
The Proapoptotic Gene Bad Regulates Brain Development via p53-Mediated Stress Signals in Zebrafish



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Experimental Fish
2.2. Apoptotic Cell Staining
2.3. RNA Extraction
2.4. Reverse-Transcription Polymerase Chain Reaction (RT-PCR) Analysis
2.5. Quantitative (q)RT-PCR
2.6. Morpholino Oligonucleotides (MOs)
2.7. Western Blotting
2.8. Whole-Mount In Situ Hybridization
2.9. Microarray
2.10. Swimming Activity Assay
2.11. Statistical Analysis
3. Results
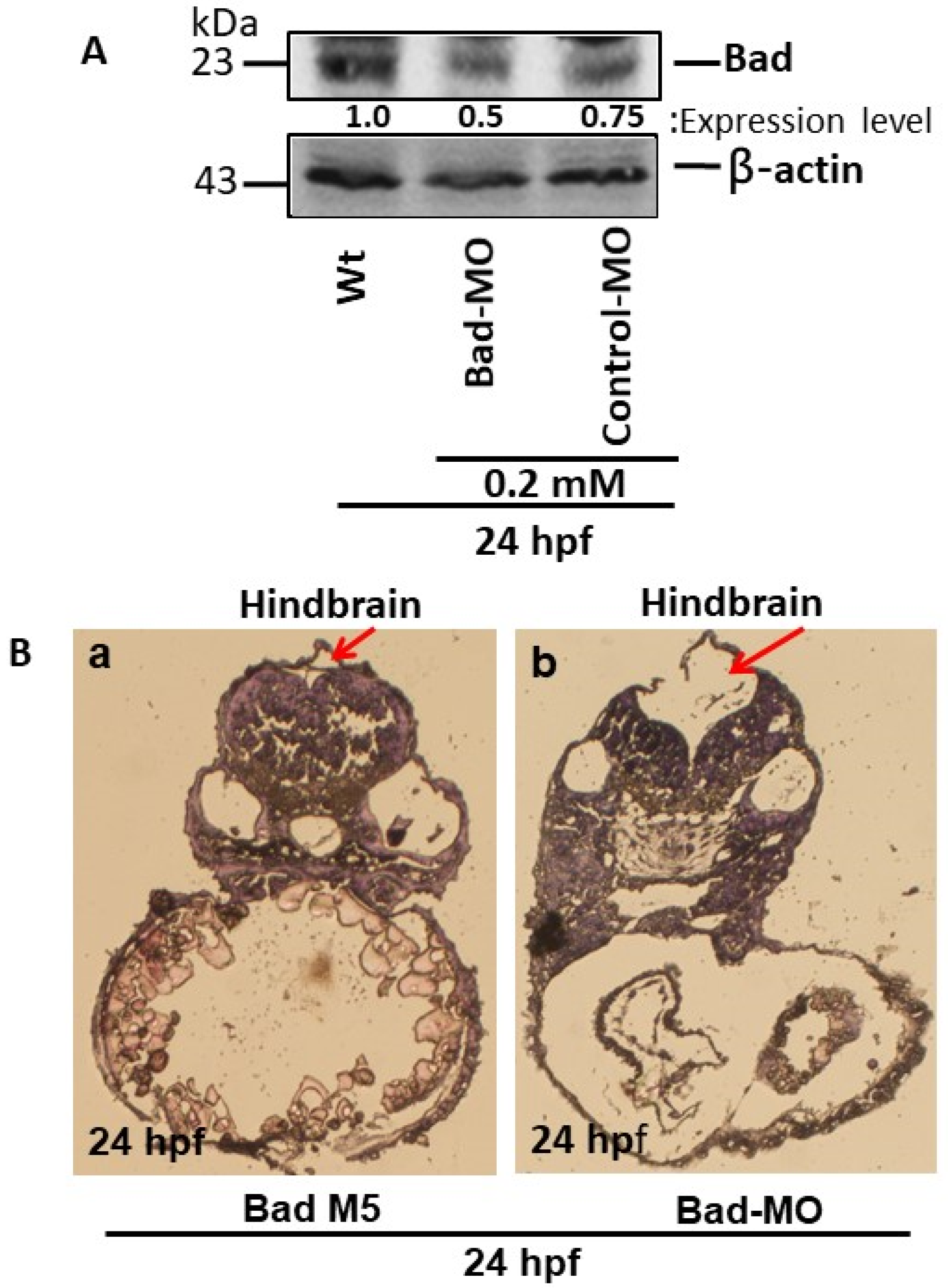
3.1. Bad-Mediated PCD-Regulated Brain Morphogenesis
3.2. Screening of Bad-Mediated PCD Death Signaling Using a Genome-Wide Approach
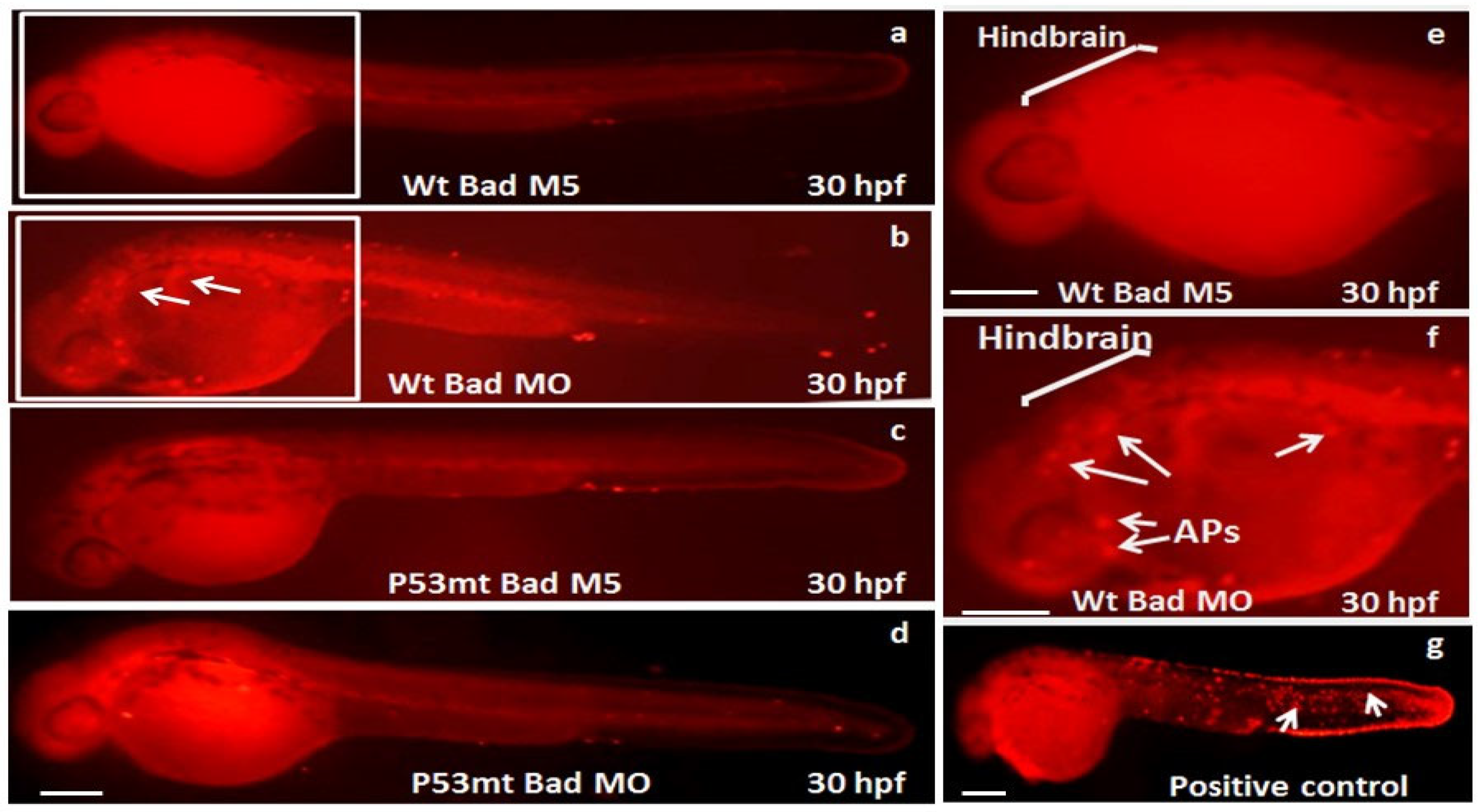
3.3. The Role of P53 Function on the Suppression of Apoptotic Cell Death during Bad Knockdown
3.4. Blockage of P53/Caspase-8-Mediated Death Signaling Induction during P53 Loss-of-Function
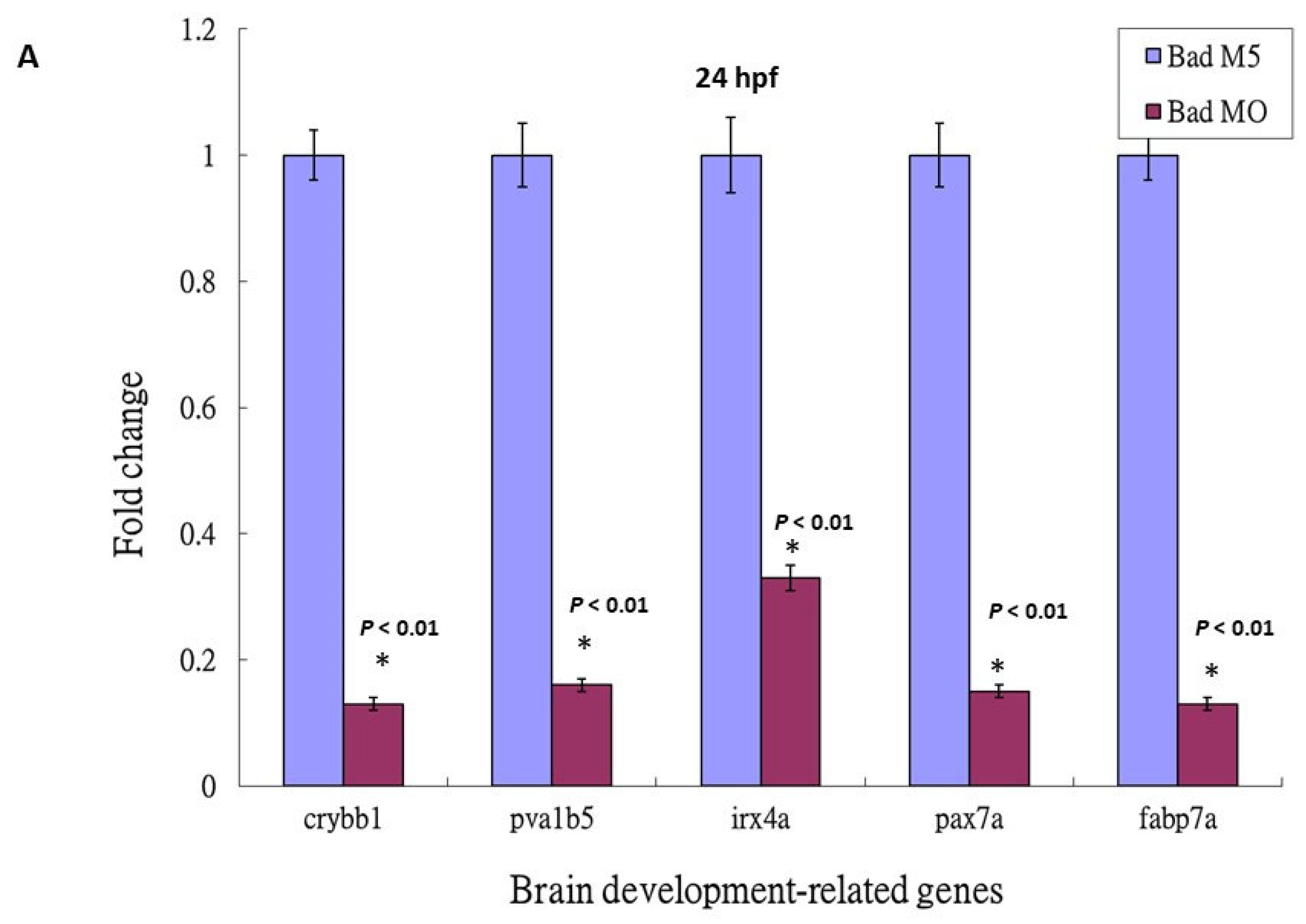
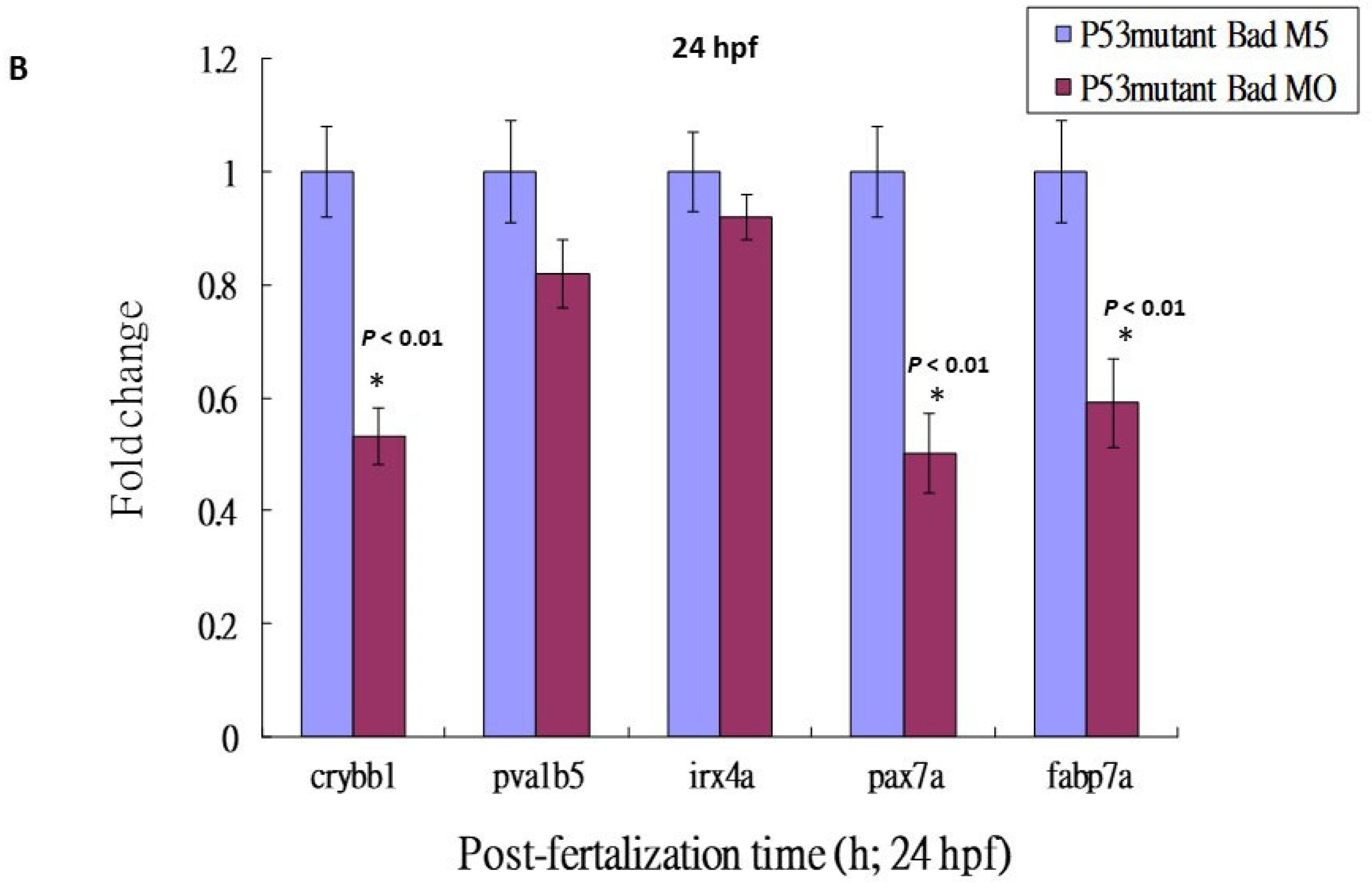
3.5. Loss-of-Function of P53 Enhanced Brain-Developmental Marker Gene Expression
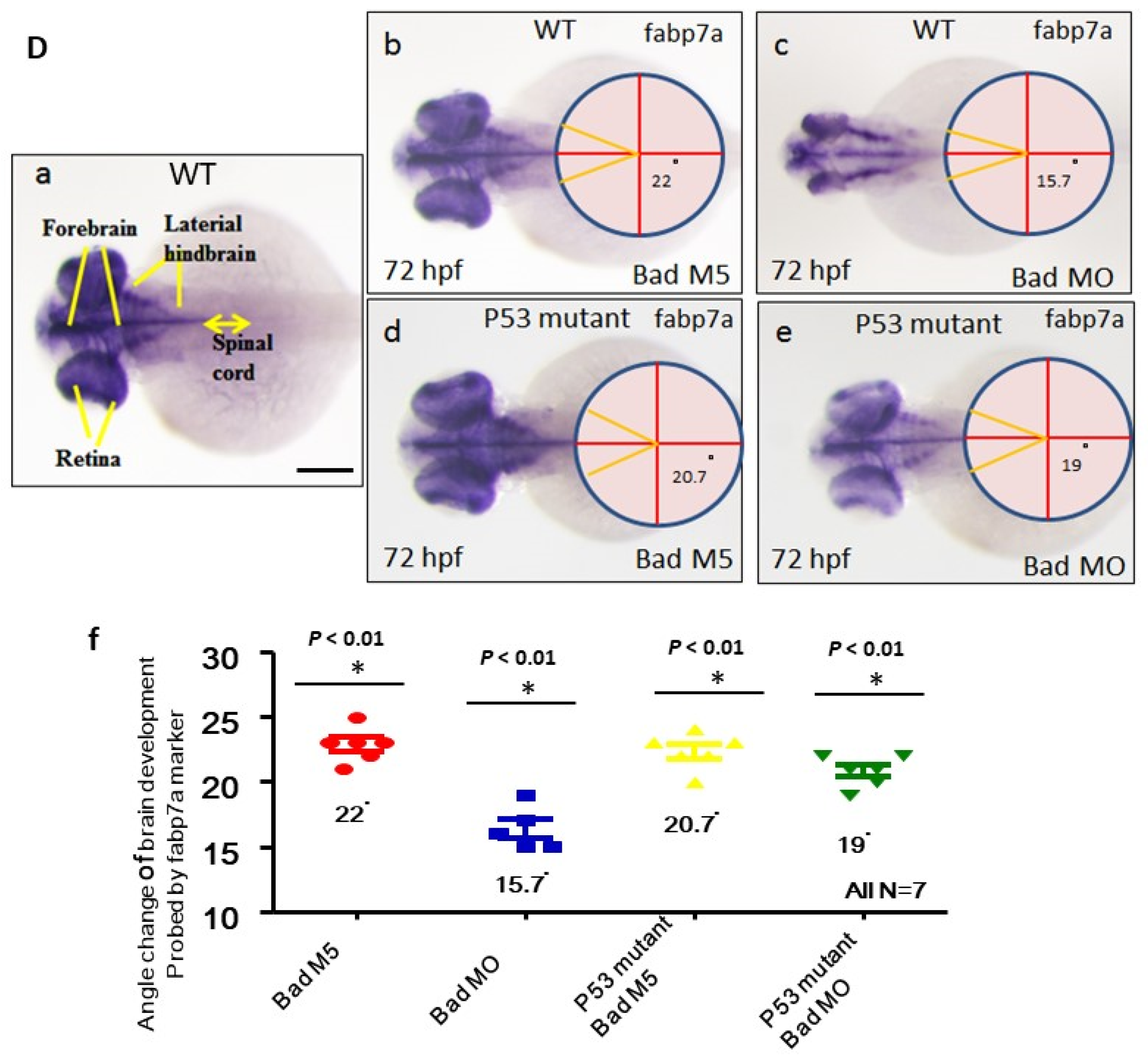
3.6. Directly Restoring Hindbrain Development in a P53 Mutant Line
3.7. Restoring Brain Function through the Swimming Ability Test
4. Discussion
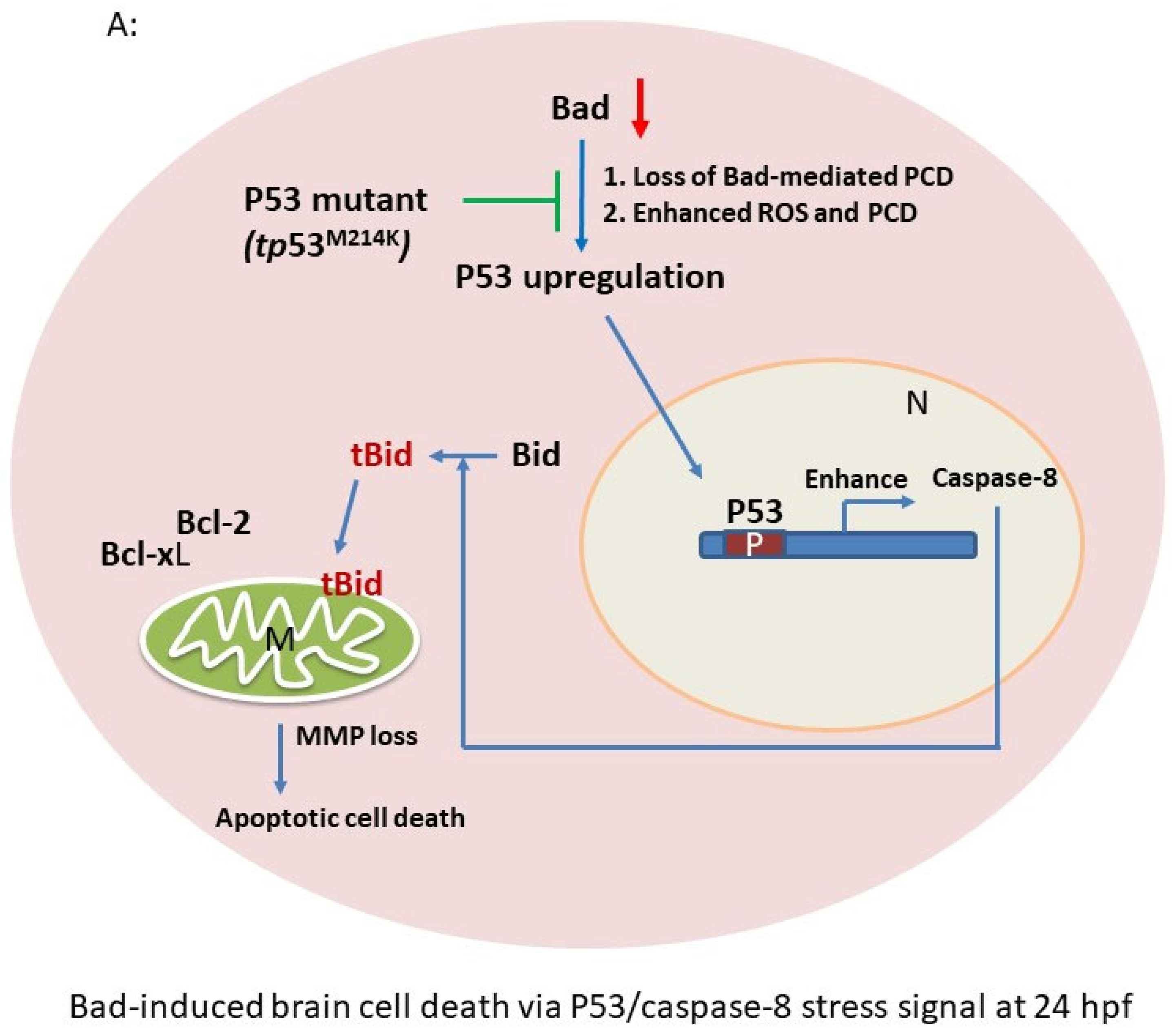
4.1. Bad Regulate Caspase-8/tBid Signaling in Brain Development
4.2. The Role of the Anti-Tumor-Suppressor P53 on Enhancing Brain Development
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Mol. Cell Biol. 2015, 16, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Prdro, J.M.; Kroemer, G. Organelle-specific of cell death. Nat. Cell Biol. 2014, 16, 728–736. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Grether, M.E.; Abrams, J.M.; Young, L.; Farrell, K.; Steller, H. Genetic control of programmed cell death in Drosophila. Science 1994, 264, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Grether, M.E.; Abrams, J.M.; Agapite, J.; White, K.; Steller, H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 1995, 9, 1694–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Nordstrom, W.; Gish, B.; Abrams, J.M. Grim, a novel cell death gene in Drosophila. Genes Dev. 1996, 10, 1773–1782. [Google Scholar] [CrossRef] [Green Version]
- Ellis, R.E.; Yuan, J.Y.; Horvitz, H.R. Mechanisms and functions of cell death. Annu. Rev. Cell Biol. 1991, 7, 663–698. [Google Scholar] [CrossRef]
- Peterson, J.S.; Barkett, M.; McCall, K. Stage-specific regulation of caspase activity in Drosophila oogenesis. Dev. Biol. 2003, 260, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2001, 147, 742–758. [Google Scholar] [CrossRef] [Green Version]
- Monier, B.; Gettings, M.; Gay, G.; Mangeat, T.; Schott, S.; Guarner, A.; Suzanne, M. Apico-basal forces exerted by apoptotic cells drive epithelium folding. Nature 2015, 518, 245–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Cheng, E.H.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar] [CrossRef]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 1996, 87, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.R.; Katsov, A.; Hu, L.; Petros, A.; Fesik, S.W.; Yaffe, M.B.; Greenberg, M.E. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 2000, 6, 41–51. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Del Peso, L.; Gonzalez-Garcia, M.; Page, C.; Herrera, R.; Nunez, G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 1997, 278, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Becknell, B.; Wilm, M.; Mann, M.; Huang, L.J.; Taylor, S.S.; Scott, J.D.; Korsmeyer, S.J. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell 1999, 3, 413–422. [Google Scholar] [CrossRef]
- Harada, H.; Andersen, J.S.; Mann, M.; Terada, N.; Korsmeyer, S.J. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. USA 2001, 98, 9666–9670. [Google Scholar] [CrossRef] [Green Version]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimamura, A.; Ballif, B.A.; Richards, S.A.; Blenis, J. Rsk1 mediates a MEK-MAP kinase cell survival signal. Curr. Biol. 2000, 10, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Ranger, A.M.; Zha, J.; Harada, H.; Datta, S.R.; Danial, N.N.; Gilmore, A.P.; Kutok, J.L.; Le Beau, M.M.; Greenberg, M.E.; Korsmeyer, S.J. Bad-deficient mice develop diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 9324–9329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, Y.C.; Chang, M.S.; Chen, J.Y.; Yen, J.Y.; Lu, I.C.; Chou, C.M.; Huang, C.J. Cloning of zebrafish BAD, a BH3-only proapoptotic protein, whose overexpression leads to apoptosis in COS-1 cells and zebrafish embryos. Biochem. Biophys. Res. Commun. 2003, 304, 667–675. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatruda, J.F.; Patton, E.E. Genetic models of cancer in zebrafish. Int. Rev. Cell. Mol. Biol. 2008, 271, 1–34. [Google Scholar]
- Jing, L.; Zon, L.I. Zebrafish as a model for normal and malignant hematopoiesis. Dis. Model. Mech. 2011, 4, 433–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, A.H.; Spaink, H.P. Host-pathogen interactions made transparent with the zebrafish model. Curr. Drug Targets 2011, 12, 1000–1017. [Google Scholar] [CrossRef]
- Mione, M.C.; Trede, N.S. The zebrafish as a model for cancer. Dis. Model. Mech. 2010, 3, 517–523. [Google Scholar] [CrossRef] [Green Version]
- Renshaw, S.A.; Trede, N.S. A model 450 million years in the making: Zebrafish and vertebrate immunity. Dis. Model. Mech. 2012, 5, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Wittbrodt, J.; Shima, A.; Schartl, M. Medaka—A model organism from the far East. Nat. Rev. Genet. 2002, 3, 53–64. [Google Scholar] [CrossRef]
- Manfred, S. Beyond the zebrafish: Diverse fish species for modeling human disease. Dis. Model. Mech. 2014, 7, 181–192. [Google Scholar]
- Shin, J.T.; Fishman, M.C. From Zebrafish to human: Modular medical models. Annu. Rev. Genom. Hum. Genet. 2002, 3, 311–340. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet 2007, 8, 353–367. [Google Scholar] [CrossRef]
- Kwan, K.M.; Fujimoto, E.; Grabher, C.; Mangum, B.D.; Hardy, M.E.; Campbell, D.S.; Parant, J.M.; Yost, H.J.; Kanki, J.P.; Chien, C.B. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 2007, 236, 3088–3099. [Google Scholar] [CrossRef] [PubMed]
- Halpern, M.E.; Rhee, J.; Goll, M.G.; Akitake, C.M.; Parsons, M.; Leach, S.D. Gal4/UAS transgenic tools and their application to zebrafish. Zebrafish 2008, 5, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Langenau, D.M.; Feng, H.; Berghmans, S.; Kanki, J.P.; Kutok, J.L.; Look, A.T. Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia. Proc. Natl Acad. Sci. USA 2005, 102, 6068–6073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, H.; Hata, K.; Matsuoka, I.; Zang, L.; Kim, Y.; Chu, D.; Juneja, L.R.; Nishimura, N.; Shimada, Y. Anti-obesity natural products tested in juvenile zebrafish obesogenic tests and mouse 3T3-L1 adipogenesis assays. Molecules 2020, 25, 5840. [Google Scholar] [CrossRef] [PubMed]
- Misselbeck, K.; Parolo, S.; Lorenzini, F.; Savoca, V.; Leonardelli, L.; Bora, P.; Morine, M.J.; Mione, M.C.; Domenici, E.; Priami, C. A network-based approach to identify deregulated pathways and drug effects in metabolic syndrome. Nat. Commun. 2019, 10, 5215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asaoka, Y.; Terai, S.; Sakaida, I.; Nishina, H. The expanding role of fish models in understanding non-alcoholic fatty liver disease. Dis. Model. Mech. 2013, 6, 905–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, H.; Shimada, Y.; Zang, L.; Terasawa, M.; Nishiura, K.; Matsuda, K.; Toombs, C.; Langdon, C.; Nishimura, N. Novel anti-obesity properties of palmaria mollis in zebrafish and mouse models. Nutrients 2018, 10, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summerton, J.; Weller, D. Morpholino antisense oligomers; design, preparation, and properties. Antisense Nucleic Drug Dev. 1997, 42, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene knockdown in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.C.; Wu, J.L.; Hong, J.R. Pro-apoptotic Bad involved in brain development, when severely defect induces dramatic malformation in zebrafish. Int. J. Mol. Sci. 2021, 22, 4832. [Google Scholar] [CrossRef] [PubMed]
- Berghmans, S.; Murphey, R.D.; Wienholds, E.; Neuberg, D.; Kutok, J.L.; Fletcher, C.D.; Morris, J.P.; Liu, T.X.; Schulte-Merker, S.; Kanki, J.P.; et al. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Brachydanio rerio), 2nd ed.; University of Oregon Press: Eugene, OR, USA, 2000; 300p. [Google Scholar]
- Hong, J.R.; Lin, G.H.; Lin, C.J.F.; Wang, W.P.; Lee, C.C.; Wu, J.L. The phosphatidylserine receptor is required for the engulfment of dead apoptotic cell and for normal development in zebrafish. Development 2004, 131, 5417–5427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.C.; Hong, J.R. Betanodavirus B2 causes ATP depletion-induced cell death via mitochondrial targeting and Complex II inhibition in vitro and in vivo. J. Biol. Chem. 2010, 285, 39801–39810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiu, W.L.; Huang, K.L.; Hung, J.C.; Li, H.C.; Wu, J.L.; Hong, J.R. Knockdown of zebrafish YY1a can downregulate the phosphatidylserine (PS) receptor expression, leading to abnormal brain and heart development. J. Biomed. Sci. 2016, 23, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.J.; Wang, S.; Cao, N.Q.; Yan, Y.B.; Zhu, S.Q. A Novel Mutation in CRYBB1 Associated with Congenital Cataract-Microcornea Syndrome: The p.Ser129Arg Mutation Destabilizes the βB1/βA3-crystallin Heteromer But Not the βB1-crystallin Homomer. Hum. Mut. 2010, 32, E2050–E2060. [Google Scholar] [CrossRef] [Green Version]
- Friedberg, F. Parvalbumin isoforms in zebrafish. Mol. Biol. Rep. 2005, 32, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Feijóo, C.G.; Saldias, M.P.; De la Paz, V.F.; Gómez-Skarmeta, J.L.; Allende, M.L. Formation of posterior cranial placode derivatives requires the Iroquois transcription factor irx4a. Mol. Cell. Neurosci. 2009, 40, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Ziman, M.; Fletcher, S.; Kay, P.H. Alternate Pax7 Transcripts are Expressed Specifically in Skeletal Muscle, Brain and Other Organs of Adult Mice. Int. J. Biochem. Cell Biol. 1997, 29, 1029–1036. [Google Scholar] [CrossRef]
- Liu, R.Z.; Denovan-Wright, E.M.; Degrave, A.; Thisse, C.; Thisse, B.; Wright, J.M. Differential expression of duplicated genes for brain-type fatty acid-binding proteins (fabp7a and fabp7b) during early development of the CNS in zebrafish (Danio rerio). Gene Exp. Patterns 2004, 4, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yin, X.M.; Chao, D.T.; Milliman, C.L.; Korsmeyer, S.J. BID: A novel BH3 domain-only death agonist. Genes Dev. 1996, 10, 2859–2869. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.R.; Wu, J.L. Induction of apoptotic death in cells via Bad gene expression by infectious pancreatic necrosis virus infection. Cell Death Differ. 2002, 9, 113–124. [Google Scholar] [CrossRef]
- Miller, F.D.; Pozniak, C.D.; Walsh, G.S. Neuronal life and death: An essential role for the p53 family. Cell Death Differ. 2000, 7, 880–888. [Google Scholar] [CrossRef]
- Levine, A.L.; Hu, W.; Feng, Z. The p53 pathway: What questions remain to be explored? Cell Death Differ. 2006, 13, 1027–1036. [Google Scholar] [CrossRef]
- Tedeschi, A.; Giovanni, S.D. The non-apoptotic role of p53 in neuronal biology: Enlightening the dark side of the moon. EMBO Rep. 2009, 10, 576–583. [Google Scholar] [CrossRef] [Green Version]
- Kantari, C.; Walczak, H. Caspase-8 and Bid: Caught in the act between death preceptors and mitochondria. Biochim. Biophys. Acta 2011, 1813, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Aloyz, R.S.; Bamji, S.X.; Pozniak, C.D.; Toma, J.G.; Atwal, J.; Kaplan, D.R.; Miller, F.D. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J. Cell Biol. 1998, 143, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.N.; Tolkovsky, A.M. A role for MAPK/ERK insympathetic neuron survival: Protection against a p53-dependent, JNK-independent induction of apoptosis by cytosine arabinoside. J. Neurosci. 1999, 19, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, J.; Galindo, M.F.; Prehn, J.H.; Weichselbaum, R.R.; Beckett, M.; Ghadge, G.D.; Roos, R.P.; Leiden, J.M.; Miller, R.J. p53 expression induces apoptosis in hippocampal pyramidal neuron cultures. J. Neurosci. 1997, 17, 1397–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slack, R.S.; Belliveau, D.J.; Rosenberg, M.; Atwal, J.; Lochmu, L.H.; Aloyz, R.; Haghighi, A.; Lach, B.; Seth, P.; Cooper, E.; et al. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J. Cell Biol. 1996, 135, 1085–1096. [Google Scholar] [CrossRef]
- Vogel, K.S.; Parada, L.F. Sympathetic neuron survival and proliferation are prolonged by loss of p53 and neurofibromin. Mol. Cell Neurosci. 1998, 11, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Chatoo, W.; Abdouh, M.; Bernier, G. p53 Pro-Oxidant Activity in the Central Nervous System: Implication in Aging and Neurodegenerative Diseases. Antioxi. Redox Signal. 2011, 15, 1729–1737. [Google Scholar] [CrossRef]
- Cole, L.K.; Ross, L.S. Apoptosis in the developing zebrafish embryo. Dev. Biol. 2001, 240, 123–142. [Google Scholar] [CrossRef] [Green Version]
- Blader, P.; Strahle, U. Zebrafish developmental genetics and central nervous system development. Hum. Mol. Genet. 2000, 9, 945–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, C.Y.; Roth, K.A.; Flavell, R.A.; Rakic, P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci 2000, 23, 291–297. [Google Scholar] [CrossRef]
- Blaschke, A.J.; Staley, K.; Chun, J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 1996, 122, 1165–1174. [Google Scholar] [CrossRef]
- Blaschke, A.J.; Weiner, J.A.; Chun, J. Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J. Comp. Neurol. 1998, 396, 39–50. [Google Scholar] [CrossRef]
- Thomaidou, D.; Mione, M.C.; Cavanagh, J.F.; Parnavelas, J.G. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J. Neurosci. 1997, 17, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziv, L.; Muto, A.; Schoonheim, P.J.; Meijsing, S.H.; Strasser, D.; Ingraham, H.A.; Schaaf, M.J.; Yamamoto, K.R.; Baier, H. An affective disorder in zebrafish with mutation of the glucocorticoid receptor. Mol. Psychiatry 2013, 18, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, B.B.; Schoonheim, P.J.; Ziv, L.; Voelker, L.; Baier, H.; Gahtan, E. A zebrafish model of glucocorticoid resistance shows serotonergic modulation of the stress response. Front. Behav. Neurosci. 2012, 6, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lee, B.; Choi, J.H.; Kim, J.H.; Kim, C.H.; Shin, H.S. Deficiency of a brain-specific chemokine-like molecule, SAM3, induces cardinal phenotypes of autism spectrum disorders in mice. Sci. Rep. 2017, 7, 16503. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.E.; Schwartz, C.E. X-linked intellectual disability: Unique vulnerability of the male genome. Dev. Disabil. Res. Rev. 2009, 15, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Khan, K.; Armfield-Uhas, K.; Srikanth, S.; Thompson, N.A.; Pardo, M.; Yu, L.; Norris, J.W.; Peng, Y.; Gripp, K.W.; et al. Mutations in FAM50A suggest that Armfield XLID syndrome is a spliceosomopathy. Nat. Commun. 2020, 11, 3698. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Sequences for pGEM-T Vector Cloning | ||
|---|---|---|
| Gene | Forward 5′ → 3′ | Reverse 5′ → 3′ |
| p53 | CTACTAAACTACATGTGCAATAGCAG | CTGAGGCAGGCACCACATCACT |
| bad | ATG GCA CAT ATG TTT AAT ATC TCT GA | CTACTCTGCGGGGCGCGA |
| fabp7a | CTCTCAACATGGTCGATGCATT | CTGGACATTATGCCTTCTCGTA |
| pax2a | CTTCTAACAGGCACATCCCAT | CATTAACCCTCACTAAAGGGAACTATCCGTTCAAAG CCCG |
| Primer Sequences | ||
|---|---|---|
| Gene | Forward 5′ → 3′ | Reverse 5′ → 3′ |
| β-actin | ACT GTA TTG TCT GGT GGT AC | TAC TCC TGC TTG CTA ATC C |
| p53 | ACC ACT GGG ACC AAA CGT AG | CAG AGT CGC TTC TTC CTT CG |
| Caspase-8 | CCA GAC AAT CTG GAT GAA CTT TAC | TGC AAA CTG CTT TAT CTC ATC T |
| pva1b5 | ATGGCACTTGCAGGAATCCTGA | TGTTGGTCTCGGCCTCTGTGAG |
| crybb1 | ATGTCTCAGACCGCCAAATCCG | GCCCTGGAAGTTCTCCTGGTCA |
| pax7a | CCAGGAACAGTTCCTCGAATGATG | CCGTGATGGGCCATTTCCAC |
| irx4a | GCGGACAAGGCTACGGGAATT | AGCGTTTTCCTG CGGGTCC |
| fabp7a | TGTGCCACTTGGAAACTGGTTGAC | CCCAGTTTGAAGGAGATCTCGGTG |
| Catalase | TAAAGGAGCAGGAGCGTTTGGCTA | TTCACTGCGAAACCACGAGGATCT |
| Mn-sod | CCGGACTATGTTAAGGCCATCT | ACACTCGGTTGCTCTCTTTTCTCT |
| Cu/Zn-sod | GTCGTCTGGCTTGTGGAGTG | TGTCAGCGGGCTAGTGCTT |
| nrf2a | GAGCGGGAGAAATCACACAGAATG | CAGGAGCTGCATGCACTCATCG |
| nrf2b | GGCAGAGGGAGGAGGAGACCAT | AAACAGCAGGGCAGACAACAAGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, J.-C.; Wu, J.-L.; Li, H.-C.; Chiu, H.-W.; Hong, J.-R. The Proapoptotic Gene Bad Regulates Brain Development via p53-Mediated Stress Signals in Zebrafish. Cells 2021, 10, 2820. https://doi.org/10.3390/cells10112820
Hung J-C, Wu J-L, Li H-C, Chiu H-W, Hong J-R. The Proapoptotic Gene Bad Regulates Brain Development via p53-Mediated Stress Signals in Zebrafish. Cells. 2021; 10(11):2820. https://doi.org/10.3390/cells10112820
Chicago/Turabian StyleHung, Jo-Chi, Jen-Leih Wu, Huei-Ching Li, Hsuan-Wen Chiu, and Jiann-Ruey Hong. 2021. "The Proapoptotic Gene Bad Regulates Brain Development via p53-Mediated Stress Signals in Zebrafish" Cells 10, no. 11: 2820. https://doi.org/10.3390/cells10112820
APA StyleHung, J.-C., Wu, J.-L., Li, H.-C., Chiu, H.-W., & Hong, J.-R. (2021). The Proapoptotic Gene Bad Regulates Brain Development via p53-Mediated Stress Signals in Zebrafish. Cells, 10(11), 2820. https://doi.org/10.3390/cells10112820