Zhang, L.; Zhang, Z.; Lu, M.; Li, Q.; Gu, S.; Ewing, J.; Chopp, M. Longitudinal Magnetic Resonance Imaging of Sildenafil Treatment of Embolic Stroke in Aged Rats. Stroke 2011, 42, 3537–3541. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, Q.; Zhang, L.; Ding, G.; Zhang, Z.G.; Li, Q.; Ewing, J.R.; Lü, M.; Panda, S.; Ledbetter, K.A.; et al. Angiogenesis and improved cerebral blood flow in the ischemic boundary area detected by MRI after administration of sildenafil to rats with embolic stroke. Brain Res. 2007, 1132, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, G.; Jiang, Q.; Li, L.; Zhang, L.; Zhang, Z.G.; Ledbetter, A.K.; Panda, S.; Nejad-Davarani, S.; Athiraman, H.; Li, Q.; et al. Magnetic Resonance Imaging Investigation of Axonal Remodeling and Angiogenesis after Embolic Stroke in Sildenafil-Treated Rats. Br. J. Pharmacol. 2008, 28, 1440–1448. [Google Scholar] [CrossRef]
- Charriaut-Marlangue, C.; Nguyen, T.; Bonnin, P.; Duy, A.P.; Leger, P.-L.; Csaba, Z.; Pansiot, J.; Bourgeois, T.; Renolleau, S.; Baud, O. Sildenafil Mediates Blood-Flow Redistribution and Neuroprotection after Neonatal Hypoxia-Ischemia. Stroke 2014, 45, 850–856. [Google Scholar] [CrossRef] [Green Version]
- Ziche, M.; Morbidelli, L.; Choudhuri, R.; Zhang, H.T.; Donnini, S.; Granger, H.J.; Bicknell, R. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J. Clin. Investig. 1997, 99, 2625–2634. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Wang, L.; Zhang, L.; Chen, J.; Zhu, Z.; Zhang, Z.; Chopp, M. Nitric Oxide Enhances Angiogenesis via the Synthesis of Vascular Endothelial Growth Factor and cGMP After Stroke in the Rat. Circ. Res. 2003, 92, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Morbidelli, L.; Chang, C.H.; Douglas, J.G.; Granger, H.J.; Ledda, F.; Ziche, M. Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. Am. J. Physiol. Circ. Physiol. 1996, 270, H411–H415. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; García-Cardeña, G.; Madri, A.J.; Sessa, W.C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Investig. 1997, 100, 3131–3139. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.-O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyriochou, A.; Zhou, Z.; Koika, V.; Petrou, C.; Cordopatis, P.; Sessa, W.C.; Papapetropoulos, A. The phosphodiesterase 5 inhibitor sildenafil stimulates angiogenesis through a protein kinase G/MAPK pathway. J. Cell. Physiol. 2007, 211, 197–204. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, R.L.; Wang, Y.; Zhang, C.; Zhang, Z.G.; Meng, H.; Chopp, M. Functional Recovery in Aged and Young Rats After Embolic Stroke: Treatment with a phosphodiesterase type 5 inhibitor. Stroke 2005, 36, 847–852. [Google Scholar] [CrossRef] [Green Version]
- Olivier, P.; Baud, O.; Bouslama, M.; Evrard, P.; Gressens, P.; Verney, C. Moderate growth restriction: Deleterious and protective effects on white matter damage. Neurobiol. Dis. 2007, 26, 253–263. [Google Scholar] [CrossRef]
- Prado, J.; Pifarre, P.; Giralt, M.; Hidalgo, J.; Garcia, A. Metallothioneins I/II are involved in the neuroprotective effect of sildenafil in focal brain injury. Neurochem. Int. 2013, 62, 70–78. [Google Scholar] [CrossRef]
- Yazdani, A.; Khoja, Z.; Johnstone, A.; Dale, L.; Rampakakis, E.; Wintermark, P. Sildenafil Improves Brain Injury Recovery following Term Neonatal Hypoxia-Ischemia in Male Rat Pups. Dev. Neurosci. 2016, 38, 251–263. [Google Scholar] [CrossRef]
- Chen, W.; Jadhav, V.; Tang, J.; Zhang, J.H. HIF-1α inhibition ameliorates neonatal brain injury in a rat pup hypoxic–ischemic model. Neurobiol. Dis. 2008, 31, 433–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzietko, M.; Derugin, N.; Wendland, M.F.; Vexler, Z.S.; Ferriero, D.M. Delayed VEGF Treatment Enhances Angiogenesis and Recovery after Neonatal Focal Rodent Stroke. Transl. Stroke Res. 2013, 4, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, H.; Lechpammer, M.; Jensen, F.E.; Warfield, S.K.; Hansen, A.H.; Kosaras, B.; Shevell, M.; Wintermark, P. Increased Brain Perfusion Persists over the First Month of Life in Term Asphyxiated Newborns Treated with Hypothermia: Does it Reflect Activated Angiogenesis? Transl. Stroke Res. 2015, 6, 224–233. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 2015, 173, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Lu, J.; Shao, A.; Zhang, J.H.; Zhang, J. Glial Cells: Role of the Immune Response in Ischemic Stroke. Front. Immunol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Jones, K.; Zouikr, I.; Patience, M.; Clarkson, A.; Isgaard, J.; Johnson, S.J.; Spratt, N.; Nilsson, M.; Walker, F.R. Chronic stress exacerbates neuronal loss associated with secondary neurodegeneration and suppresses microglial-like cells following focal motor cortex ischemia in the mouse. Brain Behav. Immun. 2015, 48, 57–67. [Google Scholar] [CrossRef]
- Langen, K.-J.; Salber, D.; Hamacher, K.; Stoffels, G.; Reifenberger, G.; Pauleit, D.; Coenen, H.H.; Zilles, K. Detection of Secondary Thalamic Degeneration After Cortical Infarction Using cis-4-18F-Fluoro- D-Proline. J. Nucl. Med. 2007, 48, 1482–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patience, M.J.; Zouikr, I.; Jones, K.; Clarkson, A.; Isgaard, J.; Johnson, S.J.; Walker, F.R.; Nilsson, M. Photothrombotic Stroke Induces Persistent Ipsilateral and Contralateral Astrogliosis in Key Cognitive Control Nuclei. Neurochem. Res. 2014, 40, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Morrison, H.W.; Filosa, J.A. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J. Neuroinflamm. 2013, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denés, A.; Vidyasagar, R.; Feng, J.; Narvainen, J.; McColl, B.W.; Kauppinen, R.A.; Allan, S.M. Proliferating Resident Microglia after Focal Cerebral Ischaemia in Mice. J. Cereb. Blood Flow Metab. 2007, 27, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.M.; Wang, C.-K.; Kuo, J.S.; Lin, T.-N. Changes in the level of glial fibrillary acidic protein (GFAP) after mild and severe focal cerebral ischemia. Chin. J. Physiol. 1999, 42, 227–235. [Google Scholar]
- Clausen, B.H.; Lambertsen, K.L.; Babcock, A.A.; Holm, T.H.; Dagnaes-Hansen, F.; Finsen, B. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J. Neuroinflamm. 2008, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Gregersen, R.; Lambertsen, K.; Finsen, B. Microglia and Macrophages Are the Major Source of Tumor Necrosis Factor in Permanent Middle Cerebral Artery Occlusion in Mice. Br. J. Pharmacol. 2000, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Dziewulska, D.; Mossakowski, M.J. Cellular expression of tumor necrosis factor a and its receptors in human ischemic stroke. Clin. Neuropathol. 2003, 22, 35–40. [Google Scholar]
- Nowicka, D.; Rogozinska, K.; Aleksy, M.; Witte, O.W.; Skangiel-Kramska, J. Spatiotemporal dynamics of astroglial and microglial responses after photothrombotic stroke in the rat brain. Acta Neurobiol. Exp. 2008, 68, 155–168. [Google Scholar]
- Venkat, P.; Chopp, M.; Zacharek, A.; Cui, C.; Landschoot-Ward, J.; Qian, Y.; Chen, Z.; Chen, J. Sildenafil treatment of vascular dementia in aged rats. Neurochem. Int. 2019, 127, 103–112. [Google Scholar] [CrossRef]
- Moretti, R.; Leger, P.-L.; Besson, V.; Csaba, Z.; Pansiot, J.; Di Criscio, L.; Gentili, A.; Titomanlio, L.; Bonnin, P.; Baud, O.; et al. Sildenafil, a cyclic GMP phosphodiesterase inhibitor, induces microglial modulation after focal ischemia in the neonatal mouse brain. J. Neuroinflamm. 2016, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Solaroglu, I.; Gürsoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 2018, 16, 1396–1415. [Google Scholar] [CrossRef]
- Chen, X.-M.; Wang, N.-N.; Zhang, T.-Y.; Wang, F.; Wu, C.-F.; Yang, J.-Y. Neuroprotection by Sildenafil: Neuronal Networks Potentiation in Acute Experimental Stroke. CNS Neurosci. Ther. 2013, 20, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Barros-Miñones, L.; Orejana, L.; Goñi-Allo, B.; Suquía, V.; Hervías, I.; Aguirre, N.; Puerta, E. Modulation of the ASK1-MKK3/6-p38/MAPK signalling pathway mediates sildenafil protection against chemical hypoxia caused by malonate. Br. J. Pharmacol. 2013, 168, 1820–1834. [Google Scholar] [CrossRef] [Green Version]
- Kilicarslan, B.; Kilicarslan, E.; Kizmazoglu, C.; Aydin, H.E.; Kaya, I.; Danyeli, A.E.; Karabekir, H.S. Evaluation of the Efficacy of Sildenafil Citrate Following Severe Head Trauma in an Experimental Rat Model. Turk. Neurosurg. 2019, 30, 501–506. [Google Scholar] [CrossRef]
- Arvidsson, A.; Collin, T.; Kirik, D.; Kokaia, Z.; Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002, 8, 963–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parent, J.M.; Vexler, Z.S.; Gong, C.; Ma, N.D.; Ferriero, D.M. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann. Neurol. 2002, 52, 802–813. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Z.; Wang, L.; Wang, Y.; Gousev, A.; Zhang, L.; Ho, K.-L.; Morshead, C.; Chopp, M. Activated Neural Stem Cells Contribute to Stroke-Induced Neurogenesis and Neuroblast Migration toward the Infarct Boundary in Adult Rats. Br. J. Pharmacol. 2004, 24, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhang, Z.; Zhang, C.; Zhang, L.; Robin, A.; Wang, Y.; Lu, M.; Chopp, M. Stroke Transiently Increases Subventricular Zone Cell Division from Asymmetric to Symmetric and Increases Neuronal Differentiation in the Adult Rat. J. Neurosci. 2004, 24, 5810–5815. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.; Zhang, L.; Zhang, Z.; Tsang, W.; Lu, M.; Zhang, L.; Chopp, M. Sildenafil (Viagra) Induces Neurogenesis and Promotes Functional Recovery after Stroke in Rats. Stroke 2002, 33, 2675–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.L.; Chopp, M.; Roberts, C.; Wei, M.; Wang, X.; Liu, X.; Lu, M.; Zhang, Z.G. Sildenafil Enhances Neurogenesis and Oligodendrogenesis in Ischemic Brain of Middle-Aged Mouse. PLoS ONE 2012, 7, e48141. [Google Scholar] [CrossRef]
- Kirton, A.; DeVeber, G. Life after Perinatal Stroke. Stroke 2013, 44, 3265–3271. [Google Scholar] [CrossRef] [Green Version]
- Al-Qazzaz, N.; Ali, S.H.M.; Ahmad, S.A.; Islam, S.; Mohamad, K. Cognitive impairment and memory dysfunction after a stroke diagnosis: A post-stroke memory assessment. Neuropsychiatr. Dis. Treat. 2014, 10, 1677–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutkenhoff, E.S.; Wright, M.J.; Shrestha, V.; Real, C.; McArthur, D.L.; Buitrago-Blanco, M.; Vespa, P.M.; Monti, M.M. The subcortical basis of outcome and cognitive impairment in TBI. Longitud. Cohort Study 2020, 95, e2398–e2408. [Google Scholar] [CrossRef]
- Korzeniewski, S.J.; Slaughter-Acey, J.; Lenski, M.; Haak, P.; Paneth, N. The complex aetiology of cerebral palsy. Nat. Rev. Neurol. 2018, 14, 528–543. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W. Perinatal Stroke in Children with Motor Impairment: A Population-Based Study. Pediatrics 2004, 114, 612–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uvebrant, P. Hemiplegic cerebral palsy aetiology and outcome. Acta Paediatr. Scand. Suppl. 1988, 77, 1–100. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, N.; Faingold, R.; Daneman, A.; Epelman, M. Intraventricular hemorrhage in term neonates with hypoxic-ischemic encephalopathy: A comparison study between neonates treated with and without hypothermia. Quant. Imaging Med. Surg. 2016, 6, 504–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvis, S.; Glinianaia, S.V.; Arnaud, C.; Fauconnier, J.; Johnson, A.; McManus, V.; Topp, M.; Uvebrant, P.; Cans, C.; Krägeloh-Mann, I. Case gender and severity in cerebral palsy varies with intrauterine growth. Arch. Dis. Child. 2005, 90, 474–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, M.V.; Hagberg, H. Sex and the pathogenesis of cerebral palsy. Dev. Med. Child Neurol. 2006, 49, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Murden, S.; Borbélyová, V.; Laštůvka, Z.; Myslivecek, J.; Otáhal, J.; Riljak, V. Gender Differences Involved in the Pathophysiology of the Perinatal Hypoxic-Ischemic Damage. Physiol. Res. 2019, 68, S207–S217. [Google Scholar] [CrossRef]
- Covassin, T.; Schatz, P.; Swanik, C.B. Sex differences in neuropsychological function and post-concussion symptoms of concussed collegiate athletes. Neurosurgery 2007, 61, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasserman, E.; Kerr, Z.Y.; Zuckerman, S.L.; Covassin, T. Epidemiology of Sports-Related Concussions in National Collegiate Athletic Association Athletes from 2009–2010 to 2013–2014: Symptom Prevalence, Symptom Resolution Time, and Return-to-Play Time. Am. J. Sports Med. 2015, 44, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Sanches, E.; Arteni, N.; Nicola, F.; Boisserand, L.; Willborn, S.; Netto, C. Early hypoxia–ischemia causes hemisphere and sex-dependent cognitive impairment and histological damage. Neuroscience 2013, 237, 208–215. [Google Scholar] [CrossRef]
- Huang, H.-Z.; Wen, X.-H.; Liu, H. Sex differences in brain MRI abnormalities and neurodevelopmental outcomes in a rat model of neonatal hypoxia-ischemia. Int. J. Neurosci. 2016, 126, 647–657. [Google Scholar] [CrossRef]
- Mayoral, S.R.; Omar, G.; Penn, A.A. Sex Differences in a Hypoxia Model of Preterm Brain Damage. Pediatr. Res. 2009, 66, 248–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, C.A.; Cernak, I.; Vink, R. Interaction between Anesthesia, Gender, and Functional Outcome Task following Diffuse Traumatic Brain Injury in Rats. J. Neurotrauma 2003, 20, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Emerson, C.S.; Headrick, J.P.; Vink, R. Estrogen improves biochemical and neurologic outcome following traumatic brain injury in male rats, but not in females. Brain Res. 1993, 608, 95–100. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Dimayuga, F.O.; Reed, J.L.; Wang, C.; Angers, R.; Wilson, M.E.; Dimayuga, V.M.; Scheff, S.W. Gender and Estrogen Manipulation Do Not Affect Traumatic Brain Injury in Mice. J. Neurotrauma 2007, 24, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Roof, R.L.; Duvdevani, R.; Stein, D.G. Gender influences outcome of brain injury: Progesterone plays a protective role. Brain Res. 1993, 607, 333–336. [Google Scholar] [CrossRef]
- Roof, R.L.; Hall, E.D. Estrogen-Related Gender Difference in Survival Rate and Cortical Blood Flow after Impact-Acceleration Head Injury in Rats. J. Neurotrauma 2000, 17, 1155–1169. [Google Scholar] [CrossRef]
- Charriaut-Marlangue, C.; Besson, V.C.; Baud, O. Sexually Dimorphic Outcomes after Neonatal Stroke and Hypoxia-Ischemia. Int. J. Mol. Sci. 2017, 19, 61. [Google Scholar] [CrossRef] [Green Version]
- Ölmestig, J.N.; Marlet, I.R.; Hainsworth, A.H.; Kruuse, C. Phosphodiesterase 5 inhibition as a therapeutic target for ischemic stroke: A systematic review of preclinical studies. Cell. Signal. 2017, 38, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, R.; Umekawa, T.; Yoshikawa, K.; Owa, T.; Magawa, S.; Furuhashi, F.; Tsuji, M.; Maki, S.; Shimada, K.; Kaneda, M.K.; et al. Tadalafil treatment in mice for preeclampsia with fetal growth restriction has neuro-benefic effects in offspring through modulating prenatal hypoxic conditions. Sci. Rep. 2019, 9, 234. [Google Scholar] [CrossRef] [PubMed]
- Taek, K.K.; Jin, C.K.; Sae, L.H.; Gyu, K.I.; Ju, K.C.; Gil, N.Y.; Hawn, K.K. Neuroprotective effects of tadalafil on gerbil dopaminergic neurons following cerebral ischemia. Neural Regen. Res. 2013, 8, 693–701. [Google Scholar] [CrossRef]
- Gulati, P.; Singh, N. Neuroprotective effect of tadalafil, a PDE-5 inhibitor, and its modulation by L-NAME in mouse model of ischemia-reperfusion injury. J. Surg. Res. 2014, 186, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.; Zhang, R.L.; Cui, Y.; LaPointe, M.C.; Silver, B.; Chopp, M. Tadalafil, a long-acting type 5 phosphodiesterase isoenzyme inhibitor, improves neurological functional recovery in a rat model of embolic stroke. Brain Res. 2006, 1118, 192–198. [Google Scholar] [CrossRef]
- Gulati, P.; Singh, N. Tadalafil enhances the neuroprotective effects of ischemic postconditioning in mice, probably in a nitric oxide associated manner. Can. J. Physiol. Pharmacol. 2014, 92, 418–426. [Google Scholar] [CrossRef]
- Ko, I.-G.; Shin, M.-S.; Kim, B.-K.; Kim, S.-E.; Sung, Y.-H.; Kim, T.-S.; Shin, M.-C.; Cho, H.-J.; Kim, S.-C.; Kim, S.H.; et al. Tadalafil improves short-term memory by suppressing ischemia-induced apoptosis of hippocampal neuronal cells in gerbils. Pharmacol. Biochem. Behav. 2009, 91, 629–635. [Google Scholar] [CrossRef]
- Gao, F.; Sugita, M.; Nukui, H. Phosphodiesterase 5 inhibitor, zaprinast, selectively increases cerebral blood flow in the ischemic penumbra in the rat brain. Neurol. Res. 2005, 27, 638–643. [Google Scholar] [CrossRef]
- Chen, X.; Wang, N.; Liu, Y.; Liu, Y.; Zhang, T.; Zhu, L.; Wang, Y.; Wu, C.; Yang, J. Yonkenafil: A novel phosphodiesterase type 5 inhibitor induces neuronal network potentiation by a cGMP-dependent Nogo-R axis in acute experimental stroke. Exp. Neurol. 2014, 261, 267–277. [Google Scholar] [CrossRef]
- Iskar, M.; Zeller, G.; Blattmann, P.; Campillos, M.; Kuhn, M.; Kaminska, K.H.; Runz, H.; Gavin, A.-C.; Pepperkok, R.; van Noort, V.; et al. Characterization of drug-induced transcriptional modules: Towards drug repositioning and functional understanding. Mol. Syst. Biol. 2013, 9, 662. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Tonai-Kachi, H.; Shinjo, K. Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett. 2006, 580, 5003–5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosi, C.; Mannaioni, G.; Cozzi, A.; Carlà, V.; Sili, M.; Cavone, L.; Maratea, D.; Moroni, F. G-protein coupled receptor 35 (GPR35) activation and inflammatory pain: Studies on the antinociceptive effects of kynurenic acid and zaprinast. Neuropharmacology 2011, 60, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Ekker, M.S.; Boot, E.M.; Singhal, A.B.; Tan, K.S.; Debette, S.; Tuladhar, A.M.; de Leeuw, F.-E. Epidemiology, aetiology, and management of ischaemic stroke in young adults. Lancet Neurol. 2018, 17, 790–801. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.-C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2019, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, M.G.; Tong, X.; Bowman, B.A. Prevalence of Cardiovascular Risk Factors and Strokes in Younger Adults. JAMA Neurol. 2017, 74, 695–703. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Pathophysiology of Cerebral Ischemia and Brain Trauma: Similarities and Differences. Br. J. Pharmacol. 2004, 24, 133–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandera, E.; Botteri, M.; Minelli, C.; Sutton, A.; Abrams, K.; Latronico, N. Cerebral Blood Flow Threshold of Ischemic Penumbra and Infarct Core in Acute Ischemic Stroke. Stroke 2006, 37, 1334–1339. [Google Scholar] [CrossRef] [Green Version]
- Rostami, E.; Nilsson, P.; Enblad, P. Cerebral Blood Flow Measurement in Healthy Children and Children Suffering Severe Traumatic Brain Injury—What Do We Know? Front. Neurol. 2020, 11, 274. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef]
- Kaur, P.; Sharma, S. Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr. Neuropharmacol. 2018, 16, 1224–1238. [Google Scholar] [CrossRef]
- Rostami, E.; Engquist, H.; Enblad, P. Imaging of Cerebral Blood Flow in Patients with Severe Traumatic Brain Injury in the Neurointensive Care. Front. Neurol. 2014, 5, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, D.; Darby, J.; Yonas, H. Acute regional cerebral blood flow changes caused by severe head injuries. J. Neurosurg. 1991, 74, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Adelson, D.; Clyde, B.; Kochanek, P.; Wisniewski, S.; Marion, D.; Yonas, H. Cerebrovascular Response in Infants and Young Children following Severe Traumatic Brain Injury: A Preliminary Report. Pediatr. Neurosurg. 1997, 26, 200–207. [Google Scholar] [CrossRef]
- Adelson, P.D.; Srinivas, R.; Chang, Y.; Bell, M.; Kochanek, P.M. Cerebrovascular response in children following severe traumatic brain injury. Child’s Nerv. Syst. 2011, 27, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.A.; Brandon, D.H. Hypoxic Ischemic Encephalopathy: Pathophysiology and Experimental Treatments. Newborn Infant Nurs. Rev. 2011, 11, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Adén, U. Neonatal Stroke Is Not a Harmless Condition. Stroke 2009, 40, 1948–1949. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.B. Perinatal Ischemic Stroke. Stroke 2007, 38, 742–745. [Google Scholar] [CrossRef] [Green Version]
- Fernández-López, D.; Natarajan, N.; Ashwal, S.; Vexler, Z.S. Mechanisms of Perinatal Arterial Ischemic Stroke. Br. J. Pharmacol. 2014, 34, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.K.; Nelson, K.B. Epidemiology of perinatal stroke. Curr. Opin. Pediatr. 2001, 13, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Raju, T.N.; Nelson, K.B.; Ferriero, D.; Lynch, J.K. Ischemic Perinatal Stroke: Summary of a Workshop Sponsored by the National Institute of Child Health and Human Development and the National Institute of Neurological Disorders and Stroke. Pediatrics 2007, 120, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Back, S.A.; Rivkees, S.A. Emerging concepts in periventricular white matter injury. Semin. Perinatol. 2004, 28, 405–414. [Google Scholar] [CrossRef] [PubMed]
- McQuillen, P.; Ferriero, D.M. Selective vulnerability in the developing central nervous system. Pediatr. Neurol. 2004, 30, 227–235. [Google Scholar] [CrossRef]
- Volpe, J.J. Brain injury in premature infants: A complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009, 8, 110–124. [Google Scholar] [CrossRef] [Green Version]
- Elbers, J.; Viero, S.; MacGregor, D.; DeVeber, G.; Moore, A.M. Placental Pathology in Neonatal Stroke. Pediatrics 2011, 127, e722–e729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, F.T. Cerebral palsy and thrombi in placental vessels of the fetus: Insights from litigation. Hum. Pathol. 1997, 28, 246–248. [Google Scholar] [CrossRef]
- Kraus, F.T.; Acheen, I.V. Fetal thrombotic vasculopathy in the placenta: Cerebral thrombi and infarcts, coagulopathies, and cerebral palsy. Hum. Pathol. 1999, 30, 759–769. [Google Scholar] [CrossRef]
- Long, M.; Brandon, D.H. Induced Hypothermia for Neonates with Hypoxic-Ischemic Encephalopathy. J. Obstet. Gynecol. Neonatal Nurs. 2007, 36, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Cotten, C.M.; Shankaran, S. Hypothermia for hypoxic–ischemic encephalopathy. Expert Rev. Obstet. Gynecol. 2010, 5, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalak, L.; Perlman, J.M. Hypoxic–ischemic brain injury in the term infant-current concepts. Early Hum. Dev. 2004, 80, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Kenney, K.; Amyot, F.; Moore, C.; Haber, M.; Turtzo, L.C.; Shenouda, C.; Silverman, E.; Gong, Y.; Qu, B.-X.; Harburg, L.; et al. Phosphodiesterase-5 inhibition potentiates cerebrovascular reactivity in chronic traumatic brain injury. Ann. Clin. Transl. Neurol. 2018, 5, 418–428. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Effect | Neonatal | Juvenile | Adult | |
|---|---|---|---|---|
| HI | TBI/Cryo | |||
| CBF | +++ | + | + | ? |
| Angiogenesis | +/−? | + | + | + |
| Inflammation | +++ | |||
| Cell death | ++ | ++ | ++ | + |
| Neurogenesis | + | ++ | ++ | ? |
| NCT/Status/Study Title | Condition | Comparator | Phase | Primary Outcome | Main Effect | Comment/Reference |
|---|---|---|---|---|---|---|
| NCT02628847 (Terminated) Sildenafil and Stroke Recovery | Adult stroke | Placebo | 1 | Motor recovery at one and three months | Unknown | Recruitment was problematic |
| NCT00452582 (Terminated) Sildenafil (Viagra) Treatment of Subacute Ischemic Stroke | Adult ischemic stroke | Usual care | 1 | The maximum tolerated dose and toxicity profile of sildenafil treatment in patients with subacute ischemic stroke | Sildenafil appeared to be safe in patients with mild to moderately severe stroke | Failure to recruit in expected time period |
| NCT03855332 (Recruiting) Oxford Haemodynamic Adaptation to Reduce Pulsatility Trial (OxHARP) | Small vessel cerebrovascular disease in adult | Cilostazol Placebo | 2 | Middle cerebral arterial pulsatility index | - | Estimated study completion date: December 2022 |
| NCT01762475 (Completed) Sildenafil for Cerebro-vascular Dysfunction in Chronic Traumatic Brain Injury | Traumatic brain injury in adults | - | 2 | Cerebrovascular reactivity | Single-dose sildenafil improves regional CVR deficits in chronic TBI patients | [116] |
| NCT02114775 (Completed) Growth Hormone or Sildenafil as Therapies for Fatigue in Mild-Traumatic-brain-injury (MTBI) | Traumatic brain injury in adults | Genotropin Placebo | 1 | Performance fatigue | Unknown | Sildenafil was found to increase protein synthesis and reduces muscle fatigue in healthy men |
| NCT04058132 (Recruiting) Cerebrovascular Reactivity Assessed With fNIRS as a Biomarker of TCVI After Acute Traumatic Brain Injury in Military | Acute/subacute traumatic brain injury in adults | None | 2 | Variation of oxyhemoglobin and deoxyhemoglobin concentration Longitudinal measure of CVR | - | Estimated study completion date: April 2021 |
| NCT02990078 (Recruiting) Non-invasive Measurement of Cerebrovascular Reactivity After Traumatic Brain Injury | Traumatic brain injury in adults | None | 1 | Change in CVR | - | Estimated study completion date: December 2026 |
| NCT03417492 (Recruiting) Cerebrovascular Reactivity in American Football Players | Traumatic brain injury in adults | None | 1 | Effect of single dose sildenafil citrate on global BOLD response to hypercapnia | - | Estimated study completion date: September 2022 |
| NCT02812433 (Active) Sildenafil Administration to Treat Neonatal Encephalopathy | Hypoxic ischemic encephalopathy in neonates | Ora-Blend | 1 | Serious adverse events | - | Estimated study completion date: June 2022 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zinni, M.; Pansiot, J.; Léger, P.-L.; El Kamouh, M.; Baud, O. Sildenafil-Mediated Neuroprotection from Adult to Neonatal Brain Injury: Evidence, Mechanisms, and Future Translation. Cells 2021, 10, 2766. https://doi.org/10.3390/cells10102766
Zinni M, Pansiot J, Léger P-L, El Kamouh M, Baud O. Sildenafil-Mediated Neuroprotection from Adult to Neonatal Brain Injury: Evidence, Mechanisms, and Future Translation. Cells. 2021; 10(10):2766. https://doi.org/10.3390/cells10102766
Chicago/Turabian StyleZinni, Manuela, Julien Pansiot, Pierre-Louis Léger, Marina El Kamouh, and Olivier Baud. 2021. "Sildenafil-Mediated Neuroprotection from Adult to Neonatal Brain Injury: Evidence, Mechanisms, and Future Translation" Cells 10, no. 10: 2766. https://doi.org/10.3390/cells10102766
APA StyleZinni, M., Pansiot, J., Léger, P.-L., El Kamouh, M., & Baud, O. (2021). Sildenafil-Mediated Neuroprotection from Adult to Neonatal Brain Injury: Evidence, Mechanisms, and Future Translation. Cells, 10(10), 2766. https://doi.org/10.3390/cells10102766