
Dysfunction of RNA/RNA-Binding Proteins in ALS Astrocytes and Microglia



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
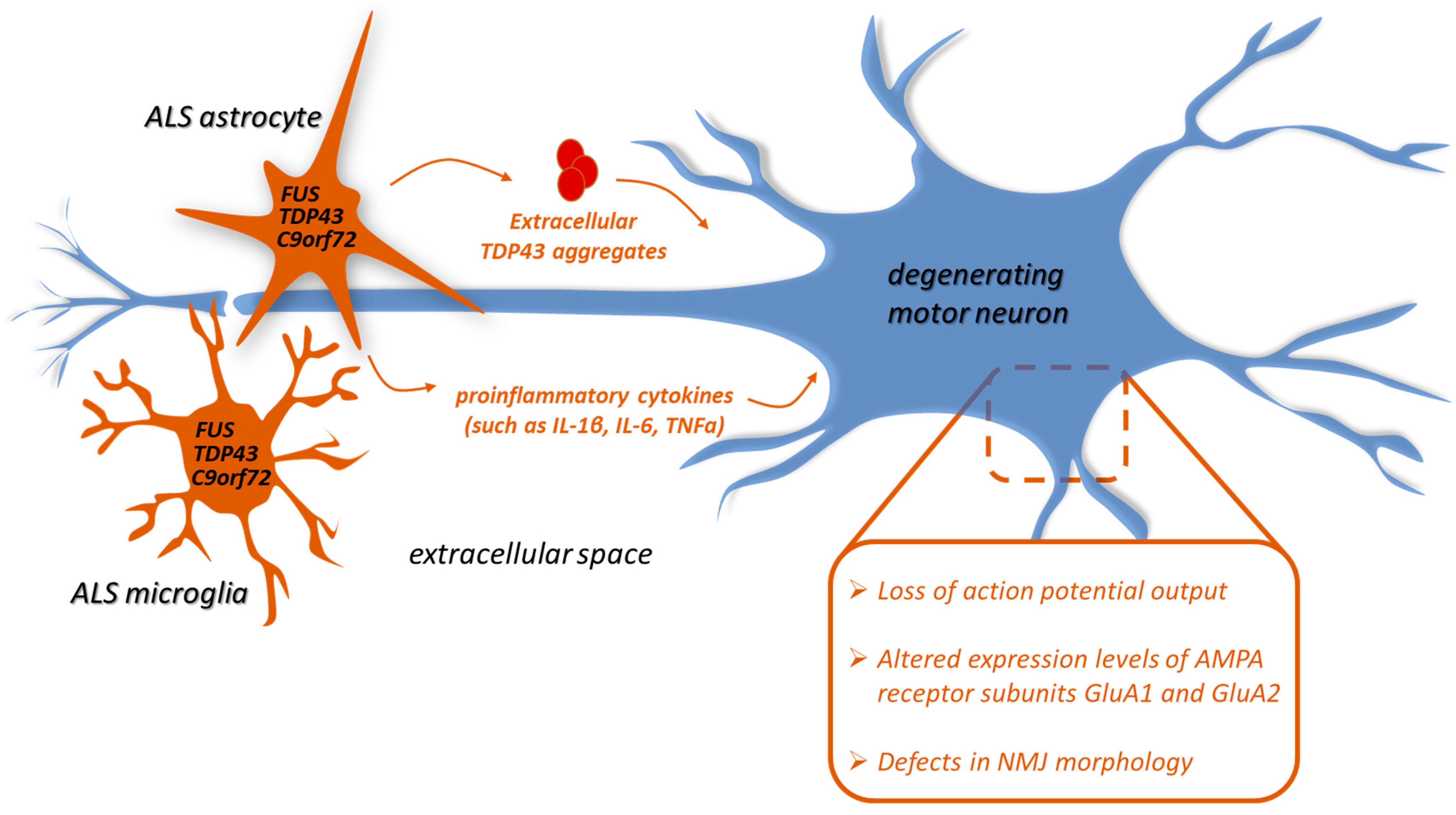
2. Glia-Mediated Neuroinflammation in TDP-43, FUS, and C9orf72 ALS
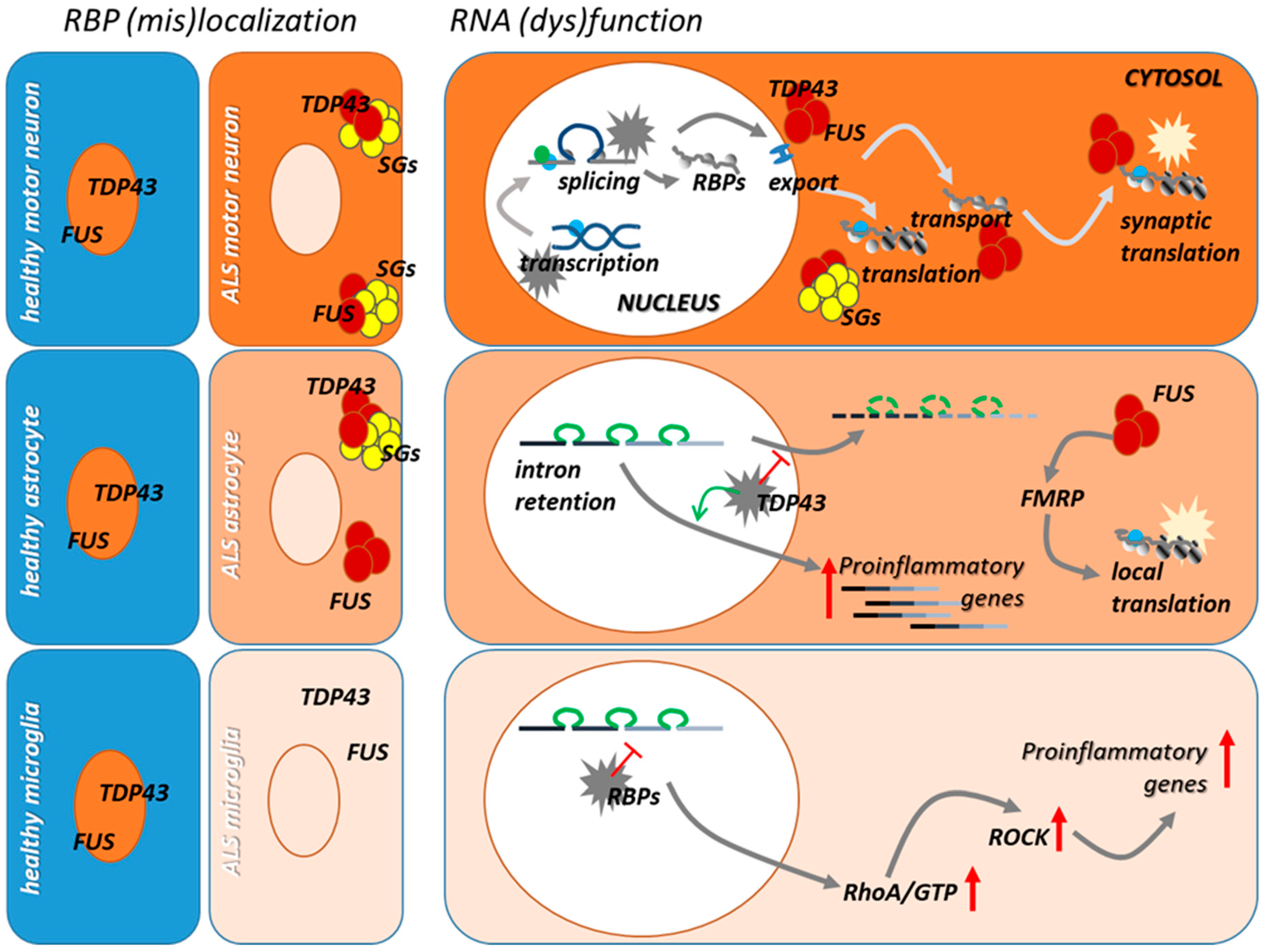
3. Dysfunction of RNA/RBPs in ALS Microglia and Astrocytes
3.1. TDP-43 and FUS
3.2. C9orf72
4. The Contribution of RNA Dysmetabolism to Neuroinflammation in ALS
4.1. Alternative Splicing in Reactive ALS Glia
4.2. RNA Trafficking and (Local) Translation in Reactive ALS Glia
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Masrori, P.; van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Van Es, M.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Brown, R.H., Jr.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS genetics: Gains, losses, and implications for future therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.; Pitout, I.L.; Fletcher, S.; Wilton, S.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Kankel, M.W.; Su, S.C.; Han, S.W.S.; Ofengeim, D. Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD. Cell Death Differ. 2018, 25, 648–662. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, S.; Cocozza, G.; Porzia, A.; Inghilleri, M.; Raspa, M.; Scavizzi, F.; Aronica, E.; Bernardini, G.; Peng, L.; Ransohoff, R.M.; et al. Natural killer cells modulate motor neuron-immune cell cross talk in models of amyotrophic lateral sclerosis. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Coque, E.; Salsac, C.; Espinosa-Carrasco, G.; Varga, B.; Degauque, N.; Cadoux, M.; Crabé, R.; Virenque, A.; Soulard, C.; Fierle, J.K.; et al. Cytotoxic CD8+ T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2312–2317. [Google Scholar] [CrossRef] [Green Version]
- Butti, Z.; Patten, S.A. RNA dysregulation in amyotrophic lateral sclerosis. Front. Genet. 2019, 9, 712. [Google Scholar] [CrossRef] [Green Version]
- Nussbacher, J.K.; Tabet, R.; Yeo, G.W.; Lagier-Tourenne, C. Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron 2019, 102, 294–320. [Google Scholar] [CrossRef] [PubMed]
- Vasile, F.; Dossi, E.; Rouach, N. Human astrocytes: Structure and functions in the healthy brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [Green Version]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.E.; Patani, R. The microglial component of amyotrophic lateral sclerosis. Brain 2020, 143, 3526–3539. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Valori, C.F.; Guidotti, G.; Brambilla, L.; Rossi, D. Astrocytes: Emerging therapeutic targets in neurological disorders. Trends Mol. Med. 2019, 25, 750–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper-Knock, J.; Hewitt, C.; Highley, R.; Brockington, A.; Milano, A.; Man, S.; Martindale, J.; Hartley, J.; Walsh, T.; Gelsthorpe, C.; et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 2012, 135, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Tauber, C.; Vercoullie, J.; Arlicot, N.; Prunier, C.; Praline, J.; Nicolas, G.; Venel, Y.; Hommet, C.; Baulieu, J.-L.; et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52941. [Google Scholar] [CrossRef]
- Engelhardt, J.I.; Appel, S.H. IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch. Neurol. 1990, 47, 1210–1216. [Google Scholar] [CrossRef]
- Henkel, J.S.; Engelhardt, J.I.; Siklós, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 2003, 55, 221–235. [Google Scholar] [CrossRef]
- Nolan, M.; Scott, C.; Gamarallage, M.P.; Lunn, D.; Carpenter, K.; McDonough, E.; Meyer, D.; Kaanumalle, S.; Santamaria-Pang, A.; Turner, M.R.; et al. Quantitative patterns of motor cortex proteinopathy across ALS genotypes. Acta Neuropathol. Commun. 2020, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J. Glial cells in amyotrophic lateral sclerosis. Exp. Neurol. 2014, 262, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Hunter, M.; Spiller, K.J.; Dominique, M.A.; Xu, H.; Hunter, F.W.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglial transcriptome analysis in the rNLS8 mouse model of TDP-43 proteinopathy reveals discrete expression profiles associated with neurodegenerative progression and recovery. Acta Neuropathol. Commun. 2021, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jara, J.H.; Gautam, M.; Kocak, N.; Xie, E.F.; Mao, Q.; Bigio, E.H.; Özdinler, P.H. MCP1-CCR2 and neuroinflammation in the ALS motor cortex with TDP-43 pathology. J. Neuroinflamm. 2019, 16, 196. [Google Scholar] [CrossRef]
- Swarup, V.; Phaneuf, D.; Bareil, C.; Robertson, J.; Rouleau, G.A.; Kriz, J.; Julien, J.-P. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 2011, 134, 2610–2626. [Google Scholar] [CrossRef]
- Tong, J.; Huang, C.; Bi, F.; Wu, Q.; Huang, B.; Liu, X.; Li, F.; Zhou, H.; Xia, X.-G. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013, 32, 1917–1926. [Google Scholar] [CrossRef]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.-F.; Zhang, Y.-J.; Lin, W.-L.; Cao, X.; Stetler, C.; Dickson, D.W.; Lewis, J.; Petrucelli, L. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol. Neurodegener. 2011, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Tong, J.; Bi, F.; Wu, Q.; Huang, B.; Zhou, H.; Xia, X.-G. Entorhinal cortical neurons are the primary targets of FUS mislocalization and ubiquitin aggregation in FUS transgenic rats. Hum. Mol. Genet. 2012, 21, 4602–4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Zhou, H.; Tong, J.; Chen, H.; Liu, Y.-J.; Wang, D.; Wei, X.; Xia, X.-G. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011, 7, e1002011. [Google Scholar] [CrossRef] [Green Version]
- Mirra, A.; Rossi, S.; Scaricamazza, S.; di Salvio, M.; Salvatori, I.; Valle, C.; Rusmini, P.; Poletti, A.; Cestra, G.; Carri, M.T.; et al. Functional interaction between FUS and SMN underlies SMA-like splicing changes in wild-type hFUS mice. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, J.C.; McGoldrick, P.; Vance, C.; Hortobagyi, T.; Sreedharan, J.; Rogelj, B.; Tudor, E.L.; Smith, B.N.; Klasen, C.; Miller, C.C.; et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013, 125, 273–288. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef]
- Brettschneider, J.; van Deerlin, V.M.; Robinson, J.L.; Kwong, L.; Lee, E.B.; Ali, Y.O.; Safren, N.; Monteiro, M.J.; Toledo, J.; Elman, L.; et al. Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol. 2012, 123, 825–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, J.; Gendron, T.F.; Prudencio, M.; Sasaguri, H.; Zhang, Y.-J.; Castanedes-Casey, M.; Lee, C.W.; Jansen-West, K.; Kurti, A.; Murray, M.E.; et al. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 2015, 348, 1151–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P.W. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schludi, M.H.; Becker, L.; Garrett, L.; Gendron, T.F.; Zhou, Q.; Schreiber, F.; Popper, B.; Dimou, L.; Strom, T.M.; Winkelmann, J.; et al. Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol. 2017, 134, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Gendron, T.F.; Ebbert, M.T.W.; O’Raw, A.; Yue, M.; Jansen-West, K.; Zhang, X.; Prudencio, M.; Chew, J.; Cook, C.N.; et al. Poly (GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 2018, 24, 1136–1142. [Google Scholar] [CrossRef]
- LaClair, K.D.; Zhou, Q.; Michaelsen, M.; Wefers, B.; Brill, M.S.; Janjic, A.; Rathkolb, B.; Farny, D.; Cygan, M.; de Angelis, M.H.; et al. Congenic expression of poly-GA but not poly-PR in mice triggers selective neuron loss and interferon responses found in C9orf72 ALS. Acta Neuropathol. 2020, 140, 121–142. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y.; Ryu, H. Astrocytes and microglia as non-cell autonomous players in the pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar] [CrossRef]
- Haidet-Phillips, A.M.; Gross, S.K.; Williams, T.; Tuteja, A.; Sherman, A.; Ko, M.; Jeong, Y.H.; Wong, P.C.; Maragakis, N.J. Altered astrocytic expression of TDP-43 does not influence motor neuron survival. Exp. Neurol. 2013, 250, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 4697–4702. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, S.; Kang, H.-Y.; Lim, H.R.; Kwon, Y.; Jo, M.; Jeon, Y.-M.; Kim, S.R.; Kim, K.; Ha, C.M.; et al. The overexpression of TDP-43 in astrocytes causes neurodegeneration via a PTP1B-mediated inflammatory response. J. Neuroinflamm. 2020, 17, 1–22. [Google Scholar] [CrossRef]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [Green Version]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.; Ferraiuolo, L.; Miranda, C.J.; Likhite, S.; McElroy, S.; Renusch, S.; Ditsworth, D.; Lagier-Tourenne, C.; Smith, R.A.; Ravits, J.; et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 829–832. [Google Scholar] [CrossRef] [Green Version]
- Qian, K.; Huang, H.; Peterson, A.; Hu, B.; Maragakis, N.J.; Ming, G.-L.; Chen, H.; Zhang, S.-C. Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Rep. 2017, 8, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Devlin, A.; Chouhan, A.K.; Selvaraj, B.T.; Stavrou, M.; Burr, K.; Brivio, V.; He, X.; Mehta, A.R.; Story, D.; et al. Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia 2019, 68, 1046–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Zheng, L.; Si, Y.; Luo, W.; Dujardin, G.; Kwan, T.; Potochick, N.R.; Thompson, S.; Schneider, D.; King, P.H. Hu antigen R (HuR) is a positive regulator of the RNA-binding proteins TDP-43 and FUS/TLS. J. Biol. Chem. 2014, 289, 31792–31804. [Google Scholar] [CrossRef] [Green Version]
- Rojas, F.; Cortes, N.; Abarzua, S.; Dyrda, A.; van Zundert, B.A. Astrocytes expressing mutant SOD1 and TDP43 trigger motoneuron death that is mediated via sodium channels and nitroxidative stress. Front. Cell. Neurosci. 2014, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Kia, A.; McAvoy, K.; Krishnamurthy, K.; Trotti, D.; Pasinelli, P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 2018, 66, 1016–1033. [Google Scholar] [CrossRef]
- Madill, M.; McDonagh, K.; Ma, J.; Vajda, A.; McLoughlin, P.; O’Brien, T.; Hardiman, O.; Shen, S. Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol. Brain 2017, 10, 1–12. [Google Scholar] [CrossRef]
- Ajmone-Cat, M.A.; Onori, A.; Toselli, C.; Stronati, E.; Morlando, M.; Bozzoni, I.; Monni, E.; Kokaia, Z.; Lupo, G.; Minghetti, L.; et al. Increased FUS levels in astrocytes leads to astrocyte and microglia activation and neuronal death. Sci. Rep. 2019, 9, 4572. [Google Scholar] [CrossRef]
- Sabatelli, M.; Moncada, A.; Conte, A.; Lattante, S.; Marangi, G.; Luigetti, M.; Lucchini, M.; Mirabella, M.; Romano, A.; del Grande, A.; et al. Mutations in the 3′ untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 4748–4755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modigliani, S.D.; Morlando, M.; Errichelli, L.; Sabatelli, M.; Bozzoni, I. An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA–FUS regulatory circuitry. Nat. Commun. 2014, 5, 4335. [Google Scholar] [CrossRef]
- Aebischer, J.; Bernard-Marissal, N.; Pettmann, B.; Raoul, C. Death receptors in the selective degeneration of motoneurons in amyotrophic lateral sclerosis. J. Neurodegener. Dis. 2013, 2013, 746845. [Google Scholar] [CrossRef]
- Correia, A.S.; Patel, P.; Dutta, K.; Julien, J.-P. Inflammation induces TDP-43 mislocalization and aggregation. PLoS ONE 2015, 10, e0140248. [Google Scholar] [CrossRef] [Green Version]
- Herman, A.M.; Khandelwal, P.J.; Rebeck, G.W.; Moussa, C.E.-H. Wild type TDP-43 induces neuro-inflammation and alters APP metabolism in lentiviral gene transfer models. Exp. Neurol. 2012, 235, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Smethurst, P.; Risse, E.; Tyzack, G.E.; Mitchell, J.S.; Taha, D.M.; Chen, Y.-R.; Newcombe, J.; Collinge, J.; Sidle, K.; Patani, R. Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis. Brain 2020, 143, 430–440. [Google Scholar] [CrossRef]
- Fomin, V.; Richard, P.; Hoque, M.; Li, C.; Gu, Z.; Fissore-O’Leary, M.; Tian, B.; Prives, C.; Manley, J.L. The C9ORF72 gene, implicated in amyotrophic lateral sclerosis and frontotemporal dementia, encodes a protein that functions in control of endothelin and glutamate signaling. Mol. Cell. Biol. 2018, 38, e00155-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Diaper, D.; Adachi, Y.; Lazarou, L.; Greenstein, M.; Simoes, F.; di Domenico, A.; Solomon, D.A.; Lowe, S.; Alsubaie, R.; Cheng, D.; et al. Drosophila TDP-43 dysfunction in glia and muscle cells cause cytological and behavioural phenotypes that characterize ALS and FTLD. Hum. Mol. Genet. 2013, 22, 3883–3893. [Google Scholar] [CrossRef] [Green Version]
- Estes, P.S.; Daniel, S.; Mccallum, A.P.; Boehringer, A.V.; Sukhina, A.S.; Zwick, R.A.; Zarnescu, D.C. Motor neurons and glia exhibit specific individualized responses to TDP-43 expression in a drosophila model of amyotrophic lateral sclerosis. Dis. Model. Mech. 2013, 6, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Cragnaz, L.; Klima, R.; de Conti, L.; Romano, G.; Feiguin, F.; Buratti, E.; Baralle, M.; Baralle, F. An age-related reduction of brain TBPH/TDP-43 levels precedes the onset of locomotion defects in a drosophila ALS model. Neuroscience 2015, 311, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, A.Y.T.; Agrawal, I.; Ho, W.Y.; Yen, Y.-C.; Pinter, A.J.; Liu, J.; Phua, Q.X.C.; Koh, K.B.; Chang, J.-C.; Sanford, E.; et al. Loss of TDP-43 in astrocytes leads to motor deficits by triggering A1-like reactive phenotype and triglial dysfunction. Proc. Natl. Acad. Sci. USA 2020, 117, 29101–29112. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; de Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Birsa, N.; Bentham, M.P.; Fratta, P. Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin. Cell Dev. Biol. 2020, 99, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507. [Google Scholar] [CrossRef] [Green Version]
- Nishihira, Y.; Tan, C.-F.; Onodera, O.; Toyoshima, Y.; Yamada, M.; Morita, T.; Nishizawa, M.; Kakita, A.; Takahashi, H. Sporadic amyotrophic lateral sclerosis: Two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol. 2008, 116, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tan, C.-F.; Mori, F.; Tanji, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2007, 115, 115–122. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Romano, G.; Appocher, C.; Scorzeto, M.; Klima, R.; Baralle, F.E.; Megighian, A.; Feiguin, F. Glial TDP-43 regulates axon wrapping, GluRIIA clustering and fly motility by autonomous and non-autonomous mechanisms. Hum. Mol. Genet. 2015, 24, 6134–6145. [Google Scholar] [CrossRef] [Green Version]
- Khalfallah, Y.; Kuta, R.; Grasmuck, C.; Prat, A.; Durham, H.D.; Velde, C.V. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci. Rep. 2018, 8, 7551. [Google Scholar] [CrossRef] [Green Version]
- Moujalled, D.; Grubman, A.; Acevedo, K.; Yang, S.; Ke, Y.; Moujalled, D.M.; Duncan, C.; Caragounis, A.; Perera, N.D.; Turner, B.; et al. TDP-43 mutations causing amyotrophic lateral sclerosis are associated with altered expression of RNA-binding protein hnRNP K and affect the Nrf2 antioxidant pathway. Hum. Mol. Genet. 2017, 26, 1732–1746. [Google Scholar] [CrossRef]
- Barton, S.K.; Lau, C.L.; Chiam, M.D.F.; Tomas, D.; Muyderman, H.; Beart, P.M.; Turner, B.J. Mutant TDP-43 expression triggers TDP-43 pathology and cell autonomous effects on primary astrocytes: Implications for non-cell autonomous pathology in ALS. Neurochem. Res. 2020, 45, 1451–1459. [Google Scholar] [CrossRef]
- Velebit, J.; Horvat, A.; Smolič, T.; Mihevc, S.P.; Rogelj, B.; Zorec, R.; Vardjan, N. Astrocytes with TDP-43 inclusions exhibit reduced noradrenergic cAMP and Ca 2+ signaling and dysregulated cell metabolism. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Swarup, V.; Phaneuf, D.; Dupré, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.-P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB–mediated pathogenic pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Hu, Q.; Wang, H.; Yang, H.; Gao, F.; Ren, H.; Chen, D.; Fu, C.; Zheng, L.; Zhen, X.; et al. Induction of COX-2-PGE2 synthesis by activation of the MAPK/ERK pathway contributes to neuronal death triggered by TDP-43-depleted microglia. Cell Death Dis. 2015, 6, e1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaRocca, T.J.; Mariani, A.; Watkins, L.R.; Link, C.D. TDP-43 knockdown causes innate immune activation via protein kinase R in astrocytes. Neurobiol. Dis. 2019, 132, 104514. [Google Scholar] [CrossRef] [PubMed]
- Kasai, T.; Tokuda, T.; Ishigami, N.; Sasayama, H.; Foulds, P.; Mitchell, D.J.; Mann, D.M.A.; Allsop, D.; Nakagawa, M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2009, 117, 55–62. [Google Scholar] [CrossRef]
- Noto, Y.-I.; Shibuya, K.; Sato, Y.; Kanai, K.; Misawa, S.; Sawai, S.; Mori, M.; Uchiyama, T.; Isose, S.; Nasu, S.; et al. Elevated CSF TDP-43 levels in amyotrophic lateral sclerosis: Specificity, sensitivity, and a possible prognostic value. Amyotroph. Lateral Scler. 2010, 12, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Hendrich, C.; Sperfeld, A.D.; Jesse, S.; von Arnim, C.A.F.; Lehnert, S.; Pabst, A.; Uttner, I.; Tumani, H.; Lee, V.M.-Y.; et al. TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch. Neurol. 2008, 65, 1481–1487. [Google Scholar] [CrossRef] [Green Version]
- Smethurst, P.; Sidle, K.C.L.; Hardy, J. Review: Prion-like mechanisms of transactive response DNA binding protein of 43 kDa (TDP-43) in amyotrophic lateral sclerosis (ALS). Neuropathol. Appl. Neurobiol. 2014, 41, 578–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal-Lasarte, M.M.; Franco, J.M.; Labrador-Garrido, A.; Pozo, D.; Roodveldt, C. Extracellular TDP-43 aggregates target MAPK/MAK/MRK overlapping kinase (MOK) and trigger caspase-3/IL-18 signaling in microglia. FASEB J. 2017, 31, 2797–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeyers, J.; Banchi, E.-G.; Latouche, M. C9ORF72: What it is, what it does, and why it matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.; Levina, V.; Halloran, M.A.; Gleeson, P.; Blair, I.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef]
- Levine, T.P.; Daniels, R.D.; Gatta, A.T.; Wong, L.H.; Hayes, M.J. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013, 29, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Sellier, C.; Campanari, M.; Corbier, C.J.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9 ORF 72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.M.; Zhou, X.; Robins, A.M.; Paushter, D.H.; Kim, D.; Smolka, M.B.; Hu, F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 2016, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK 1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef]
- Webster, C.P.; Smith, E.F.; Grierson, A.J.; de Vos, K.J. C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases 2018, 9, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Liang, C.; Swaminathan, K.; Herrlinger, S.; Lai, F.; Shiekhattar, R.; Chen, J.-F. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv. 2016, 2, e1601167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Iyer, L.M.; He, F.; Aravind, L. Discovery of novel DENN proteins: Implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front. Genet. 2012, 3, 283. [Google Scholar] [CrossRef] [Green Version]
- Atanasio, A.; Decman, V.; White, D.; Ramos, M.; Ikiz, B.; Lee, H.-C.; Siao, C.-J.; Brydges, S.; LaRosa, E.; Bai, Y.; et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production and glomerulonephropathy in mice. Sci. Rep. 2016, 6, 23204. [Google Scholar] [CrossRef] [PubMed]
- Burberry, A.; Suzuki, N.; Wang, J.-Y.; Moccia, R.; Mordes, D.A.; Stewart, M.H.; Suzuki-Uematsu, S.; Ghosh, S.; Singh, A.; Merkle, F.T.; et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 2016, 8, 347ra93. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.M.G.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, J.G.; Bogdanik, L.; Muhammad, A.; Gendron, T.F.; Kim, K.J.; Austin, A.; Cady, J.; Liu, E.Y.; Zarrow, J.; Grant, S.; et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron 2015, 88, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Peters, O.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.-H.; Hung, S.-T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- Langseth, A.; Kim, J.; Ugolino, J.E.; Shah, Y.; Hwang, H.-Y.; Wang, J.; Bergles, D.E.; Brown, S.P. Cell-type specific differences in promoter activity of the ALS-linked C9orf72 mouse ortholog. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Braems, E.; Swinnen, B.; Bosch, L.V.D. C9orf72 loss-of-function: A trivial, stand-alone or additive mechanism in C9 ALS/FTD? Acta Neuropathol. 2020, 140, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarraj, S.; King, A.; Troakes, C.; Smith, B.; Maekawa, S.; Bodi, I.; Rogelj, B.; Al-Chalabi, A.; Hortobágyi, T.; Shaw, C.E. P62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011, 122, 691–702. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Rostalski, H.; Leskelä, S.; Huber, N.; Katisko, K.; Cajanus, A.; Solje, E.; Marttinen, M.; Natunen, T.; Remes, A.M.; Hiltunen, M.; et al. Astrocytes and microglia as potential contributors to the pathogenesis of C9orf72 repeat expansion-associated FTLD and ALS. Front. Neurosci. 2019, 13, 486. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Highley, R.; Dickman, M.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.; Kirby, J.; et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.-W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hautbergue, G.M.; Castelli, L.M.; Ferraiuolo, L.; Sanchez-Martinez, A.; Cooper-Knock, J.; Higginbottom, A.; Lin, Y.-H.; Bauer, C.S.; Dodd, J.E.; Myszczynska, M.; et al. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat. Commun. 2017, 8, 16063. [Google Scholar] [CrossRef]
- Mori, K.; Lammich, S.; MacKenzie, I.R.A.; Forné, I.; Zilow, S.; Kretzschmar, H.; Edbauer, D.; Janssens, J.; Kleinberger, G.; Cruts, M.; et al. HnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013, 125, 413–423. [Google Scholar] [CrossRef]
- Ramesh, N.; Daley, E.L.; Gleixner, A.M.; Mann, J.R.; Kour, S.; Mawrie, D.; Anderson, E.N.; Kofler, J.; Donnelly, C.J.; Kiskinis, E.; et al. RNA dependent suppression of C9orf72 ALS/FTD associated neurodegeneration by matrin-3. Acta Neuropathol. Commun. 2020, 8, 1–20. [Google Scholar] [CrossRef]
- Rossi, S.; Serrano, A.; Gerbino, V.; Giorgi, A.; di Francesco, L.; Nencini, M.; Bozzo, F.; Schininà, M.E.; Bagni, C.; Cestra, G.; et al. Nuclear accumulation of mRNAs underlies G4C2 repeat-induced translational repression in a cellular model of C9orf72 ALS. J. Cell Sci. 2015, 128, 1787–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sareen, D.; O’Rourke, J.G.; Meera, P.; Muhammad, A.K.M.G.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 2013, 5, 208ra149. [Google Scholar] [CrossRef] [Green Version]
- Conlon, E.G.; Lu, L.; Sharma, A.; Yamazaki, T.; Tang, T.; Shneider, N.A.; Manley, J.L. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. eLife 2016, 5, e17820. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-B.; Chen, H.-J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Štalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conlon, E.G.; Fagegaltier, D.; Agius, P.; Davis-Porada, J.; Gregory, J.; Hubbard, I.; Kang, K.; Kim, D.; Phatnani, H.; Kwan, J.; et al. Unexpected similarities between C9ORF72 and sporadic forms of ALS/FTD suggest a common disease mechanism. eLife 2018, 7, e37754. [Google Scholar] [CrossRef]
- Prudencio, M.; Belzil, V.V.; Batra, R.; Ross, C.A.; Gendron, T.F.; Pregent, L.J.; Murray, M.E.; Overstreet, K.K.; Piazza-Johnston, A.E.; Desaro, P.; et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 2015, 18, 1175–1182. [Google Scholar] [CrossRef]
- Goodman, L.; Bonini, N.M. Repeat-associated non-AUG (RAN) translation mechanisms are running into focus for GGGGCC-repeat associated ALS/FTD. Prog. Neurobiol. 2019, 183, 101697. [Google Scholar] [CrossRef]
- Schmitz, A.; Marques, J.P.; Oertig, I.; Maharjan, N.; Saxena, S. Emerging perspectives on dipeptide repeat proteins in C9ORF72 ALS/FTD. Front. Cell. Neurosci. 2021, 15, 637548. [Google Scholar] [CrossRef]
- Saberi, S.; Stauffer, J.E.; Jiang, J.; Garcia, S.D.; Taylor, A.E.; Schulte, D.; Ohkubo, T.; Schloffman, C.L.; Maldonado, M.; Baughn, M.; et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 459–474. [Google Scholar] [CrossRef]
- Schipper, L.J.; Raaphorst, J.; Aronica, E.; Baas, F.; de Haan, R.J.; de Visser, M.; Troost, D. Prevalence of brain and spinal cord inclusions, including dipeptide repeat proteins, in patients with the C9ORF72 hexanucleotide repeat expansion: A systematic neuropathological review. Neuropathol. Appl. Neurobiol. 2016, 42, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.; Hall, B.; Castelli, L.M.; Francis, L.; Woof, R.; Siskos, A.; Kouloura, E.; Gray, E.; Thompson, A.G.; Talbot, K.; et al. Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 2019, 142, 586–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, S.P.; Hall, B.; Woof, R.; Francis, L.; Gatto, N.; Shaw, A.C.; Myszczynska, M.; Hemingway, J.; Coldicott, I.; Willcock, A.; et al. C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 2019, 142, 3771–3790. [Google Scholar] [CrossRef]
- Zaepfel, B.L.; Rothstein, J.D. RNA is a double-edged sword in ALS pathogenesis. Front. Cell. Neurosci. 2021, 15, 708181. [Google Scholar] [CrossRef]
- Arnold, E.S.; Ling, S.-C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 110, E736–E745. [Google Scholar] [CrossRef] [Green Version]
- Donde, A.; Sun, M.; Ling, J.; Braunstein, K.E.; Pang, B.; Wen, X.; Cheng, X.; Chen, L.; Wong, P.C. Splicing repression is a major function of TDP-43 in motor neurons. Acta Neuropathol. 2019, 138, 813–826. [Google Scholar] [CrossRef]
- Fratta, P.; Sivakumar, P.; Humphrey, J.; Lo, K.; Ricketts, T.; Oliveira, H.; Brito-Armas, J.M.; Kalmar, B.; Ule, A.; Yu, Y.; et al. Mice with endogenous TDP -43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Jutzi, D.; Campagne, S.; Schmidt, R.; Reber, S.; Mechtersheimer, J.; Gypas, F.; Schweingruber, C.; Colombo, M.; von Schroetter, C.; Loughlin, F.E.; et al. Aberrant interaction of FUS with the U1 snRNA provides a molecular mechanism of FUS induced amyotrophic lateral sclerosis. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.P.; Pletnikova, O.; Troncoso, J.C.; Wong, P.C. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 2015, 349, 650–655. [Google Scholar] [CrossRef] [Green Version]
- Reber, S.; Stettler, J.; Filosa, G.; Colombo, M.; Jutzi, D.; Lenzken, S.C.; Schweingruber, C.; Bruggmann, R.; Bachi, A.; Barabino, S.M.; et al. Minor intron splicing is regulated by FUS and affected by ALS -associated FUS mutants. EMBO J. 2016, 35, 1504–1521. [Google Scholar] [CrossRef]
- Sun, S.; Ling, S.-C.; Qiu, J.; Albuquerque, C.P.; Zhou, Y.; Tokunaga, S.; Li, H.; Qiu, H.; Bui, A.; Yeo, E.; et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Conlon, E.G.; Manley, J.L.; Rio, D.C. Widespread intron retention impairs protein homeostasis in C9orf72 ALS brains. Genome Res. 2020, 30, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Kim, E.; Duffy, A.; Adalbert, R.; Phillips, B.U.; Peters, O.M.; Stephenson, J.; Yang, S.; Massenzio, F.; Lin, Z.; et al. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat. Neurosci. 2018, 21, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Lopez-Gonzalez, R.; Kunz, R.C.; Gangopadhyay, J.; Borufka, C.; Gygi, S.P.; Gao, F.-B.; Reed, R. Evidence that C9ORF72 dipeptide repeat proteins associate with U2 snRNP to cause mis-splicing in ALS/FTD Patients. Cell Rep. 2017, 19, 2244–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-L.G.; Bristol, L.A.; Jin, L.; Dykes-Hoberg, M.; Crawford, T.; Clawson, L.; Rothstein, J.D. Aberrant RNA processing in a neurodegenerative disease: The cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 1998, 20, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, J.; Birsa, N.; Milioto, C.; McLaughlin, M.; Ule, A.M.; Robaldo, D.; Eberle, A.B.; Kräuchi, R.; Bentham, M.; Brown, A.-L.; et al. FUS ALS-causative mutations impair FUS autoregulation and splicing factor networks through intron retention. Nucleic Acids Res. 2020, 48, 6889–6905. [Google Scholar] [CrossRef]
- Fratta, P.; Isaacs, A.M. The snowball effect of RNA binding protein dysfunction in amyotrophic lateral sclerosis. Brain 2018, 141, 1236–1238. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Ziff, O.J.; Taha, D.M.; Crerar, H.; Clarke, B.E.; Chakrabarti, A.M.; Kelly, G.; Neeves, J.; Tyzack, G.E.; Luscombe, N.M.; Patani, R. Reactive astrocytes in ALS display diminished intron retention. Nucleic Acids Res. 2021, 49, 3168–3184. [Google Scholar] [CrossRef] [PubMed]
- Luisier, R.; Tyzack, G.E.; Hall, C.E.; Mitchell, J.S.; Devine, H.; Taha, D.M.; Malik, B.; Meyer, I.; Greensmith, L.; Newcombe, J.; et al. Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Tyzack, G.E.; Neeves, J.; Crerar, H.; Klein, P.; Ziff, O.; Taha, D.M.; Luisier, R.; Luscombe, N.M.; Patani, R. Aberrant cytoplasmic intron retention is a blueprint for RNA binding protein mislocalization in VCP-related amyotrophic lateral sclerosis. Brain 2021, 144, 1985–1993. [Google Scholar] [CrossRef]
- Lee, J.; Villarreal, O.D.; Chen, X.; Zandee, S.; Young, Y.K.; Torok, C.; Lamarche-Vane, N.; Prat, A.; Rivest, S.; Gosselin, D.; et al. QUAKING regulates microexon alternative splicing of the rho GTPase pathway and controls microglia homeostasis. Cell Rep. 2020, 33, 108560. [Google Scholar] [CrossRef]
- Cestra, G.; Rossi, S.; di Salvio, M.; Cozzolino, M. Control of mRNA translation in ALS proteinopathy. Front. Mol. Neurosci. 2017, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Lehmkuhl, E.M.; Zarnescu, D.C. Lost in translation: Evidence for protein synthesis deficits in ALS/FTD and related neurodegenerative diseases. Adv. Neurobiol. 2018, 20, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Frottin, F.; Pérez-Berlanga, M.; Hartl, F.U.; Hipp, M.S. Multiple pathways of toxicity induced by C9orf72 dipeptide repeat aggregates and G4C2 RNA in a cellular model. eLife 2021, 10, e62718. [Google Scholar] [CrossRef]
- Hartmann, H.; Hornburg, D.; Czuppa, M.; Bader, J.; Michaelsen, M.; Farny, D.; Arzberger, T.; Mann, M.; Meissner, F.; Edbauer, D. Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci. Alliance 2018, 1, e201800070. [Google Scholar] [CrossRef] [Green Version]
- Moens, T.G.; Niccoli, T.; Wilson, K.M.; Atilano, M.; Birsa, N.; Gittings, L.; Holbling, B.V.; Dyson, M.; Thoeng, A.; Neeves, J.; et al. C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 2019, 137, 487–500. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Eshov, A.; Zhou, J.; Isiktas, A.U.; Guo, J.U. C9orf72 arginine-rich dipeptide repeats inhibit UPF1-mediated RNA decay via translational repression. Nat. Commun. 2020, 11, 3354. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Yagi, T.; Cammack, A.J.; Mahadevan, J.; Kuroda, M.; Harms, M.B.; Miller, T.M.; Urano, F. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum. Mol. Genet. 2016, 25, 1803–1813. [Google Scholar] [CrossRef] [Green Version]
- López-Erauskin, J.; Tadokoro, T.; Baughn, M.W.; Myers, B.; McAlonis-Downes, M.; Chillon-Marinas, C.; Asiaban, J.N.; Artates, J.; Bui, A.T.; Vetto, A.P.; et al. ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron 2020, 106, 354. [Google Scholar] [CrossRef] [PubMed]
- Birsa, N.; Ule, A.M.; Garone, M.G.; Tsang, B.; Mattedi, F.; Chong, P.A.; Humphrey, J.; Jarvis, S.; Pisiren, M.; Wilkins, O.G.; et al. FUS-ALS mutants alter FMRP phase separation equilibrium and impair protein translation. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Garone, M.G.; Birsa, N.; Rosito, M.; Salaris, F.; Mochi, M.; de Turris, V.; Nair, R.R.; Cunningham, T.J.; Fisher, E.M.C.; Morlando, M.; et al. ALS-related FUS mutations alter axon growth in motoneurons and affect HuD/ELAVL4 and FMRP activity. Commun. Biol. 2021, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Valdez-Sinon, A.N.; Bassell, G.J. Regulation of RNA granules by FMRP and implications for neurological diseases. Traffic 2020, 21, 454–462. [Google Scholar] [CrossRef]
- Higashimori, H.; Morel, L.; Huth, J.; Lindemann, L.; Dulla, C.; Taylor, A.; Freeman, M.; Yang, Y. Astroglial FMRP-dependent translational down-regulation of mGluR5 underlies glutamate transporter GLT1 dysregulation in the fragile X mouse. Hum. Mol. Genet. 2013, 22, 2041–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakers, K.; Lake, A.M.; Khazanchi, R.; Ouwenga, R.; Vasek, M.J.; Dani, A.; Dougherty, J.D. Astrocytes locally translate transcripts in their peripheral processes. Proc. Natl. Acad. Sci. USA 2017, 114, E3830–E3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, S.; Cozzolino, M. Dysfunction of RNA/RNA-Binding Proteins in ALS Astrocytes and Microglia. Cells 2021, 10, 3005. https://doi.org/10.3390/cells10113005
Rossi S, Cozzolino M. Dysfunction of RNA/RNA-Binding Proteins in ALS Astrocytes and Microglia. Cells. 2021; 10(11):3005. https://doi.org/10.3390/cells10113005
Chicago/Turabian StyleRossi, Simona, and Mauro Cozzolino. 2021. "Dysfunction of RNA/RNA-Binding Proteins in ALS Astrocytes and Microglia" Cells 10, no. 11: 3005. https://doi.org/10.3390/cells10113005
APA StyleRossi, S., & Cozzolino, M. (2021). Dysfunction of RNA/RNA-Binding Proteins in ALS Astrocytes and Microglia. Cells, 10(11), 3005. https://doi.org/10.3390/cells10113005