
Pulmonary Alveolar Stem Cell Senescence, Apoptosis, and Differentiation by p53-Dependent and -Independent Mechanisms in Telomerase-Deficient Mice

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Respiration Function Assay
2.3. Fibrosis-Associated Masson’s Staining, Collagen Volume Fraction
2.4. In Situ Senescence-Associated (SA)-β-Gal Staining in Lung Tissues
2.5. Double Immunofluorescence Staining Analysis
2.6. Isolation of AEC2 Cells from Lung Tissues by Fluorescence-Activated Cell Sorting (FACS)
2.7. Real-Time Quantitative PCR
2.8. Statistical Analysis
3. Results
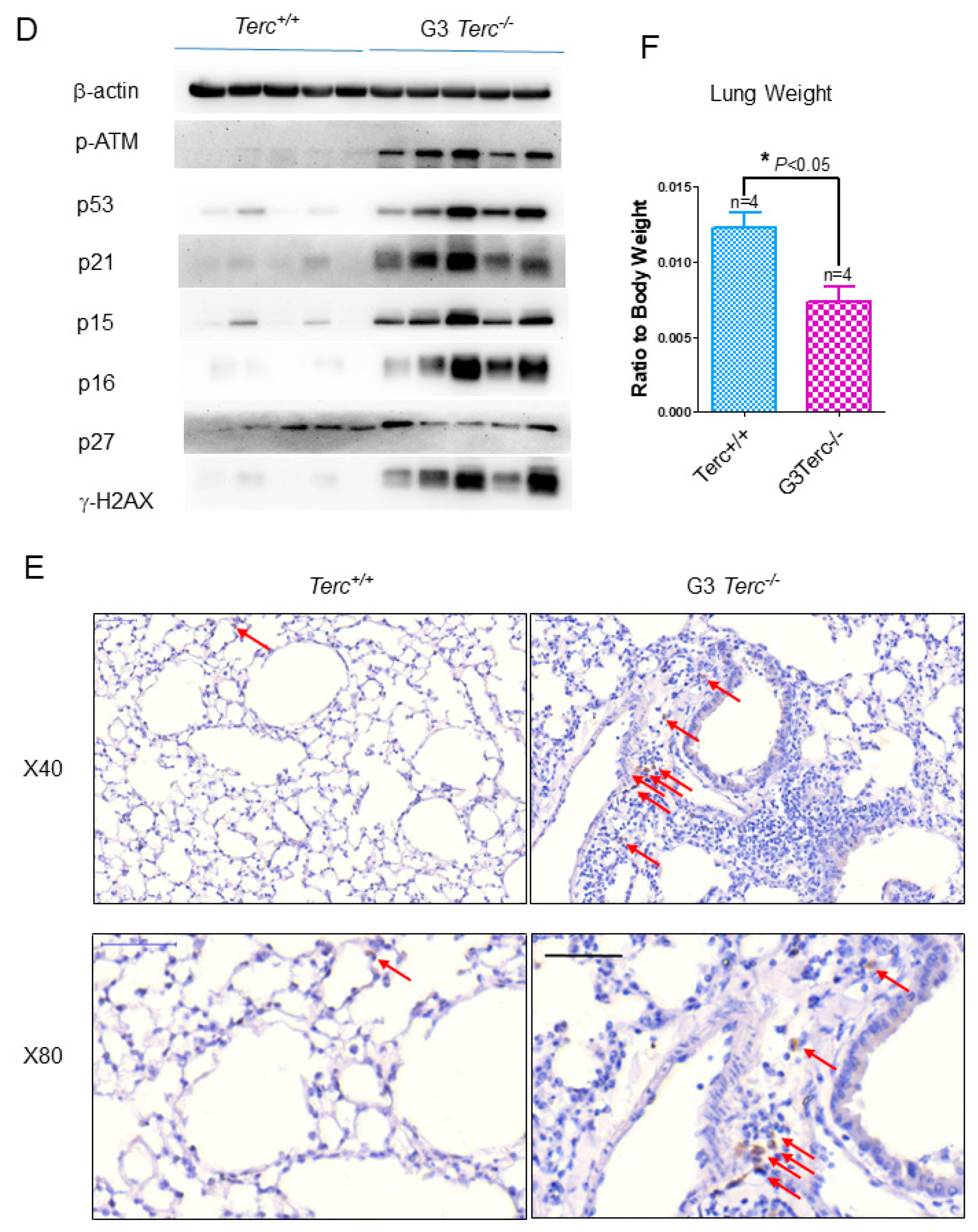
3.1. Age-Related Activation of the p53 Signalling Pathway in Pulmonary Fibrosis in Mice
3.2. The Third Generation of Telomerase RNA Subunit Deficiency Causes Pulmonary Senescence and Low-Grade Inflammation
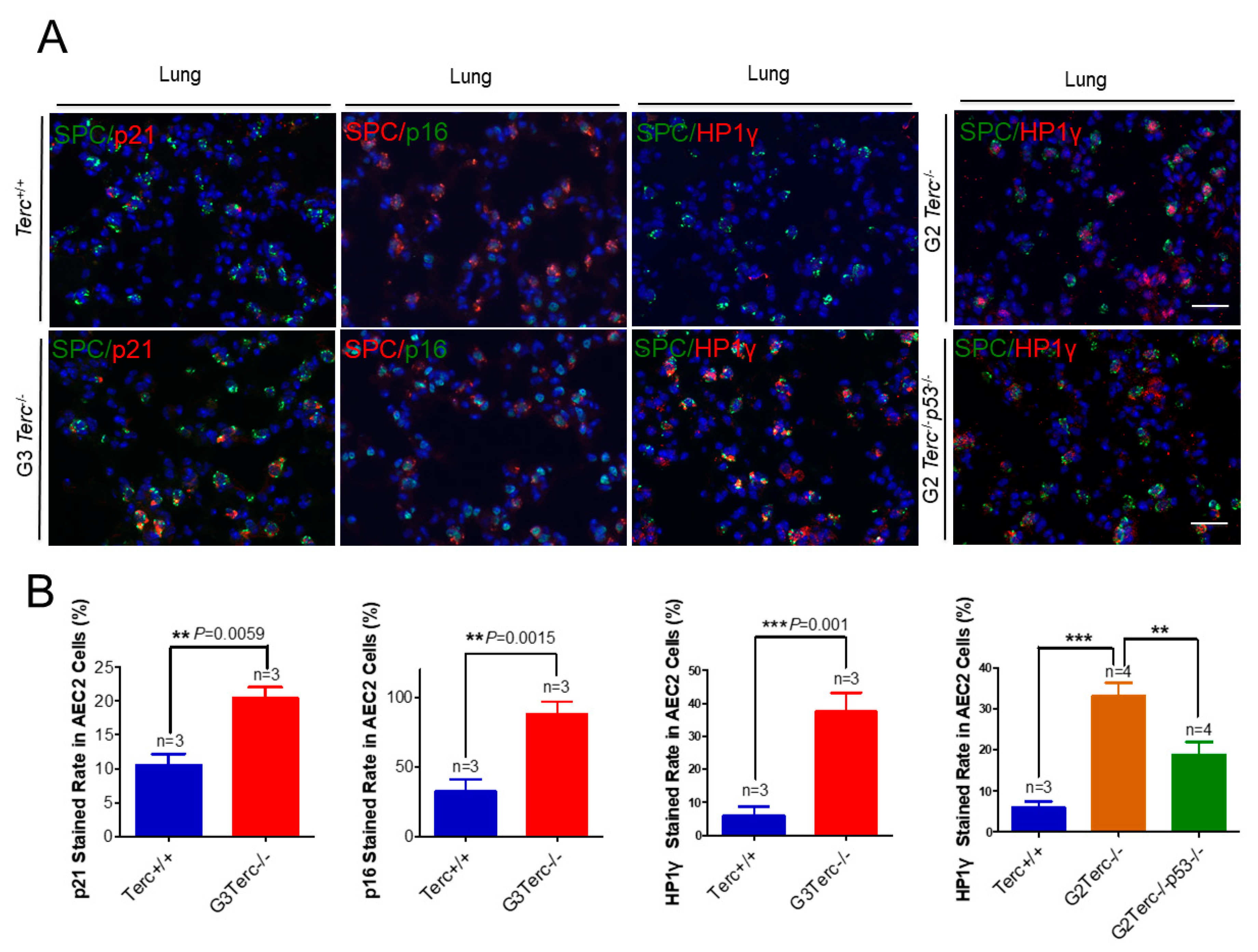
3.3. Pulmonary AEC2 Stem Cells Undergo Senescence by Mechanisms Involving p21 in Mice Deficient of the Telomerase RNA Component
3.4. Marked Increase in Apoptosis-Related Cleaved Caspase-3 Staining in AEC2 Cells from G3 TERC Knockout Mouse Lungs
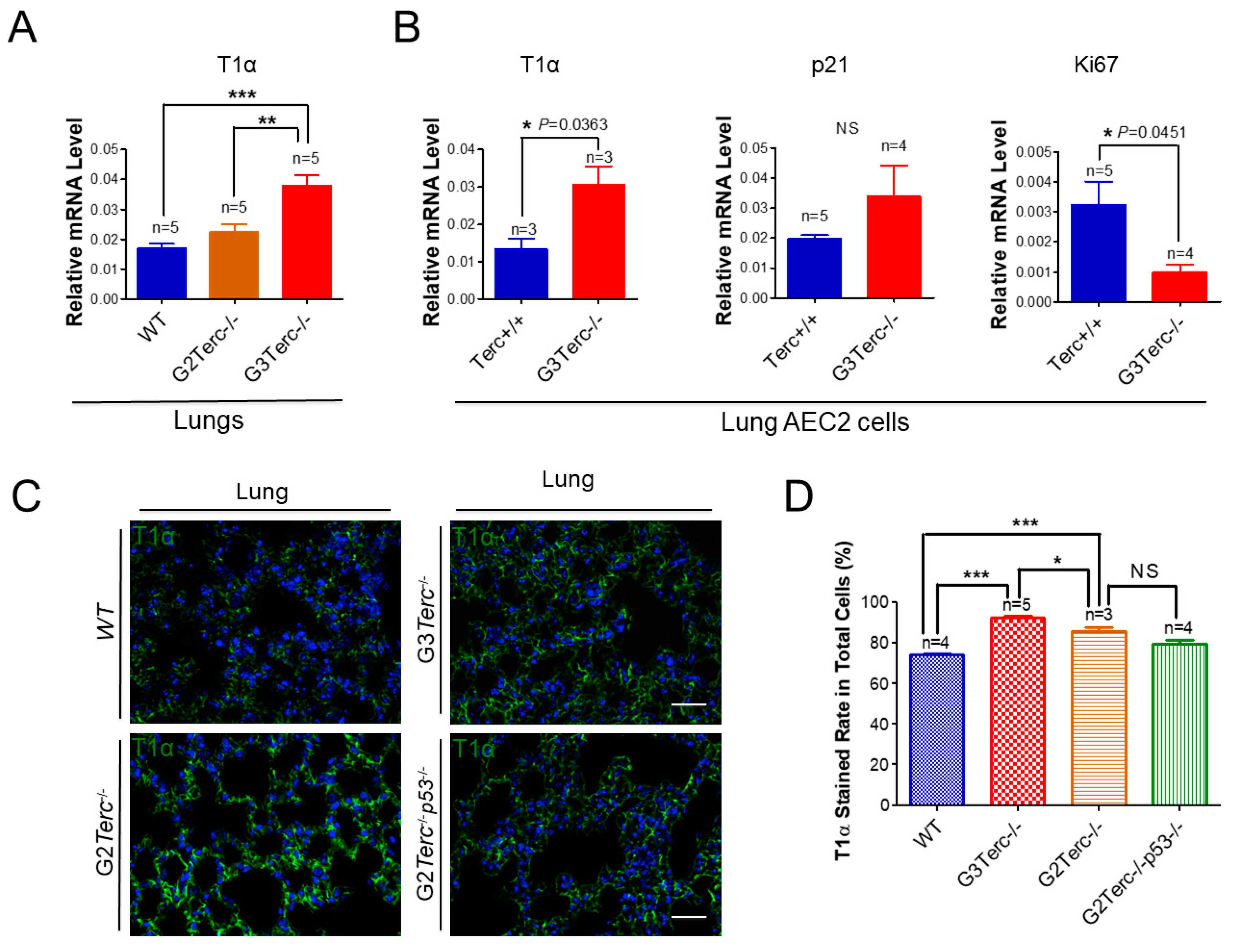
3.5. AEC1 Cell-Associated T1α Is Significantly Elevated in Both G3 TERC Knockout Mouse Lungs and AEC2 Cells
3.6. Rescue of Alveolar Stem Cell Senescence and Apoptosis in the Late Generation of TERC KO Mice by p53 Deficiency
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Borok, Z. Alveolar epithelium: Beyond the barrier. Am. J. Respir. Cell Mol. Biol. 2014, 50, 853–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Fehrenbach, H. Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir. Res. 2001, 2, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, R.; Li, G.; Wang, Z.; Liu, J.; Liang, Y.; Liu, J.P. FBW7 Mediates Senescence and Pulmonary Fibrosis through Telomere Uncapping. Cell Metab. 2020, 32, 860–877.e869. [Google Scholar] [CrossRef]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [Green Version]
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104. [Google Scholar] [CrossRef] [Green Version]
- Dyer, C. The interaction of ageing and lung disease. Chron. Respir. Dis. 2012, 9, 63–67. [Google Scholar] [CrossRef]
- Zeki, A.A.; Schivo, M.; Chan, A.L.; Hardin, K.A.; Kenyon, N.J.; Albertson, T.E.; Rosenquist, G.L.; Louie, S. Geoepidemiology of COPD and idiopathic pulmonary fibrosis. J. Autoimmun. 2010, 34, J327–J338. [Google Scholar] [CrossRef]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [Green Version]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, A.R.; Wyatt, H.D.; Ting, N.S.; Beattie, T.L. hTERT mutations associated with idiopathic pulmonary fibrosis affect telomerase activity, telomere length, and cell growth by distinct mechanisms. Aging Cell 2012, 11, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Dressen, A.; Abbas, A.R.; Cabanski, C.; Reeder, J.; Ramalingam, T.R.; Neighbors, M.; Bhangale, T.R.; Brauer, M.J.; Hunkapiller, J.; Reeder, J.; et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: A candidate gene sequencing study. Lancet Respir. Med. 2018, 6, 603–614. [Google Scholar] [CrossRef]
- Alder, J.K.; Guo, N.; Kembou, F.; Parry, E.M.; Anderson, C.J.; Gorgy, A.I.; Walsh, M.F.; Sussan, T.; Biswal, S.; Mitzner, W.; et al. Telomere length is a determinant of emphysema susceptibility. Am. J. Respir. Crit. Care Med. 2011, 184, 904–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Sandford, A.J.; Connett, J.E.; Yan, J.; Mui, T.; Li, Y.; Daley, D.; Anthonisen, N.R.; Brooks-Wilson, A.; Man, S.F.; et al. The relationship between telomere length and mortality in chronic obstructive pulmonary disease (COPD). PLoS ONE 2012, 7, e35567. [Google Scholar] [CrossRef]
- Rode, L.; Bojesen, S.E.; Weischer, M.; Vestbo, J.; Nordestgaard, B.G. Short telomere length, lung function and chronic obstructive pulmonary disease in 46,396 individuals. Thorax 2013, 68, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Blasco, M.A.; Funk, W.; Villeponteau, B.; Greider, C.W. Functional characterization and developmental regulation of mouse telomerase RNA. Science 1995, 269, 1267–1270. [Google Scholar] [CrossRef] [Green Version]
- Blasco, M.A.; Lee, H.W.; Hande, M.P.; Samper, E.; Lansdorp, P.M.; DePinho, R.A.; Greider, C.W. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997, 91, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhang, K.; Chen, H.; Zhao, X.; Wang, J.; Li, L.; Cong, Y.; Ju, Z.; Xu, D.; Williams, B.R.; et al. Telomerase Deficiency Causes Alveolar Stem Cell Senescence-associated Low-grade Inflammation in Lungs. J. Biol. Chem. 2015, 290, 30813–30829. [Google Scholar] [CrossRef] [Green Version]
- Rubtsova, M.; Naraykina, Y.; Vasilkova, D.; Meerson, M.; Zvereva, M.; Prassolov, V.; Lazarev, V.; Manuvera, V.; Kovalchuk, S.; Anikanov, N.; et al. Protein encoded in human telomerase RNA is involved in cell protective pathways. Nucleic Acids Res. 2018, 46, 8966–8977. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, A.R.; Ju, Z.; Djojosubroto, M.W.; Schienke, A.; Lechel, A.; Schaetzlein, S.; Jiang, H.; Stepczynska, A.; Wang, C.; Buer, J.; et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat. Genet. 2007, 39, 99–105. [Google Scholar] [CrossRef]
- Sperka, T.; Song, Z.; Morita, Y.; Nalapareddy, K.; Guachalla, L.M.; Lechel, A.; Begus-Nahrmann, Y.; Burkhalter, M.D.; Mach, M.; Schlaudraff, F.; et al. Puma and p21 represent cooperating checkpoints limiting self-renewal and chromosomal instability of somatic stem cells in response to telomere dysfunction. Nat. Cell Biol. 2011, 14, 73–79. [Google Scholar] [CrossRef]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, Z.; Liu, J.P. Identification of peptidomimetic telomere dysfunction inhibitor (TELODIN) through telomere dysfunction-induced foci (TIF) assay. STAR Protoc. 2021, 2, 100620. [Google Scholar] [CrossRef]
- Wang, L.; Yu, X.; Liu, J.P. Telomere Damage Response and Low-Grade Inflammation. Adv. Exp. Med. Biol. 2017, 1024, 213–224. [Google Scholar] [CrossRef]
- Razdan, N.; Vasilopoulos, T.; Herbig, U. Telomere dysfunction promotes transdifferentiation of human fibroblasts into myofibroblasts. Aging Cell 2018, 17, e12838. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, J.; Ju, Z.; Rudolph, K.L. Telomere dysfunctional environment induces loss of quiescence and inherent impairments of hematopoietic stem cell function. Aging Cell 2012, 11, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yi, W.; Morita, Y.; Wang, H.; Cong, Y.; Liu, J.P.; Xiao, Z.; Rudolph, K.L.; Cheng, T.; Ju, Z. Wip1 deficiency impairs haematopoietic stem cell function via p53 and mTORC1 pathways. Nat. Commun. 2015, 6, 6808. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Rueda, B.R.; Tilly, K.I.; Botros, I.W.; Jolly, P.D.; Hansen, T.R.; Hoyer, P.B.; Tilly, J.L. Increased bax and interleukin-1beta-converting enzyme messenger ribonucleic acid levels coincide with apoptosis in the bovine corpus luteum during structural regression. Biol. Reprod. 1997, 56, 186–193. [Google Scholar] [CrossRef]
- Lv, X.; Cai, Z.; Li, S. Increased apoptosis rate of human decidual cells and cytotrophoblasts in patients with recurrent spontaneous abortion as a result of abnormal expression of CDKN1A and Bax. Exp. Ther. Med. 2016, 12, 2865–2868. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.I.; Millien, G.; Hinds, A.; Cao, Y.; Seldin, D.C.; Williams, M.C. T1alpha, a lung type I cell differentiation gene, is required for normal lung cell proliferation and alveolus formation at birth. Dev. Biol. 2003, 256, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissleder, C.; Barry, G.; Fung, S.J.; Wong, M.W.; Double, K.L.; Webster, M.J.; Weickert, C.S. Reduction in IGF1 mRNA in the Human Subependymal Zone During Aging. Aging Dis. 2019, 10, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, E.H. Structure and function of telomeres. [Review]. Nature 1991, 350, 569–573. [Google Scholar] [CrossRef]
- Blackburn, E.H. Telomeres and their synthesis. Science 1990, 249, 489–490. [Google Scholar] [CrossRef]
- Lee, J.; Reddy, R.; Barsky, L.; Scholes, J.; Chen, H.; Shi, W.; Driscoll, B. Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L57–L70. [Google Scholar] [CrossRef]
- Jackson, S.R.; Lee, J.; Reddy, R.; Williams, G.N.; Kikuchi, A.; Freiberg, Y.; Warburton, D.; Driscoll, B. Partial pneumonectomy of telomerase null mice carrying shortened telomeres initiates cell growth arrest resulting in a limited compensatory growth response. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L898–L909. [Google Scholar] [CrossRef]
- Flores, I.; Blasco, M.A. A p53-dependent response limits epidermal stem cell functionality and organismal size in mice with short telomeres. PLoS ONE 2009, 4, e4934. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Muller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Shao, R.; Li, F.; Monteiro, M.; Liu, J.P.; Xu, Z.P.; Gu, W. PI3K/Akt/mTOR Pathway Dual Inhibitor BEZ235 Suppresses the Stemness of Colon Cancer Stem Cells. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Robin, J.D.; Ludlow, A.T.; Batten, K.; Magdinier, F.; Stadler, G.; Wagner, K.R.; Shay, J.W.; Wright, W.E. Telomere position effect: Regulation of gene expression with progressive telomere shortening over long distances. Genes Dev. 2014, 28, 2464–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colla, S.; Ong, D.S.; Ogoti, Y.; Marchesini, M.; Mistry, N.A.; Clise-Dwyer, K.; Ang, S.A.; Storti, P.; Viale, A.; Giuliani, N.; et al. Telomere dysfunction drives aberrant hematopoietic differentiation and myelodysplastic syndrome. Cancer Cell 2015, 27, 644–657. [Google Scholar] [CrossRef] [Green Version]
- Fali, T.; Fabre-Mersseman, V.; Yamamoto, T.; Bayard, C.; Papagno, L.; Fastenackels, S.; Zoorab, R.; Koup, R.A.; Boddaert, J.; Sauce, D.; et al. Elderly human hematopoietic progenitor cells express cellular senescence markers and are more susceptible to pyroptosis. JCI Insight 2018, 3, e95319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotorynski, E. Telomere crisis activates autophagic death. Nat. Rev. Mol. Cell Biol. 2019, 20, 133. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; Wang, L.; Hong, X.; Chen, H.; Shi, Y.; Liu, Y.; Liu, J.; Liu, J.-P. Pulmonary Alveolar Stem Cell Senescence, Apoptosis, and Differentiation by p53-Dependent and -Independent Mechanisms in Telomerase-Deficient Mice. Cells 2021, 10, 2892. https://doi.org/10.3390/cells10112892
Zhang K, Wang L, Hong X, Chen H, Shi Y, Liu Y, Liu J, Liu J-P. Pulmonary Alveolar Stem Cell Senescence, Apoptosis, and Differentiation by p53-Dependent and -Independent Mechanisms in Telomerase-Deficient Mice. Cells. 2021; 10(11):2892. https://doi.org/10.3390/cells10112892
Chicago/Turabian StyleZhang, Kexiong, Lihui Wang, Xiaojing Hong, Hao Chen, Yao Shi, Yingying Liu, Jun Liu, and Jun-Ping Liu. 2021. "Pulmonary Alveolar Stem Cell Senescence, Apoptosis, and Differentiation by p53-Dependent and -Independent Mechanisms in Telomerase-Deficient Mice" Cells 10, no. 11: 2892. https://doi.org/10.3390/cells10112892
APA StyleZhang, K., Wang, L., Hong, X., Chen, H., Shi, Y., Liu, Y., Liu, J., & Liu, J. -P. (2021). Pulmonary Alveolar Stem Cell Senescence, Apoptosis, and Differentiation by p53-Dependent and -Independent Mechanisms in Telomerase-Deficient Mice. Cells, 10(11), 2892. https://doi.org/10.3390/cells10112892