
Altered Mucus Barrier Integrity and Increased Susceptibility to Colitis in Mice upon Loss of Telocyte Bone Morphogenetic Protein Signalling

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Induction and Assessment of DSS-Induced Colitis and Recovery
2.3. Histological Analysis and Grading of Colitis
2.4. Intestinal Permeability Assay
2.5. Electron Microscopy
2.6. Immunofluorescence and Fluorescence In Situ Hybridisation
2.7. Analysis of Post-Translational Modifications in Goblet Cell and Mucins
2.8. Quantification of Cell Number, Cell Distribution, Vesicle and Goblet Cell Post-Translational Modifications
2.9. RNA Extraction and Gene Expression
2.10. Statistical Analysis
3. Results
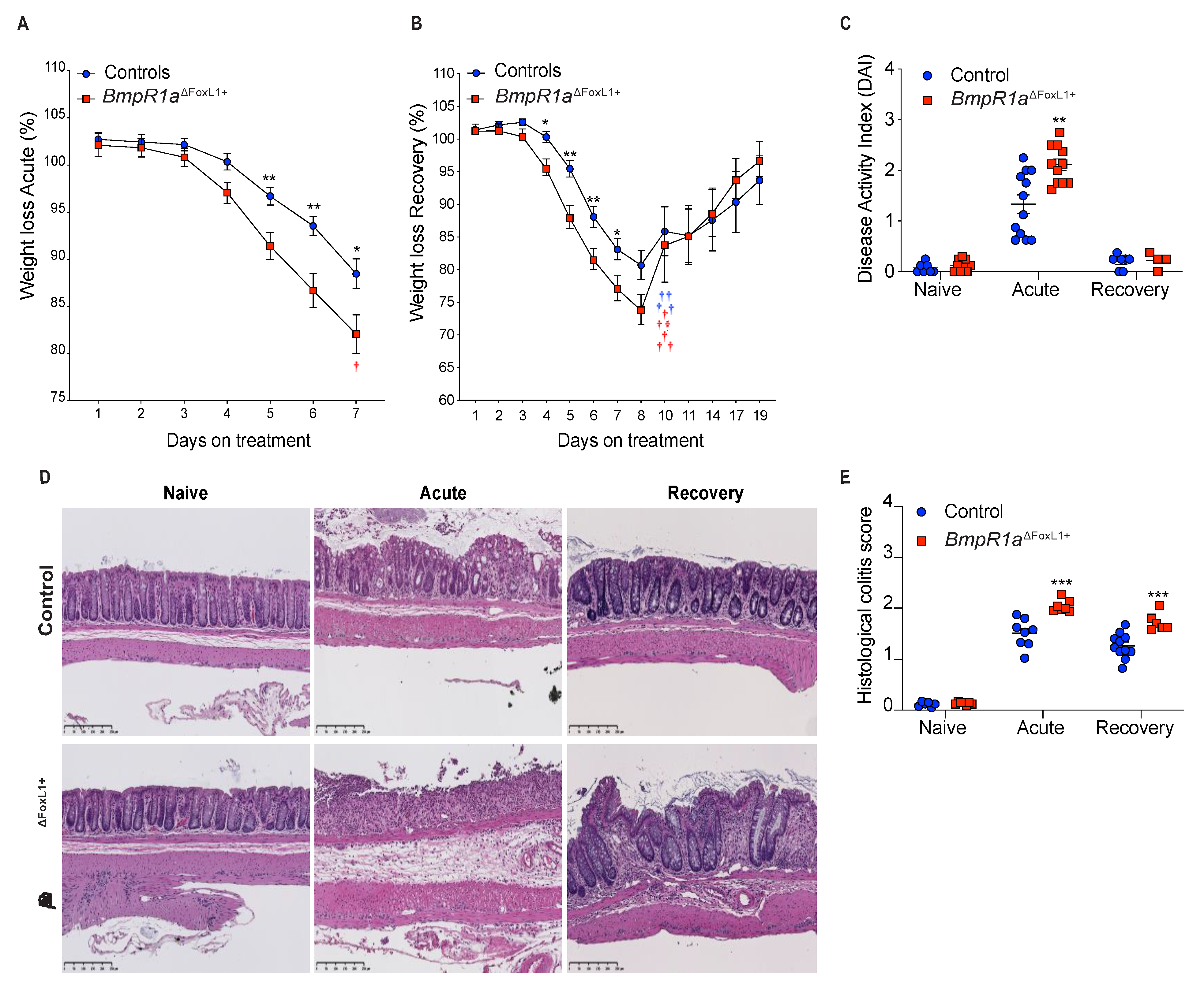
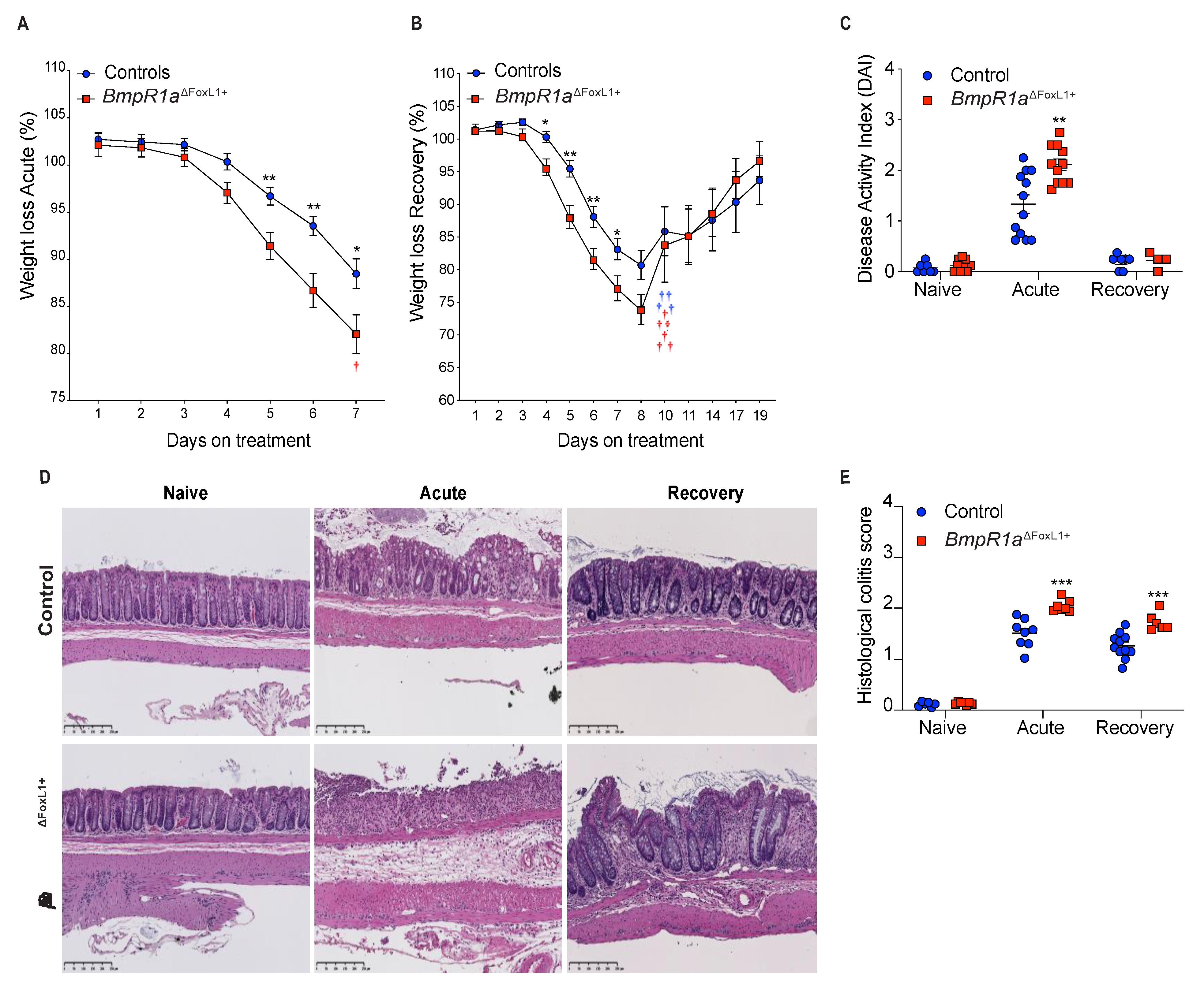
3.1. BmpR1aΔFoxL1+ Mice Have Increased Susceptibility to Experimental Colitis and Delayed Wound Healing
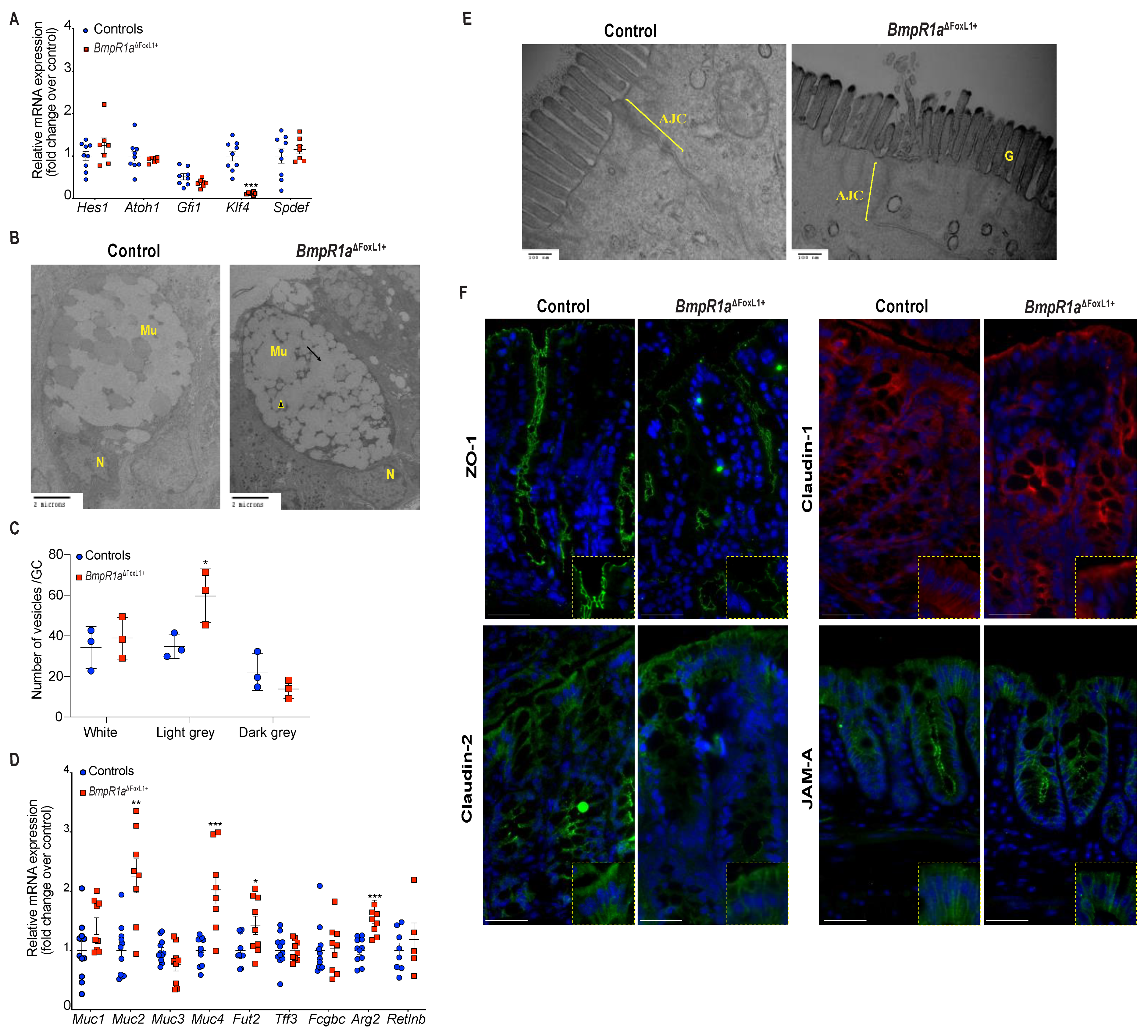
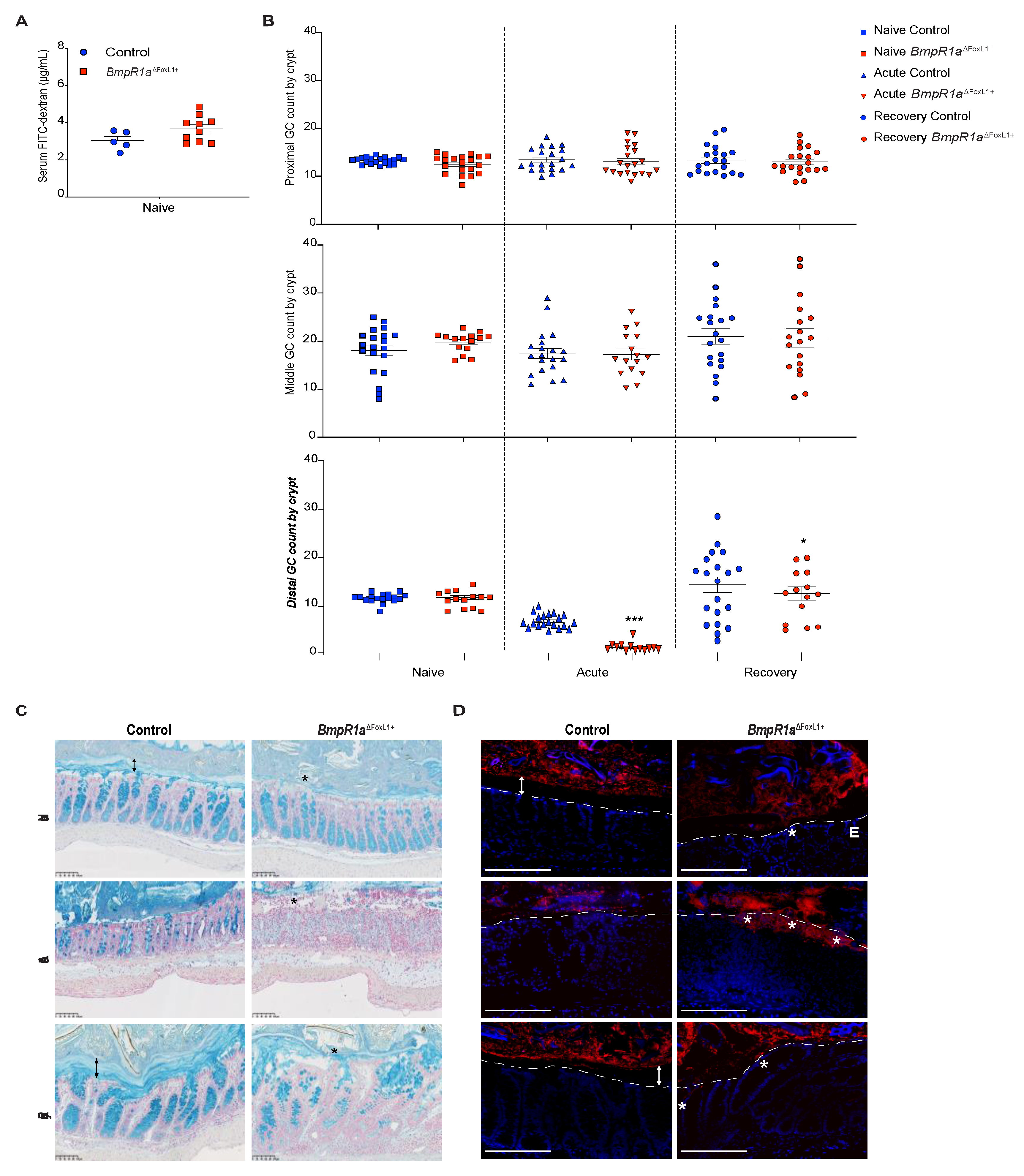
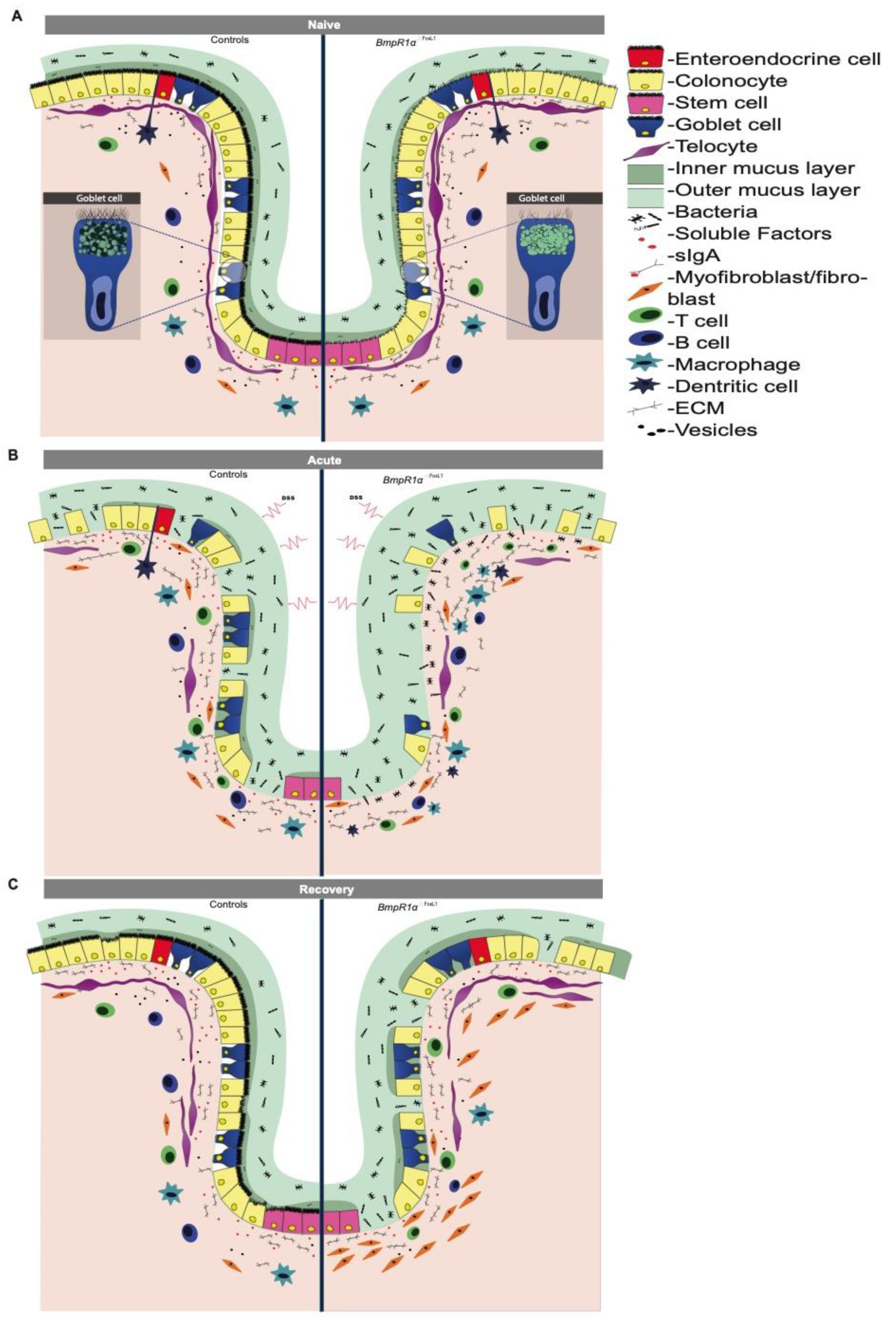
3.2. Defective Telocytes Lead to the Development of Compromised Mucus Layers and Abnormal Bacterial Infiltration
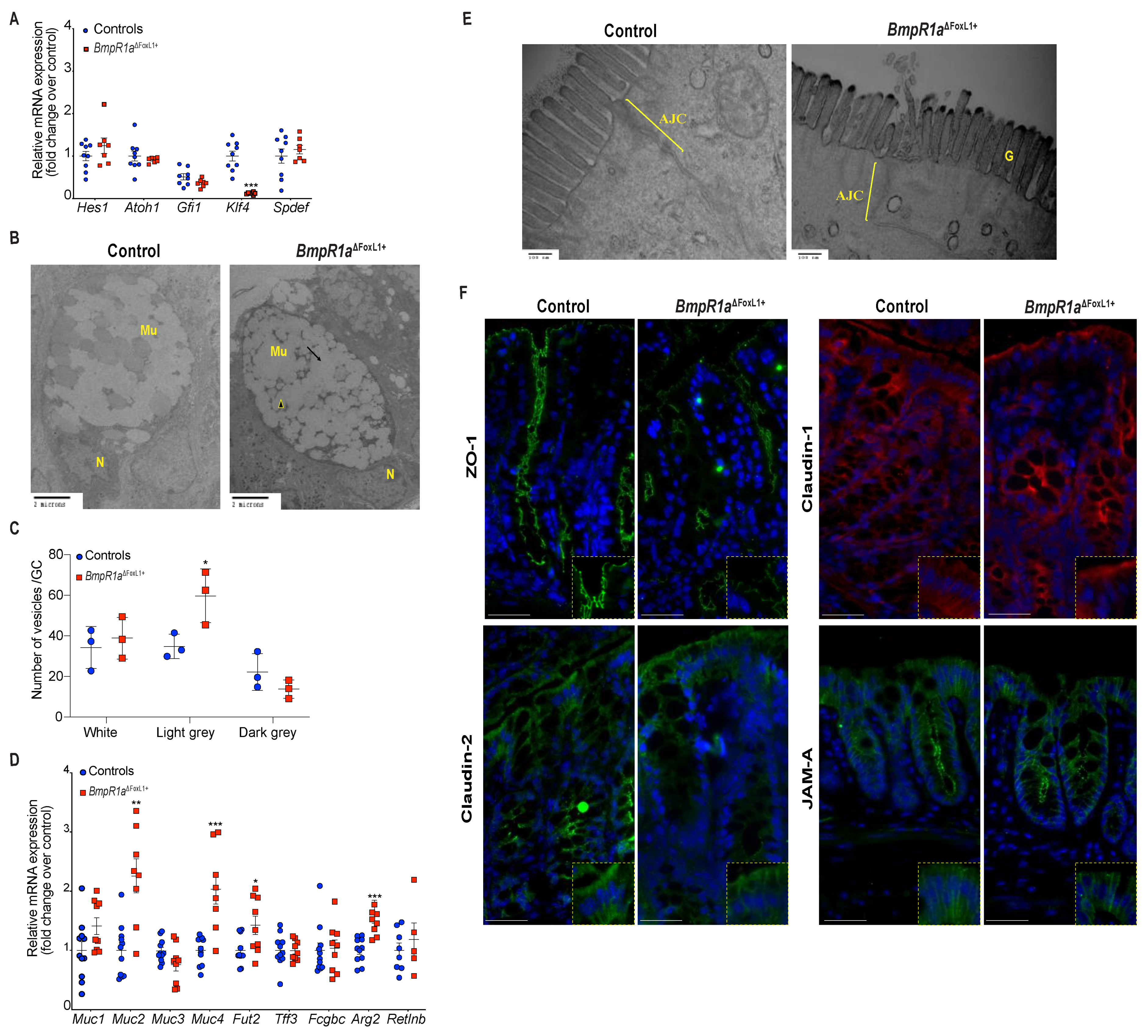
3.3. BMP-Associated Signalling in Telocytes Supports the Maturation of Colonic Goblet Cells
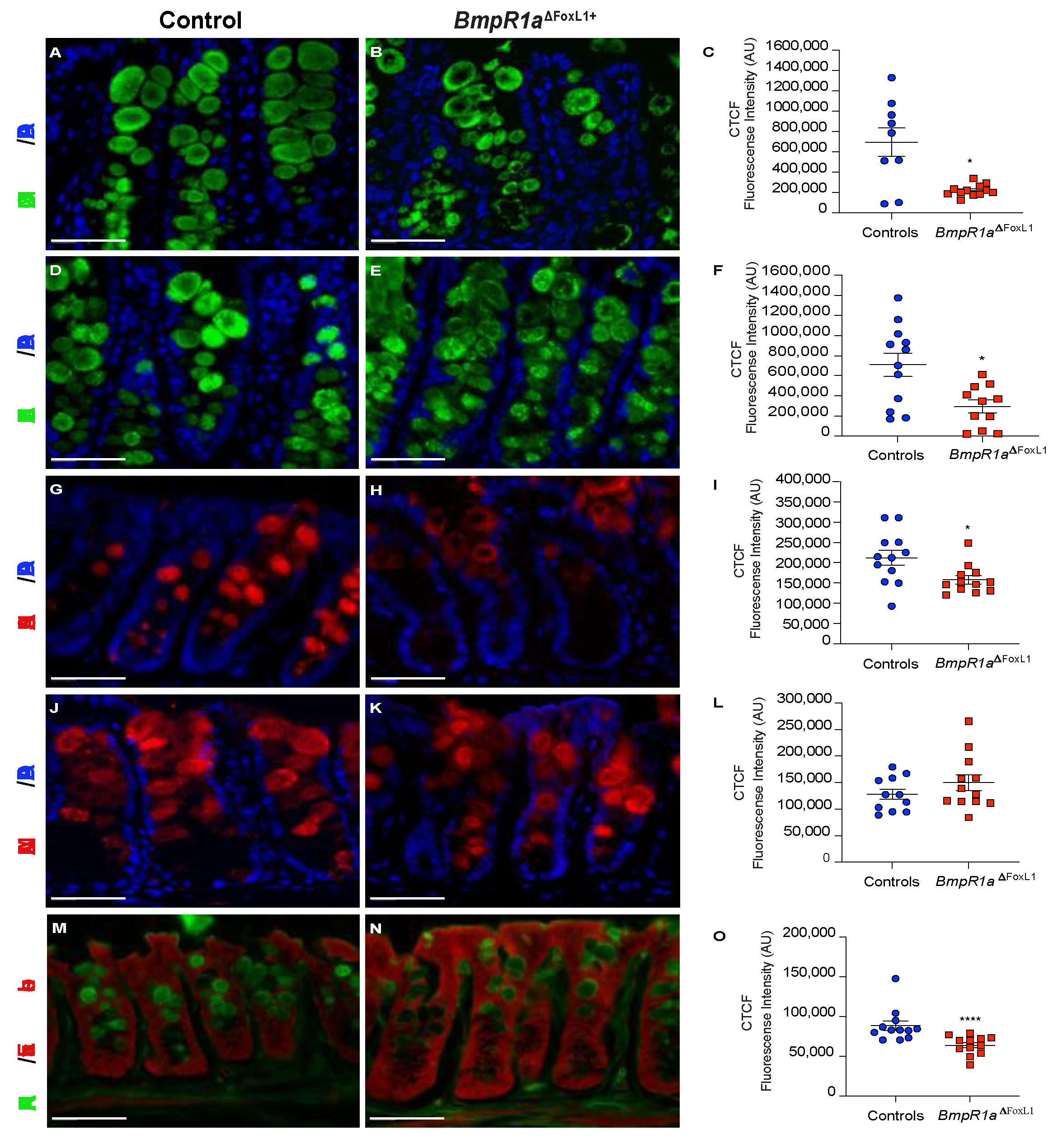
3.4. BMP-Associated Signalling in Telocytes Affects Post-Translational Modifications of Mucins
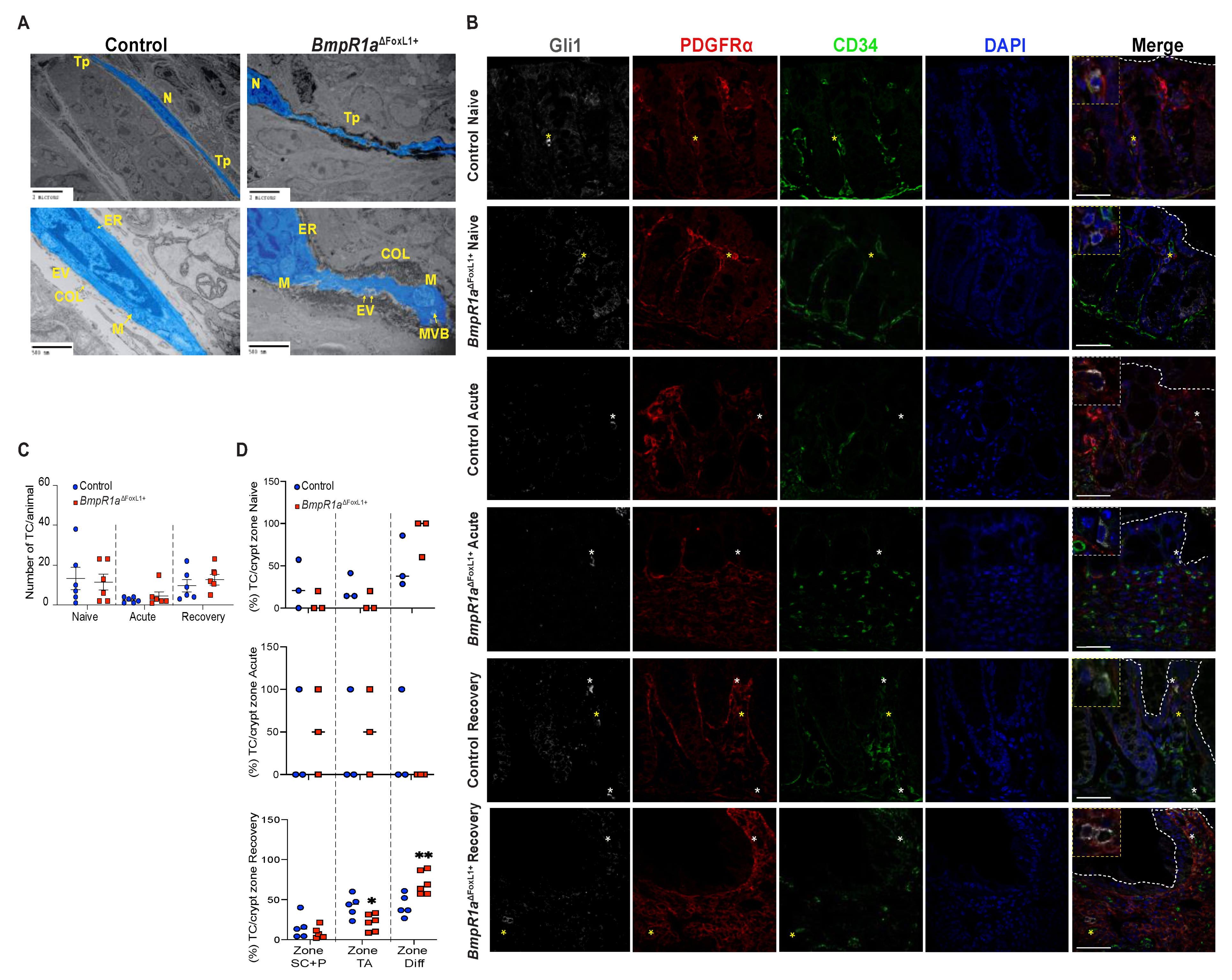
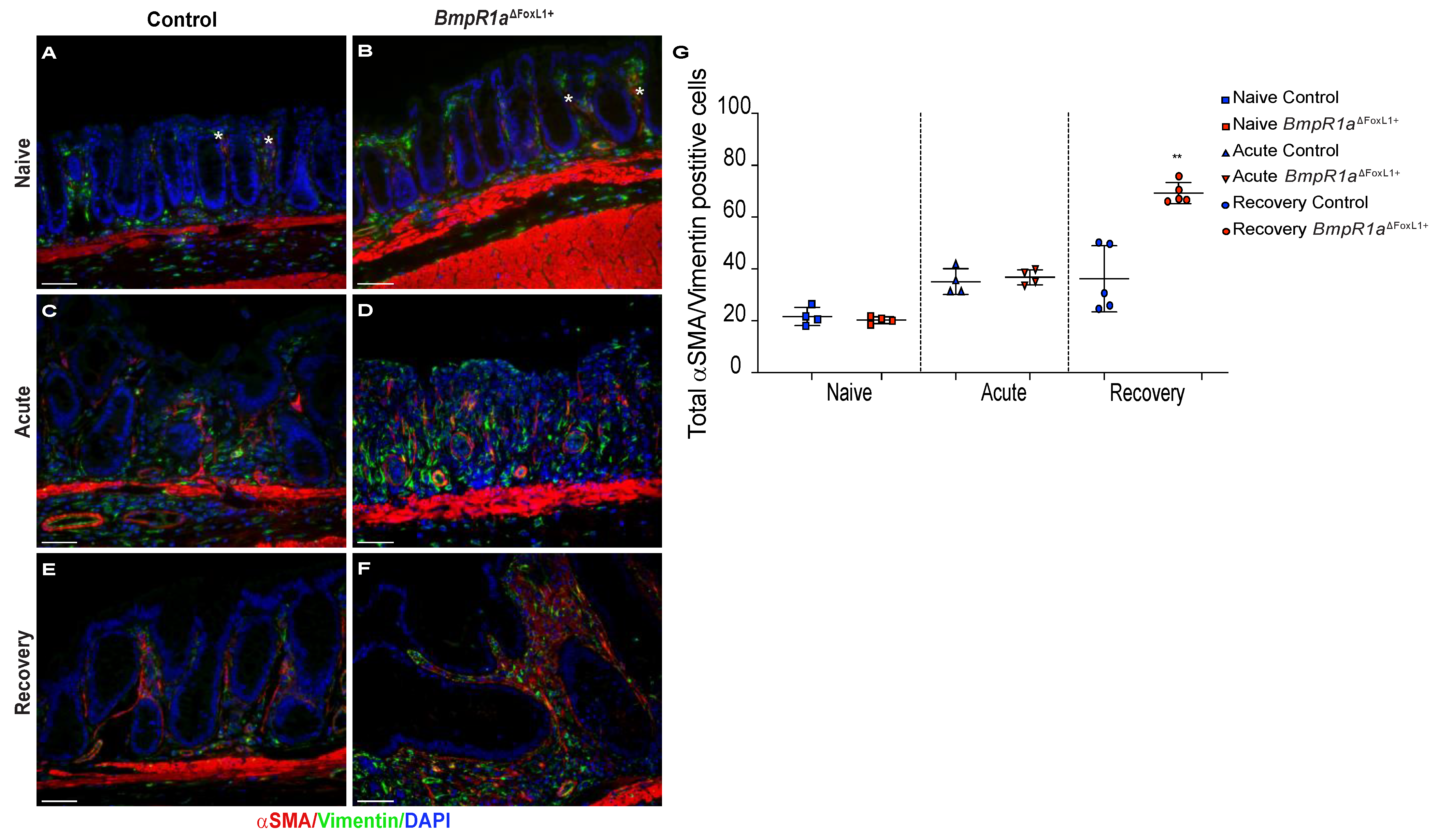
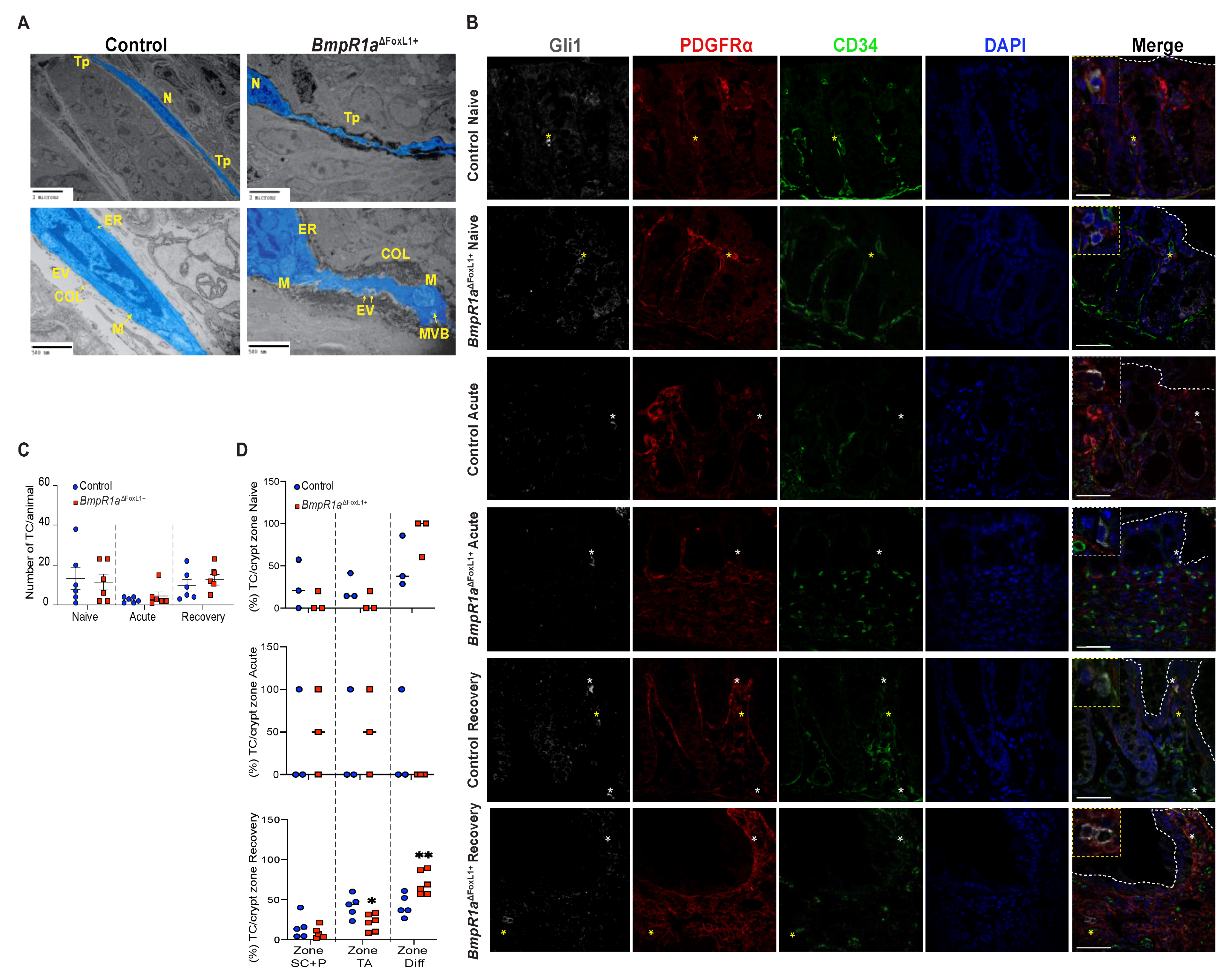
3.5. Impaired BMP Signalling Does Not Affect Telocytes’ Number but Their Localisation toward the crypt axis following Inflammatory Stress
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BMPs | Bone morphogenetic proteins |
| BmpR1a | Bone morphogenetic protein receptor type IA |
| TCFoxL1+ | FoxL1+-Telocytes |
| IBD | inflammatory bowel disease |
| DAI | disease activity index |
| ECM | extracellular matrix |
| Sma | smooth muscle actin |
References
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Allaire, J.M.; Darsigny, M.; Marcoux, S.S.; Roy, S.A.; Schmouth, J.F.; Umans, L.; Zwijsen, A.; Boudreau, F.; Perreault, N. Loss of Smad5 leads to the disassembly of the apical junctional complex and increased susceptibility to experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G586–G597. [Google Scholar] [CrossRef] [Green Version]
- Kinchen, J.; Chen, H.H.; Parikh, K.; Antanaviciute, A.; Jagielowicz, M.; Fawkner-Corbett, D.; Ashley, N.; Cubitt, L.; Mellado-Gomez, E.; Attar, M.; et al. Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 2018, 175, 372–386.e317. [Google Scholar] [CrossRef] [Green Version]
- Stzepourginski, I.; Nigro, G.; Jacob, J.M.; Dulauroy, S.; Sansonetti, P.J.; Eberl, G.; Peduto, L. CD34+ mesenchymal cells are a major component of the intestinal stem cells niche at homeostasis and after injury. Proc. Natl. Acad. Sci. USA 2017, 114, E506–E513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowarski, R.; Jackson, R.; Flavell, R.A. The Stromal Intervention: Regulation of Immunity and Inflammation at the Epithelial-Mesenchymal Barrier. Cell 2017, 168, 362–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshkes-Carmel, M.; Wang, Y.J.; Wangensteen, K.J.; Toth, B.; Kondo, A.; Massasa, E.E.; Itzkovitz, S.; Kaestner, K.H. Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 2018, 557, 242–246. [Google Scholar] [CrossRef]
- McCarthy, N.; Kraiczy, J.; Shivdasani, R.A. Cellular and molecular architecture of the intestinal stem cell niche. Nat. Cell Biol. 2020, 22, 1033–1041. [Google Scholar] [CrossRef]
- Allaire, J.M.; Roy, S.A.; Ouellet, C.; Lemieux, E.; Jones, C.; Paquet, M.; Boudreau, F.; Perreault, N. Bmp signaling in colonic mesenchyme regulates stromal microenvironment and protects from polyposis initiation. Int. J. Cancer 2016, 138, 2700–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, D.W.; Pinchuk, I.V.; Saada, J.I.; Chen, X.; Mifflin, R.C. Mesenchymal cells of the intestinal lamina propria. Annu. Rev. Physiol. 2011, 73, 213–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.A.; Allaire, J.M.; Ouellet, C.; Maloum-Rami, F.; Pomerleau, V.; Lemieux, E.; Babeu, J.P.; Rousseau, J.; Paquet, M.; Garde-Granger, P.; et al. Loss of mesenchymal bone morphogenetic protein signaling leads to development of reactive stroma and initiation of the gastric neoplastic cascade. Sci. Rep. 2016, 6, 32759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greicius, G.; Kabiri, Z.; Sigmundsson, K.; Liang, C.; Bunte, R.; Singh, M.K.; Virshup, D.M. PDGFRalpha(+) pericryptal stromal cells are the critical source of Wnts and RSPO3 for murine intestinal stem cells in vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E3173–E3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auclair, B.A.; Benoit, Y.D.; Rivard, N.; Mishina, Y.; Perreault, N. Bone morphogenetic protein signaling is essential for terminal differentiation of the intestinal secretory cell lineage. Gastroenterology 2007, 133, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Antoni, L.; Nuding, S.; Wehkamp, J.; Stange, E.F. Intestinal barrier in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Faderl, M.; Noti, M.; Corazza, N.; Mueller, C. Keeping bugs in check: The mucus layer as a critical component in maintaining intestinal homeostasis. IUBMB Life 2015, 67, 275–285. [Google Scholar] [CrossRef]
- Howe, J.R.; Sayed, M.G.; Ahmed, A.F.; Ringold, J.; Larsen-Haidle, J.; Merg, A.; Mitros, F.A.; Vaccaro, C.A.; Petersen, G.M.; Giardiello, F.M.; et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J. Med. Genet. 2004, 41, 484–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sackett, S.D.; Fulmer, J.T.; Friedman, J.R.; Kaestner, K.H. Foxl1-Cre BAC transgenic mice: A new tool for gene ablation in the gastrointestinal mesenchyme. Genesis 2007, 45, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Mishina, Y.; Hanks, M.C.; Miura, S.; Tallquist, M.D.; Behringer, R.R. Generation of Bmpr/Alk3 conditional knockout mice. Genesis 2002, 32, 69–72. [Google Scholar] [CrossRef]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 1993, 69, 238–249. [Google Scholar] [PubMed]
- Dieleman, L.A.; Elson, C.O.; Tennyson, G.S.; Beagley, K.W. Kinetics of cytokine expression during healing of acute colitis in mice. Am. J. Physiol. 1996, 271, G130–G136. [Google Scholar] [CrossRef] [PubMed]
- Zarepour, M.; Bhullar, K.; Montero, M.; Ma, C.; Huang, T.; Velcich, A.; Xia, L.; Vallance, B.A. The mucin Muc2 limits pathogen burdens and epithelial barrier dysfunction during Salmonella enterica serovar Typhimurium colitis. Infect. Immun. 2013, 81, 3672–3683. [Google Scholar] [CrossRef] [Green Version]
- Liquori, G.E.; Mastrodonato, M.; Mentino, D.; Scillitani, G.; Desantis, S.; Portincasa, P.; Ferri, D. In situ characterization of O-linked glycans of Muc2 in mouse colon. Acta. Histochem. 2012, 114, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Gagne-Sansfacon, J.; Allaire, J.M.; Jones, C.; Boudreau, F.; Perreault, N. Loss of Sonic hedgehog leads to alterations in intestinal secretory cell maturation and autophagy. PLoS ONE 2014, 9, e98751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, K.; Maiden, L.; Bjarnason, I. Genetic aspects of intestinal permeability in inflammatory bowel disease. Novartis Found. Symp. 2004, 263, 151–158, discussion 159–163, 211–158. [Google Scholar] [PubMed]
- Bergstrom, K.; Shan, X.; Casero, D.; Batushansky, A.; Lagishetty, V.; Jacobs, J.P.; Hoover, C.; Kondo, Y.; Shao, B.; Gao, L.; et al. Proximal colon-derived O-glycosylated mucus encapsulates and modulates the microbiota. Science 2020, 370, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.P.; Perreault, N.; Goldstein, B.G.; Lee, C.S.; Labosky, P.A.; Yang, V.W.; Kaestner, K.H. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development 2002, 129, 2619–2628. [Google Scholar] [CrossRef]
- Noah, T.K.; Donahue, B.; Shroyer, N.F. Intestinal development and differentiation. Exp. Cell Res. 2011, 317, 2702–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodach, L.L.; Wiercinska, E.; de Miranda, N.F.; Bleuming, S.A.; Musler, A.R.; Peppelenbosch, M.P.; Dekker, E.; van den Brink, G.R.; van Noesel, C.J.; Morreau, H.; et al. The bone morphogenetic protein pathway is inactivated in the majority of sporadic colorectal cancers. Gastroenterology 2008, 134, 1332–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceafalan, L.; Gherghiceanu, M.; Popescu, L.M.; Simionescu, O. Telocytes in human skin—Are they involved in skin regeneration? J. Cell. Mol. Med. 2012, 16, 1405–1420. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lin, M.; Li, L.; Wang, R.; Zhang, C.; Qi, G.; Xu, M.; Rong, R.; Zhu, T. Renal telocytes contribute to the repair of ischemically injured renal tubules. J. Cell. Mol. Med. 2014, 18, 1144–1156. [Google Scholar] [CrossRef]
- Diaz-Flores, L.; Gutierrez, R.; Gonzalez-Gomez, M.; Diaz-Flores, L., Jr.; Valladares, F.; Rancel, N.; Saez, F.J.; Madrid, J.F. Telocyte Behaviour During Inflammation, Repair and Tumour Stroma Formation. Adv. Exp. Med. Biol. 2016, 913, 177–191. [Google Scholar] [CrossRef]
- Popescu, L.M.; Faussone-Pellegrini, M.S. TELOCYTES—A case of serendipity: The winding way from Interstitial Cells of Cajal (ICC), via Interstitial Cajal-Like Cells (ICLC) to TELOCYTES. J. Cell. Mol. Med. 2010, 14, 729–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolescu, M.I.; Bucur, A.; Dinca, O.; Rusu, M.C.; Popescu, L.M. Telocytes in parotid glands. Anat. Rec. 2012, 295, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Niculite, C.M.; Regalia, T.M.; Gherghiceanu, M.; Huica, R.; Surcel, M.; Ursaciuc, C.; Leabu, M.; Popescu, L.M. Dynamics of telopodes (telocyte prolongations) in cell culture depends on extracellular matrix protein. Mol. Cell Biochem. 2015, 398, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Edelstein, L.; Fuxe, K.; Levin, M.; Popescu, B.O.; Smythies, J. Telocytes in their context with other intercellular communication agents. Semin. Cell Dev. Biol. 2016, 55, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Albulescu, R.; Tanase, C.; Codrici, E.; Popescu, D.I.; Cretoiu, S.M.; Popescu, L.M. The secretome of myocardial telocytes modulates the activity of cardiac stem cells. J. Cell. Mol. Med. 2015, 19, 1783–1794. [Google Scholar] [CrossRef]
- Manetti, M.; Rosa, I.; Messerini, L.; Ibba-Manneschi, L. Telocytes are reduced during fibrotic remodelling of the colonic wall in ulcerative colitis. J. Cell. Mol. Med. 2015, 19, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.V.; Mifflin, R.C.; Saada, J.I.; Powell, D.W. Intestinal mesenchymal cells. Curr. Gastroenterol. Rep. 2010, 12, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.E.; Ambort, D.; Pelaseyed, T.; Schutte, A.; Gustafsson, J.K.; Ermund, A.; Subramani, D.B.; Holmen-Larsson, J.M.; Thomsson, K.A.; Bergstrom, J.H.; et al. Composition and functional role of the mucus layers in the intestine. Cell. Mol. Life Sci. 2011, 68, 3635–3641. [Google Scholar] [CrossRef]
- Bergstrom, J.H.; Berg, K.A.; Rodriguez-Pineiro, A.M.; Stecher, B.; Johansson, M.E.; Hansson, G.C. AGR2, an endoplasmic reticulum protein, is secreted into the gastrointestinal mucus. PLoS ONE 2014, 9, e104186. [Google Scholar] [CrossRef]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008, 5, e54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, B.O. Fight them or feed them: How the intestinal mucus layer manages the gut microbiota. Gastroenterol. Rep. (Oxf) 2019, 7, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corfield, A.P. Mucins: A biologically relevant glycan barrier in mucosal protection. Biochim. Biophys. Acta. 2015, 1850, 236–252. [Google Scholar] [CrossRef]
- Jensen, P.H.; Kolarich, D.; Packer, N.H. Mucin-type O-glycosylation—Putting the pieces together. FEBS J. 2010, 277, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.R.; Curtis, M.M.; Ritchie, J.M.; Munera, D.; Waldor, M.K.; Moreira, C.G.; Sperandio, V. Fucose sensing regulates bacterial intestinal colonization. Nature 2012, 492, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Almagro-Moreno, S.; Boyd, E.F. Bacterial catabolism of nonulosonic (sialic) acid and fitness in the gut. Gut Microbes 2010, 1, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Mastrodonato, M.; Mentino, D.; Liquori, G.E.; Ferri, D. Histochemical characterization of the sialic acid residues in mouse colon mucins. Microsc. Res. Tech. 2013, 76, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Schwegmann, C.; Zimmer, G.; Yoshino, T.; Enss, M.; Herrler, G. Comparison of the sialic acid binding activity of transmissible gastroenteritis coronavirus and E. coli K99. Virus Res. 2001, 75, 69–73. [Google Scholar] [CrossRef]
- Pham, T.A.; Clare, S.; Goulding, D.; Arasteh, J.M.; Stares, M.D.; Browne, H.P.; Keane, J.A.; Page, A.J.; Kumasaka, N.; Kane, L.; et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe 2014, 16, 504–516. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes Nicolás, V.; Allaire, J.M.; Alfonso, A.B.; Pupo Gómez, D.; Pomerleau, V.; Giroux, V.; Boudreau, F.; Perreault, N. Altered Mucus Barrier Integrity and Increased Susceptibility to Colitis in Mice upon Loss of Telocyte Bone Morphogenetic Protein Signalling. Cells 2021, 10, 2954. https://doi.org/10.3390/cells10112954
Reyes Nicolás V, Allaire JM, Alfonso AB, Pupo Gómez D, Pomerleau V, Giroux V, Boudreau F, Perreault N. Altered Mucus Barrier Integrity and Increased Susceptibility to Colitis in Mice upon Loss of Telocyte Bone Morphogenetic Protein Signalling. Cells. 2021; 10(11):2954. https://doi.org/10.3390/cells10112954
Chicago/Turabian StyleReyes Nicolás, Vilcy, Joannie M. Allaire, Alain B. Alfonso, Dianne Pupo Gómez, Véronique Pomerleau, Véronique Giroux, François Boudreau, and Nathalie Perreault. 2021. "Altered Mucus Barrier Integrity and Increased Susceptibility to Colitis in Mice upon Loss of Telocyte Bone Morphogenetic Protein Signalling" Cells 10, no. 11: 2954. https://doi.org/10.3390/cells10112954
APA StyleReyes Nicolás, V., Allaire, J. M., Alfonso, A. B., Pupo Gómez, D., Pomerleau, V., Giroux, V., Boudreau, F., & Perreault, N. (2021). Altered Mucus Barrier Integrity and Increased Susceptibility to Colitis in Mice upon Loss of Telocyte Bone Morphogenetic Protein Signalling. Cells, 10(11), 2954. https://doi.org/10.3390/cells10112954