
Rac GTPase Signaling in Immune-Mediated Mechanisms of Atherosclerosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
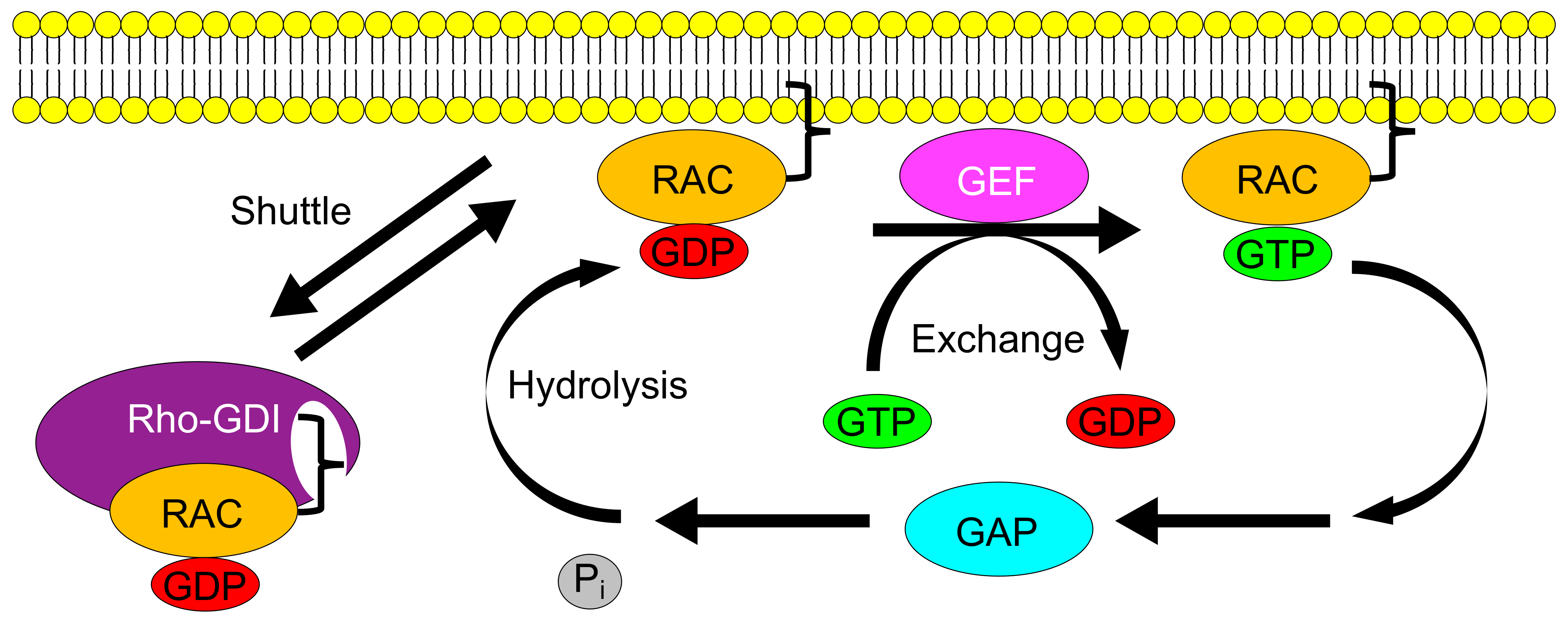
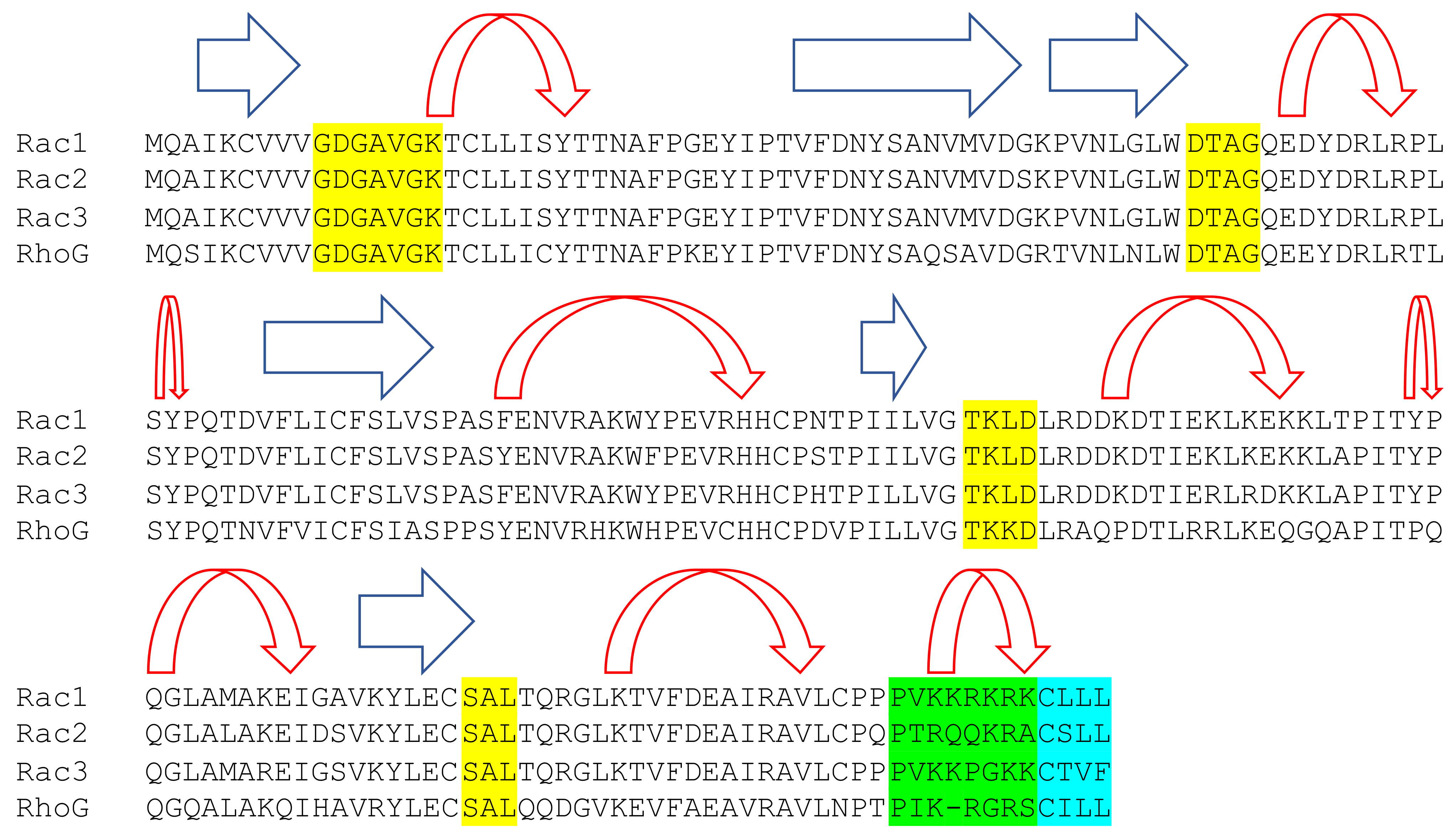
2. Rac GTPases
3. Overview of Atherosclerosis
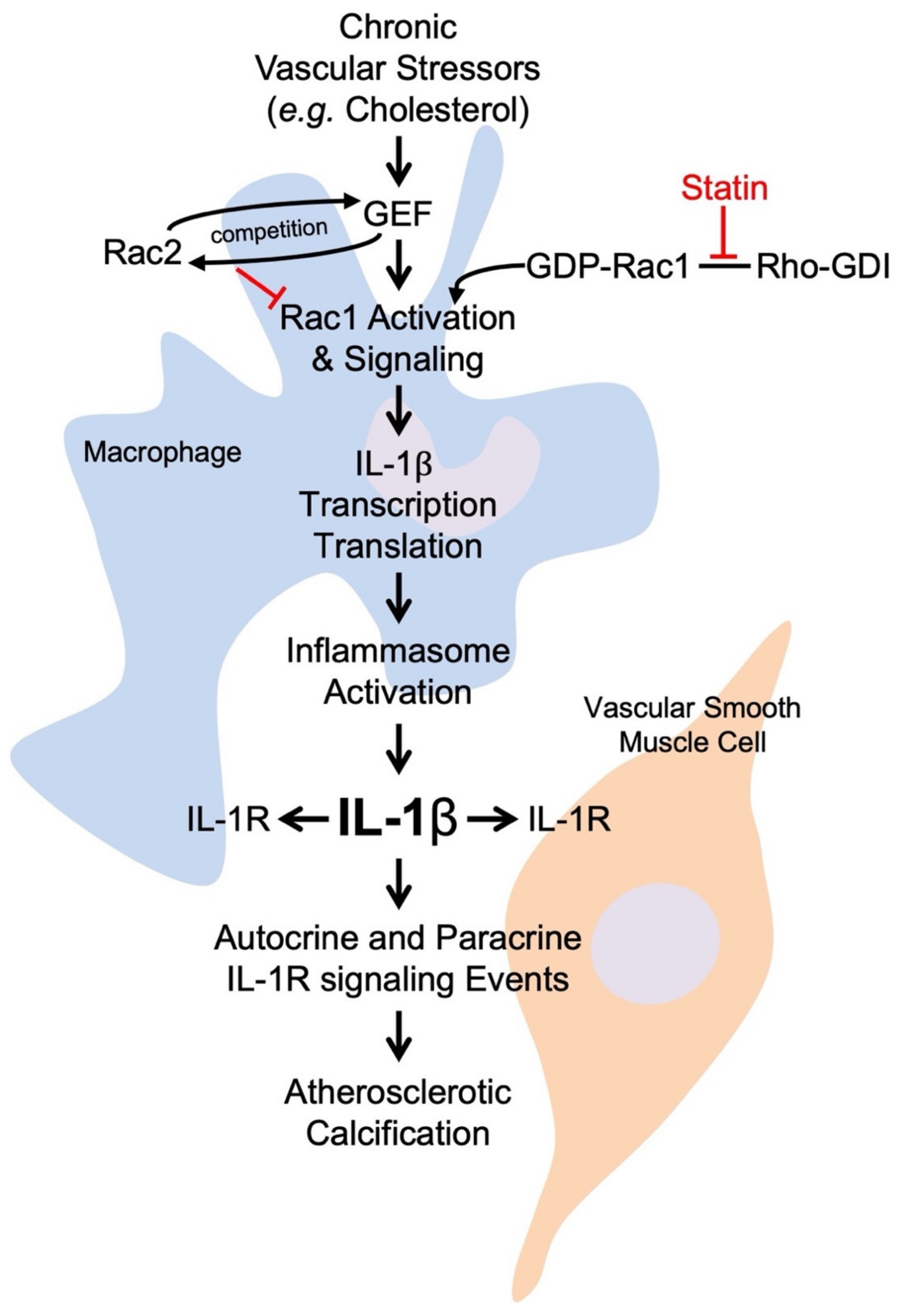
4. Rac Observations in Inflammatory Atherosclerosis
5. Current Therapy: Statins
6. Rac and Immune-Targeted Treatments
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bosco, E.E.; Mulloy, J.C.; Zheng, Y. Rac1 GTPase: A “Rac” of all trades. Cell. Mol. Life Sci. 2009, 66, 370–374. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Flentje, A.; Kalsi, R.; Monahan, T.S. Small gtpases and their role in vascular disease. Int. J. Mol. Sci. 2019, 20, 917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Sánchez, R.M.; Bustelo, X.R. Structural basis for the signaling specificity of RhoG and Rac1 GTPases. J. Biol. Chem. 2003, 278, 37916–37925. [Google Scholar] [CrossRef] [Green Version]
- Bustelo, X.R.; Sauzeau, V.; Berenjeno, I.M. GTP-binding proteins of the Rho/Rac family: Regulation, effectors and functions in vivo. BioEssays 2007, 29, 356–370. [Google Scholar] [CrossRef] [Green Version]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The “invisible hand”: Regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011, 12, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Didsbury, J.; Weber, R.F.; Bokoch, G.M.; Evans, T.; Snyderman, R. Rac, a Novel Ras-Related Family of Proteins That Are Botulinum Toxin Substrates. J. Biol. Chem. 1989, 264, 16378–16382. [Google Scholar] [CrossRef]
- Didsbury, J.R.; Uhing, R.J.; Snyderman, R. Isoprenylation of the low molecular mass GTP-binding proteins rac 1 and rac 2: Possible role in membrane localization. Biochem. Biophys. Res. Commun. 1990, 171, 804–812. [Google Scholar] [CrossRef]
- Just, I.; Rohrbeck, A.; Huelsenbeck, S.C.; Hoeltje, M. Therapeutic effects of Clostridium botulinum C3 exoenzyme. Naunyn. Schmiedebergs. Arch. Pharmacol. 2011, 383, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.; Malliri, A. Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases 2017, 8, 139–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worthylake, D.K.; Rossman, K.L.; Sondek, J. Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature 2000, 408, 682–688. [Google Scholar] [CrossRef]
- Bellanger, J.M.; Astier, C.; Sardet, C.; Ohta, Y.; Stossel, T.P.; Debant, A. The Rac1- and RhoG-specific GEF domain of trio targets filamin to remodel cytoskeletal actin. Nat. Cell Biol. 2000, 2, 888–892. [Google Scholar] [CrossRef]
- Rapley, J.; Tybulewicz, V.L.J.; Rittinger, K. Crucial structural role for the PH and C1 domains of the Vav1 exchange factor. EMBO Rep. 2008, 9, 655–661. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Rossman, K.L.; Liu, B.; Ritola, K.D.; Chiang, D.; Campbell, S.L.; Burridge, K.; Der, C.J. Vav2 is an activator of Cdc42, Rac1, and RhoA. J. Biol. Chem. 2000, 275, 10141–10149. [Google Scholar] [CrossRef] [Green Version]
- Balamatsias, D.; Kong, A.M.; Waters, J.E.; Sriratana, A.; Gurung, R.; Bailey, C.G.; Rasko, J.E.J.; Tiganis, T.; Macaulay, S.L.; Mitchell, C.A. Identification of P-Rex1 as a novel Rac1-guanine nucleotide exchange factor (GEF) that promotes actin remodeling and GLUT4 protein trafficking in adipocytes. J. Biol. Chem. 2011, 286, 43229–43240. [Google Scholar] [CrossRef] [Green Version]
- Healy, A.; Berus, J.M.; Christensen, J.L.; Lee, C.; Mantsounga, C.; Dong, W.; Watts, J.P.; Assali, M.; Ceneri, N.; Nilson, R.; et al. Statins disrupt macrophage rac1 regulation leading to increased atherosclerotic plaque calcification. Arterioscler. Thromb. Vasc. Biol. 2020, 714–732. [Google Scholar] [CrossRef] [PubMed]
- Wertheimer, E.; Gutierrez-Uzquiza, A.; Rosemblit, C.; Lopez-Haber, C.; Sosa, M.S.; Kazanietz, M.G. Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell Signal. 2012, 24, 353–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canagarajah, B.; Leskow, F.C.; Ho, J.Y.S.; Mischak, H.; Saidi, L.F.; Kazanietz, M.G.; Hurley, J.H. Structural mechanism for lipid activation of the Rac-specific GAP, β2-chimaerin. Cell 2004, 119, 407–418. [Google Scholar] [CrossRef] [Green Version]
- Dovas, A.; Couchman, J.R. RhoGDI: Multiple functions in the regulation of Rho family GTPase activities. Biochem. J. 2005, 390, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R.; Ojeda, V.; Barreira, M.; Sauzeau, V.; Castro-Castro, A. Rac-ing to the plasma membrane. Small GTPases 2012, 3, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Haataja, L.; Groffen, J.; Heisterkamp, N. Characterization of RAC3, a novel member of the Rho family. J. Biol. Chem. 1997, 272, 20384–20388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippi, M.D.; Harris, C.E.; Meller, J.; Gu, Y.; Zheng, Y.; Williams, D.A. Localization of Rac2 via the C terminus and aspartic acid 150 specifies superoxide generation, actin polarity and chemotaxis in neutrophils. Nat. Immunol. 2004, 5, 744–751. [Google Scholar] [CrossRef]
- Sun, C.X.; Downey, G.P.; Zhu, F.; Koh, A.L.Y.; Thang, H.; Glogauer, M. Rac1 is the small GTPase responsible for regulating the neutrophil chemotaxis compass. Blood 2004, 104, 3758–3765. [Google Scholar] [CrossRef]
- Ceneri, N.; Zhao, L.; Young, B.D.; Healy, A.; Coskun, S.; Vasavada, H.; Yarovinsky, T.O.; Ike, K.; Pardi, R.; Qin, L.; et al. Rac2 modulates atherosclerotic calcification by regulating macrophage interleukin-1β production. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 328–340. [Google Scholar] [CrossRef] [Green Version]
- Courjal, F.; Chuchana, P.; Theillet, C.; Fort, P. Structure and chromosomal assignment to 22q12 and 17qter of the ras- related Rac2 and Rac3 human genes. Genomics 1997, 44, 242–246. [Google Scholar] [CrossRef]
- Vigorito, E.; Billadeu, D.D.; Savoy, D.; McAdam, S.; Doody, G.; Fort, P.; Turner, M. RhoG regulates gene expression and the actin cytoskeleton in lymphocytes. Oncogene 2003, 22, 330–342. [Google Scholar] [CrossRef] [Green Version]
- Gauthier-Rouvière, C.; Vignal, E.; Mériane, M.; Roux, P.; Montcourier, P.; Fort, P. RhoG GTPase controls a pathway that independently activates Rac1 and Cdc42Hs. Mol. Biol. Cell 1998, 9, 1379–1394. [Google Scholar] [CrossRef] [Green Version]
- Vincent, S.; Jeanteur, P.; Fort, P. Growth-regulated expression of rhoG, a new member of the ras homolog gene family. Mol. Cell. Biol. 1992, 12, 3138–3148. [Google Scholar] [CrossRef] [Green Version]
- Dever, T.E.; Glynias, M.J.; Merrick, W.C. GTP-binding domain: Three consensus sequence elements with distinct spacing. Proc. Natl. Acad. Sci. USA 1987, 84, 1814–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mccormick, F.; Clark, B.F.C.; La Cour, T.F.M.; Kjeldgaard, M.; Norskov-Lauritsen, L.; Nyborg, J. A model for the tertiary structure of p21, the product of the ras oncogene. Science 1985, 230, 78–82. [Google Scholar] [CrossRef]
- Hirshberg, M.; Stockley, R.W.; Dodson, G.; Webb, M.R. The crystal structure of human rac1, a member of the rho-family complexed with a GTP analogue. Nat. Struct. Biol. 1997, 4, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Boulter, E.; Garcia-Mata, R.; Guilluy, C.; Dubash, A.; Rossi, G.; Brennwald, P.J.; Burridge, K. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDINat. Cell Biol. 2010, 12, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Seabra, M.C. Membrane association and targeting of prenylated Ras-like GTPases. Cell. Signal. 1998, 10, 167–172. [Google Scholar] [CrossRef]
- Fu, H.; Alabdullah, M.; Großmann, J.; Spieler, F.; Abdosh, R.; Lutz, V.; Kalies, K.; Knöpp, K.; Rieckmann, M.; Koch, S.; et al. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. Int. J. Mol. Sci. 2019, 21, 1–16. [Google Scholar] [CrossRef] [Green Version]
- McTaggart, S.J. Isoprenylated proteins. Cell. Mol. Life Sci. 2006, 63, 255–267. [Google Scholar] [CrossRef]
- Running, M.P. The role of lipid post-translational modification in plant developmental processes. Front. Plant Sci. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelson, D.; Silletti, J.; Murphy, G.; D’Eustachio, P.; Rush, M.; Philips, M.R. Differential localization of Rho GTPases in live cells: Regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 2001, 152, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajdo-Milašinović, A.; Ellenbroek, S.I.J.; van Es, S.; van der Vaart, B.; Collard, J.G. Rac1 and rac3 have opposing functions in cell adhesion and differentiation of neuronal cells. J. Cell Sci. 2007, 120, 555–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, A.; Cordelières, F.P.; Cherfils, J.; Olofsson, B. RhoGDI3 and RhoG: Vesicular trafficking and interactions with the Sec3 Exocyst subunit. Small GTPases 2010, 1, 142–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Sánchez, R.M.; Berenjeno, I.M.; Bustelo, X.R. Involvement of the Rho/Rac family member RhoG in caveolar endocytosis. Oncogene 2006, 25, 2961–2973. [Google Scholar] [CrossRef] [Green Version]
- Hancock, J.F.; Magee, A.I.; Childs, J.E.; Marshall, C.J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989, 57, 1167–1177. [Google Scholar] [CrossRef]
- Navarro-Lérida, I.; Sánchez-Perales, S.; Calvo, M.; Rentero, C.; Zheng, Y.; Enrich, C.; Del Pozo, M.A. A palmitoylation switch mechanism regulates Rac1 function and membrane organization. EMBO J. 2012, 31, 534–551. [Google Scholar] [CrossRef] [Green Version]
- Abdrabou, A.; Wang, Z. Post-Translational Modification and Subcellular Distribution of Rac1: Update. Cells 2018, 7, 263. [Google Scholar] [CrossRef] [Green Version]
- Lanning, C.C.; Daddona, J.L.; Ruiz-Velasco, R.; Shafer, S.H.; Williams, C.L. The Rac1 C-terminal polybasic region regulates the nuclear localization and protein degradation of Rac1. J. Biol. Chem. 2004, 279, 44197–44210. [Google Scholar] [CrossRef] [Green Version]
- Laporte, J.; Blondeau, F.; Gansmuller, A.; Lutz, Y.; Vonesch, J.L.; Mandel, J.L. The Ptdlns3P phosphate myotubularin is a cytoplasmic protein that also localizes to Rac1-inducible plasma membrane ruffles. J. Cell Sci. 2002, 115, 3105–3117. [Google Scholar] [CrossRef]
- Molnár, G.; Dagher, M.C.; Geiszt, M.; Settleman, J.; Ligeti, E. Role of prenylation in the interaction of RHO-family small GTPases with GTPpase activating proteins. Biochemistry 2001, 40, 10542–10549. [Google Scholar] [CrossRef]
- Kinsella, B.T.; Erdman, R.A.; Maltese, W.A. Carboxyl-terminal isoprenylation of ras-related GTP-binding proteins encoded by rac1, rac2, and ralA. J. Biol. Chem. 1991, 266, 9786–9794. [Google Scholar] [CrossRef]
- Joyce, P.L.; Cox, A.D. Rac1 and Rac3 Are Targets for Geranylgeranyltransferase I Inhibitor-Mediated Inhibition of Signaling, Transformation, and Membrane Ruffling. Cancer Res. 2003, 63, 7959–7967. [Google Scholar] [PubMed]
- Manser, E.; Leung, T.; Salihuddin, H.; Zhao, Z.S.; Lim, L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 1994, 367, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Mao, R.; Zhou, Q.; Yang, Y.; Cao, J.; Ding, Y.; Yang, Y.; Zhang, X.; Li, L.; Xu, L. Inhibition of Rac1 Activity in the Hippocampus Impairs the Forgetting of Contextual Fear Memory. Mol. Neurobiol. 2016, 53, 1247–1253. [Google Scholar] [CrossRef]
- Khan, O.M.; Ibrahim, M.X.; Jonsson, I.M.; Karlsson, C.; Liu, M.; Sjogren, A.K.M.; Olofsson, F.J.; Brisslert, M.; Andersson, S.; Ohlsson, C.; et al. Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J. Clin. Investig. 2011, 121, 628–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Lérida, I.; Pellinen, T.; Sanchez, S.A.; Guadamillas, M.C.; Wang, Y.; Mirtti, T.; Calvo, E.; DelPozo, M.A. Rac1 nucleocytoplasmic shuttling drives nuclear shape changes and tumor invasion. Dev. Cell 2015, 32, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Pop, M.; Aktories, K.; Schmidt, G. Isotype-specific degradation of Rac activated by the cytotoxic necrotizing factor 1. J. Biol. Chem. 2004, 279, 35840–35848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.L. The polybasic region of Ras and Rho family small GTPases: A regulator of protein interactions and membrane association and a site of nuclear localization signal sequences. Cell. Signal. 2003, 15, 1071–1080. [Google Scholar] [CrossRef]
- Justilien, V.; Fields, A.P. Ect2 links the PKC-Par6α complex to Rac1 activation and cellular transformation. Oncogene 2009, 28, 3597–3607. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, T.; Bokoch, G.M.; Waterman-Storer, C.M. Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J. Cell Biol. 2003, 161, 845–851. [Google Scholar] [CrossRef]
- Carrizzo, A.; Vecchione, C.; Damato, A.; di Nonno, F.; Ambrosio, M.; Pompeo, F.; Cappello, E.; Capocci, L.; Peruzzi, M.; Valenti, V.; et al. Rac1 pharmacological inhibition rescues human endothelial dysfunction. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hordijk, P.L. Regulation of NADPH oxidases: The role of Rac proteins. Circ. Res. 2006, 98, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Meier, B.; Radeke, H.H.; Selle, S.; Younes, M.; Sies, H.; Resch, K.; Habermehl, G.G. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-α. Biochem. J. 1989, 263, 539–545. [Google Scholar] [CrossRef]
- Gregg, D.; Rauscher, F.M.; Goldschmidt-Clermont, P.J. Rac regulates cardiovascular superoxide through diverse molecular interactions: More than a binary GTP switch. Am. J. Physiol. Cell Physiol. 2003, 285. [Google Scholar] [CrossRef] [Green Version]
- Tzima, E.; Del Pozo, M.A.; Kiosses, W.B.; Mohamed, S.A.; Li, S.; Chien, S.; Schwartz, M.A. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 2002, 21, 6791–6800. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.M.; Lali, F.; Willetts, K.; Balague, C.; Godessart, N.; Brennan, F.; Feldmann, M.; Foxwell, B.M.J. Rac mediates TNF-induced cytokine production via modulation of NF-κB. Mol. Immunol. 2008, 45, 2446–2454. [Google Scholar] [CrossRef]
- Waterman-Storer, C.M.; Worthylake, R.A.; Liu, B.P.; Burridget, K.; Salmon, E.D. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat. Cell Biol. 1999, 1, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, J.S.; Hansen, M.D.H.; Nelson, W.J. Spatio-temporal regulation of Rac1 localization and lamellipodia dynamics during epithelial cell-cell adhesion. Dev. Cell 2002, 3, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, A.P.; Wells, C.M.; Smith, S.D.; Vega, F.M.; Henderson, R.B.; Tybulewicz, V.L.; Ridley, A.J. Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. J. Cell Sci. 2006, 119, 2749–2757. [Google Scholar] [CrossRef] [Green Version]
- Wells, C.M.; Walmsley, M.; Ooi, S.; Tybulewicz, V.; Ridley, A.J. Rac1-deficient macrophages exhibit defects in cell spreading and membrane ruffling but not migration. J. Cell Sci. 2004, 117, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Filić, V.; Marinović, M.; Faix, J.; Weber, I. A dual role for Rac1 GTPases in the regulation of cell motility. J. Cell Sci. 2012, 125, 387–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippi, M.D.; Szczur, K.; Harris, C.E.; Berclaz, P.Y. Rho GTPase Rac1 is critical for neutrophil migration into the lung. Blood 2007, 109, 1257–1264. [Google Scholar] [CrossRef] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, A.; González-Billault, C. Crosstalk between Rac1-mediated actin regulation and ROS production. Free Radic. Biol. Med. 2018, 116, 101–113. [Google Scholar] [CrossRef]
- Cheng, G.; Diebold, B.A.; Hughes, Y.; Lambeth, J.D. Nox1-dependent reactive oxygen generation is regulated by Rac1. J. Biol. Chem. 2006, 281, 17718–17726. [Google Scholar] [CrossRef] [Green Version]
- Ando, S.; Kaibuchi, K.; Sasaki, T.; Hiraoka, K.; Nishiyama, T.; Mizuno, T.; Asada, M.; Nunoi, H.; Matsuda, I.; Matsuura, Y.; et al. Post-translational processing of rac p21s is important both for their interaction with the GDP/GTP exchange proteins and for their activation of NADPH oxidase. J. Biol. Chem. 1992, 267, 25709–25713. [Google Scholar] [CrossRef]
- Ambruso, D.R.; Knall, C.; Abell, A.N.; Panepinto, J.; Kurkchubasche, A.; Thurman, G.; Gonzalez-Aller, C.; Hiester, A.; DeBoer, M.; Harbeck, R.J.; et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 4654–4659. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yamauchi, A.; Marchal, C.C.; Molitoris, J.K.; Quilliam, L.A.; Dinauer, M.C. Chemoattractant-Stimulated Rac Activation in Wild-Type and Rac2-Deficient Murine Neutrophils: Preferential Activation of Rac2 and Rac2 Gene Dosage Effect on Neutrophil Functions. J. Immunol. 2002, 169, 5043–5051. [Google Scholar] [CrossRef] [Green Version]
- Schnelzer, A.; Prechtel, D.; Knaus, U.; Dehne, K.; Gerhard, M.; Graeff, H.; Harbeck, N.; Schmitt, M.; Lengyel, E. Rac1 in human breast cancer: Overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000, 19, 3013–3020. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.A.; Meredith, J.E.; Kiosses, W.B. An activated Rac mutant functions as a dominant negative for membrane ruffling. Oncogene 1998, 17, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Karnoub, A.E.; Palmby, T.R.; Lengyel, E.; Sondek, J.; Der, C.J. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 2004, 23, 9369–9380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, C.; Hass, R.; Lehnert, H.; Ungefroren, H. RAC1B: A Rho GTPase with Versatile Functions in Malignant Transformation and Tumor Progression. Cells 2019, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esufali, S.; Charames, G.S.; Pethe, V.V.; Buongiorno, P.; Bapat, B. Activation of tumor-specific splice variant Rac1b by dishevelled promotes canonical Wnt signaling and decreased adhesion of colorectal cancer cells. Cancer Res. 2007, 67, 2469–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlad-Fiegen, A.; Langerak, A.; Eberth, S.; Müller, O. The Wnt pathway destabilizes adherens junctions and promotes cell migration via β-catenin and its target gene cyclin D1. FEBS Open Bio 2012, 2, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Després, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, e29–e39. [Google Scholar] [CrossRef] [Green Version]
- Yazdanyar, A.; Newman, A.B. The burden of cardiovascular disease in the elderly: Morbidity, mortality, and costs. Clin. Geriatr. Med. 2009, 25, 563–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 1–18. [Google Scholar] [CrossRef]
- Kramsch, D.M.; Franzblau, C.; Hollander, W. The protein and lipid composition of arterial elastin and its relationship to lipid accumulation in the atherosclerotic plaque. J. Clin. Investig. 1971, 50, 1666–1677. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Vendrov, A.; Runge, M.S. Oxidative Stress and Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Kzhyshkowska, J.; Neyen, C.; Gordon, S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology 2012, 217, 492–502. [Google Scholar] [CrossRef]
- Podrez, E.A.; Febbraio, M.; Sheibani, N.; Schmitt, D.; Silverstein, R.L.; Hajjar, D.P.; Cohen, P.A.; Frazier, W.A.; Hoff, H.F.; Hazen, S.L. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J. Clin. Investig. 2000, 105, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.C.; Glass, C.K. The macrophage foam cell as a target for therapeutic intervention. Nat. Med. 2002, 8, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nũez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.J.; Phillips, J.E.; Mason, R.P.; Casscells, S.W. Cholesterol crystallization and macrophage apoptosis: Implication for atherosclerotic plaque instability and rupture. Biochem. Pharmacol. 2003, 66, 1485–1492. [Google Scholar] [CrossRef]
- Rajam̈aki, K.; Lappalainen, J.; Öörni, K.; Välimäki, E.; Matikainen, S.; Kovanen, P.T.; Kari, E.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- Budoff, M.J.; Mao, S.; Zalace, C.P.; Bakhsheshi, H.; Oudiz, R. Comparison of spiral and electron beam tomography in the evaluation of coronary calcification in asymptomatic persons. Int. J. Cardiol. 2001, 77, 181–188. [Google Scholar] [CrossRef]
- Rennenberg, R.J.; Kessels, A.G.; Schurgers, L.J.; van Engelshoven, J.M.; de Leeuw, P.W.; Kroon, A.A. Vascular calcifications as a marker of increased cardiovascular risk: A meta-analysis. Vascular health and risk managem. Vasc. Health Risk Manag. 2009, 5, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Dweck, M.R.; Aikawa, E.; Newby, D.E.; Tarkin, J.M.; Rudd, J.H.F.; Narula, J.; Fayad, Z.A. Noninvasive Molecular Imaging of Disease Activity in Atherosclerosis. Circ. Res. 2016, 119, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Shioi, A.; Ikari, Y. Plaque calcification during atherosclerosis progression and regression. J. Atheroscler. Thromb. 2018, 25, 294–303. [Google Scholar] [CrossRef] [Green Version]
- Irkle, A.; Vesey, A.T.; Lewis, D.Y.; Skepper, J.N.; Bird, J.L.E.; Dweck, M.R.; Joshi, F.R.; Gallagher, F.A.; Warburton, E.A.; Bennett, M.R.; et al. Identifying active vascular microcalcification by 18F-sodium fluoride positron emission tomography. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Criqui, M.H.; Denenberg, J.O.; Ix, J.H.; McClelland, R.L.; Wassel, C.L.; Rifkin, D.E.; Carr, J.J.; Budoff, M.J.; Allison, M.A. Calcium density of coronary artery plaque and risk of incident cardiovascular events. JAMA-J. Am. Med. Assoc. 2014, 311, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Montanaro, M.; Scimeca, M.; Anemona, L.; Servadei, F.; Giacobbi, E.; Bonfiglio, R.; Bonanno, E.; Urbano, N.; Ippoliti, A.; Santeusanio, G.; et al. The paradox effect of calcification in carotid atherosclerosis: Microcalcification is correlated with plaque instability. Int. J. Mol. Sci. 2021, 22, 395. [Google Scholar] [CrossRef] [PubMed]
- De Jong, P.A.; Hellings, W.E.; Takx, R.A.P.; Išgum, I.; Van Herwaarden, J.A.; Mali, W.P.T.M. Computed tomography of aortic wall calcifications in aortic dissection patients. PLoS ONE 2014, 9, e102036. [Google Scholar] [CrossRef] [Green Version]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Donkor, E.S. Stroke in the 21st Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Ioacara, S.; Popescu, A.C.; Tenenbaum, J.; Dimulescu, D.R.; Popescu, M.R.; Sirbu, A.; Fica, S. Acute myocardial infarction mortality rates and trends in Romania between 1994 and Int. J. Environ. Res. Public Health 2020, 17, 285. [Google Scholar] [CrossRef] [Green Version]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Galea, J.; Armstrong, J.; Gadsdon, P.; Holden, H.; Francis, S.E.; Holt, C.M. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arter. Thromb Vasc. Biol. 1996, 16, 1000–1006. [Google Scholar] [CrossRef]
- Dewberry, R.; Holden, H.; Crossman, D.; Francis, S. Interleukin-1 receptor antagonist expression in human endothelial cells and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2394–2400. [Google Scholar] [CrossRef] [Green Version]
- Bevilacqua, M.P.; Pober, J.S.; Wheeler, M.E. Interleukin-1 activation of vascular endothelium. Effects on procoagulant activity and leukocyte adhesion. Am. J. Pathol. 1985, 121, 393–403. [Google Scholar]
- Libby, P.; Warner, S.J.C.; Friedman, G.B. Interleukin 1: A mitogen for human vascular smooth muscle cells that induces the release of growth-inhibitory prostanoids. J. Clin. Investig. 1988, 81, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Eun, S.Y.; Ko, Y.S.; Park, S.W.; Chang, K.C.; Kim, H.J. IL-1β enhances vascular smooth muscle cell proliferation and migration via P2Y2 receptor-mediated RAGE expression and HMGB1 release. Vascul. Pharmacol. 2015, 72, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Tintut, Y.; Patel, J.; Territo, M.; Saini, T.; Parhami, F.; Linda, L.; Vitro, C.I. Monocyte/Macrophage Regulation of Vascular Calcification In Vitro. Circulation 2002, 105, 650–655. [Google Scholar]
- Al-Aly, Z.; Shao, J.S.; Lai, C.F.; Huang, E.; Cai, J.; Behrmann, A.; Cheng, S.L.; Towler, D.A. Aortic Msx2-Wnt calcification cascade is regulated by TNF-α-dependent signals in diabetic Ldlr-/- mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2589–2596. [Google Scholar] [CrossRef] [Green Version]
- Sage, A.P.; Tintut, Y.; Demer, L.L. Regulatory mechanisms in vascular calcification. Nat. Rev. Cardiol. 2010, 7, 528–536. [Google Scholar] [CrossRef]
- Ramel, D.; Gayral, S.; Sarthou, M.K.; Augé, N.; Nègre-Salvayre, A.; Laffargue, M. Immune and smooth muscle cells interactions in atherosclerosis: How to target a breaking bad dialogue? Front. Pharmacol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, D.; Baylis, R.A.; Durgin, B.G.; Newman, A.A.C.; Alencar, G.F.; Mahan, S.; Hilaire, C.S.; Müller, W.; Waisman, A.; Francis, S.E.; et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 2018, 24, 1418–1429. [Google Scholar] [CrossRef]
- Hou, T.; Tieu, B.; Ray, S.; Recinos III, A.; Cui, R.; Tilton, R.; Brasier, A. Roles of IL-6-gp130 Signaling in Vascular Inflammation. Curr. Cardiol. Rev. 2008, 4, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Weber, D.S.; Taniyama, Y.; Rocic, P.; Seshiah, P.N.; Dechert, M.A.; Gerthoffer, W.T.; Griendling, K.K. Phosphoinositide-dependent kinase 1 and p21-activated protein kinase mediate reactive oxygen species-dependent regulation of platelet-derived growth factor-induced smooth muscle cell migration. Circ. Res. 2004, 94, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.H.; Fanaroff, A.C.; Sharma, K.C.; Smith, L.S.; Brian, L.; Eipper, B.A.; Mains, R.E.; Freedman, N.J.; Zhang, L. Kalirin promotes neointimal hyperplasia by activating rac in smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 702–708. [Google Scholar] [CrossRef] [Green Version]
- Talwar, S.; Kant, A.; Xu, T.; Shenoy, V.B.; Assoian, R.K. Mechanosensitive smooth muscle cell phenotypic plasticity emerging from a null state and the balance between Rac and Rho. Cell Rep. 2021, 35, 109019. [Google Scholar] [CrossRef]
- Ohkawara, H.; Ishibashi, T.; Shiomi, M.; Sugimoto, K.; Uekita, H.; Kamioka, M.; Takuwa, Y.; Teramoto, T.; Maruyama, Y.; Takeishi, Y. RhoA and Rac1 changes in the atherosclerotic lesions of WHHLMI rabbits. J. Atheroscler. Thromb. 2009, 16, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Bandaru, S.; Ala, C.; Ekstrand, M.; Akula, M.K.; Pedrelli, M.; Liu, X.; Bergström, G.; Håversen, L.; Borén, J.; Bergo, M.O.; et al. Lack of RAC1 in macrophages protects against atherosclerosis. PLoS ONE 2020, 15, e0239284. [Google Scholar] [CrossRef] [PubMed]
- Eitel, J.; Meixenberger, K.; van Laak, C.; Orlovski, C.; Hocke, A.; Schmeck, B.; Hippenstiel, S.; N’Guessan, P.D.; Suttorp, N.; Opitz, B. Rac1 regulates the NLRP3 inflammasome which mediates IL-1beta production in Chlamydophila pneumoniae infected human mononuclear cells. PLoS ONE 2012, 7, e30379. [Google Scholar] [CrossRef] [PubMed]
- Page, M.M.; Watts, G.F. PCSK9 inhibitors-mechanisms of action. Aust. Prescr. 2016, 39, 164–167. [Google Scholar] [CrossRef] [Green Version]
- Sizar, O.; Khare, S.; Jamil, R.T.; Talati, R. Statin Medications. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Davignon, J. Beneficial cardiovascular pleiotropic effects of statins. Circulation 2004, 109, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.; Armitage, J.; Parish, S.; Sleight, P.; Peto, R. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20 536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 7–22. [Google Scholar] [CrossRef]
- Jain, M.K.; Ridker, P.M. Anti-inflammatory effects of statins: Clinical evidence and basic mechanisms. Nat. Rev. Drug Discov. 2005, 4, 977–987. [Google Scholar] [CrossRef]
- White, H.D.; Simes, R.J.; Anderson, N.E.; Hankey, G.J.; Watson, J.D.G.; Hunt, D.; Colquhoun, D.M.; Glasziou, P.; MacMahon, S.; Kirby, A.C.; et al. Pravastatin Therapy and the Risk of Stroke . N. Engl. J. Med. 2000, 343, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, A.G.; Ganz, P.; Oliver, M.F.; Waters, D.; Zeiher, A.; Chaitman, B.R.; Leslie, S.; Stern, T. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes. Curr. Cardiol. Rep. 2001, 3, 384. [Google Scholar]
- Achenbach, S.; Ropers, D.; Pohle, K.; Leber, A.; Thilo, C.; Knez, A.; Menendez, T.; Maeffert, R.; Kusus, M.; Regenfus, M.; et al. Influence of lipid-lowering therapy on the progression of coronary artery calcification: A prospective evaluation. Circulation 2002, 106, 1077–1082. [Google Scholar] [CrossRef] [Green Version]
- Arad, Y.; Spadaro, L.A.; Roth, M.; Newstein, D.; Guerci, A.D. Treatment of asymptomatic adults with elevated coronary calcium scores with atorvastatin, vitamin C, and vitamin E: The St. Francis heart study randomized clinical trial. J. Am. Coll. Cardiol. 2005, 46, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Callister, T.Q.; Raggi, P.; Cooil, B.; Lippolis, N.J.; Russo, D. Effect of HMG-CoA reductase inhibitors on coronary artery disease as assessed by electron-beam computed tomography. N. Engl. J. Med. 1998, 31, 1972–1978. [Google Scholar] [CrossRef]
- Hecht, H.S.; Harman, S.M. Relation of aggressiveness of lipid-lowering treatment to changes in calcified plaque burden by electron beam tomography. Am. J. Cardiol. 2003, 92, 334–336. [Google Scholar] [CrossRef]
- Raggi, P.; Davidson, M.; Callister, T.Q.; Welty, F.K.; Bachmann, G.A.; Hecht, H.; Rumberger, J.A. Aggressive versus moderate lipid-lowering therapy in hypercholesterolemic postmenopausal women: Beyond endorsed lipid lowering with EBT scanning (BELLES). Circulation 2005, 112, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Houslay, E.S.; Cowell, S.J.; Prescott, R.J.; Reid, J.; Burton, J.; Northridge, D.B.; Boon, N.A.; Newby, D.E. Progressive coronary calcification despite intensive lipid-lowering treatment: A randomised controlled trial. Heart 2006, 92, 1207–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banach, M.; Serban, C.; Sahebkar, A.; Mikhailidis, D.P.; Ursoniu, S.; Ray, K.K.; Rysz, J.; Toth, P.P.; Muntner, P.; Mosteoru, S.; et al. Impact of statin therapy on coronary plaque composition: A systematic review and meta-analysis of virtual histology intravascular ultrasound studies. BMC Med. 2015, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, R.; Nicholls, S.J.; Shao, M.; Kataoka, Y.; Uno, K.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E. Impact of statins on serial coronary calcification during atheroma progression and regression. J. Am. Coll. Cardiol. 2015, 65, 1273–1282. [Google Scholar] [CrossRef]
- Kavalipati, N.; Shah, J.; Ramakrishan, A.; Vasnawala, H. Pleiotropic effects of statins. Indian J. Endocrinol. Metab. 2015, 19, 554–562. [Google Scholar] [CrossRef]
- Li, J.; Zhu, H.; Shen, E.; Wan, L.; Arnold, J.M.O.; Peng, T. Deficiency of Rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 2010, 59, 2033–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbar, H.; Duan, X.; Piatt, R.; Saleem, S.; Davis, A.K.; Tandon, N.N.; Bergmeier, W.; Zheng, Y. Small molecule targeting the Rac1-NOX2 interaction prevents collagen-related peptide and thrombin-induced reactive oxygen species generation and platelet activation. J. Thromb. Haemost. 2018, 16, 2083–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marei, H.; Malliri, A. GEFs: Dual regulation of Rac1 signaling. Small GTPases 2017, 8, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Ferri, N.; Colombo, G.; Ferrandi, C.; Raines, E.W.; Levkau, B.; Corsini, A. Simvastatin reduces MMP1 expression in human smooth muscle cells cultured on polymerized collagen by inhibiting Rac1 activation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1043–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.I.; Fukumoto, Y.; Nochioka, K.; Minami, T.; Kudo, S.; Shiba, N.; Takai, Y.; Williams, C.L.; Liao, J.K.; Shimokawa, H. Statins exert the pleiotropic effects through small gtp-binding protein dissociation stimulator upregulation with a resultant rac1 degradation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1591–1600. [Google Scholar] [CrossRef] [Green Version]
- Kotelevets, L.; Chastre, E. Rac1 signaling: From intestinal homeostasis to colorectal cancer metastasis. Cancers 2020, 12, 665. [Google Scholar] [CrossRef] [Green Version]
- Rikitake, Y.; Liao, J.K. Rho GTPases, statins, and nitric oxide. Circ. Res. 2005, 97, 1232–1235. [Google Scholar] [CrossRef] [Green Version]
- Rashid, M.; Tawara, S.; Fukumoto, Y.; Seto, M.; Yano, K.; Shimokawa, H. Importance of racl signaling pathway inhibition in the pleiotropic effects of HMG-CoA reductase inhibitors. Circ. J. 2009, 73, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Laufs, U.; Kilter, H.; Konkol, C.; Wassmann, S.; Böhm, M.; Nickenig, G. Impact of HMG CoA reductase inhibition on small GTPases in the heart. Cardiovasc. Res. 2002, 53, 911–920. [Google Scholar] [CrossRef] [Green Version]
- Akula, M.K.; Ibrahim, M.X.; Ivarsson, E.G.; Khan, O.M.; Kumar, I.T.; Erlandsson, M.; Karlsson, C.; Xu, X.; Brisslert, M.; Brakebusch, C.; et al. Protein prenylation restrains innate immunity by inhibiting Rac1 effector interactions. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, O.M.; Akula, M.K.; Skålen, K.; Karlsson, C.; Ståhlman, M.; Young, S.G.; Borén, J.; Bergo, M.O. Targeting GGTase-I activates RHOA, increases macrophage reverse cholesterol transport, and reduces atherosclerosis in mice. Circulation 2013, 127, 782–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.F.; Carley, R.E.; Butler, C.A.; Morrison, A.R. Rac GTPase Signaling in Immune-Mediated Mechanisms of Atherosclerosis. Cells 2021, 10, 2808. https://doi.org/10.3390/cells10112808
Lee CF, Carley RE, Butler CA, Morrison AR. Rac GTPase Signaling in Immune-Mediated Mechanisms of Atherosclerosis. Cells. 2021; 10(11):2808. https://doi.org/10.3390/cells10112808
Chicago/Turabian StyleLee, Cadence F., Rachel E. Carley, Celia A. Butler, and Alan R. Morrison. 2021. "Rac GTPase Signaling in Immune-Mediated Mechanisms of Atherosclerosis" Cells 10, no. 11: 2808. https://doi.org/10.3390/cells10112808