
Willin/FRMD6: A Multi-Functional Neuronal Protein Associated with Alzheimer’s Disease


Abstract
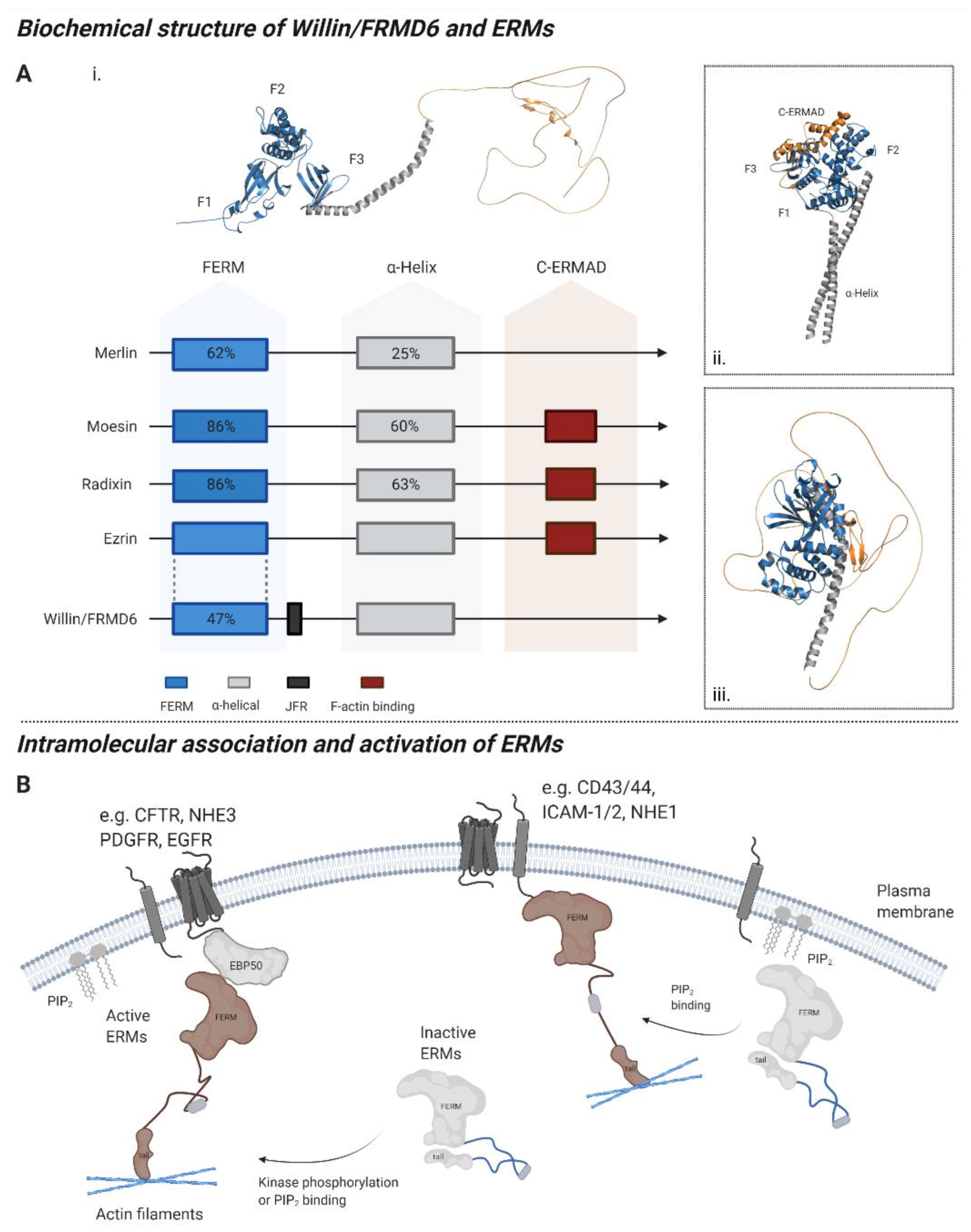
:1. The Willin/FRMD6 Protein
- What is the expression pattern of Willin/FRMD6 in neurons and the nervous system?
- What are the physiological functions of Willin/FRMD6?
- Is Willin/FRMD6 dysregulated in AD?
- If so, which mechanisms may explain its dysregulation?
- What would be the functional consequences AD-induced Willin/FRMD6 dysregulation?
2. Expression of Willin/FRMD6
2.1. Regulation of Willin/FRMD6 Expression
2.2. Willin/FRMD6 Sub-Cellular Expression in Mitotic Cells
2.3. Willin/FRMD6 Expression in the Nervous System
3. Willin/FRMD6 under Normal Physiological Conditions
3.1. Functions of Willin/FRMD6 in Neural Tissues
3.2. Willin/FRMD6 Is an Upstream Regulator of Hippo Signaling
3.3. ERK, mTOR, and c-Myc Signaling Are Involved in Willin/FRMD6 Functional Output
3.4. Willin/FRMD6 in Mechanical Signaling and Cell-Cell Junctions
4. A Link between Willin/FRMD6 and Alzheimer’s Disease
4.1. Single Nucleotide Polymorphisms in the Willin/FRMD6 Gene Are Associated with AD
4.2. Does Willin/FRMD6 Dysregulation Occur in AD?
5. Functional Consequences of AD-Induced Downregulation of Willin/FRMD6
5.1. Signaling Outputs in Oxidative Stress, Autophagy, and Neuroinflammation
5.2. Changes in the Extracellular Environment
5.3. Blood–Brain Barrier Dysfunction
5.4. Dysregulation of the Actin Cytoskeleton and Its Dependent Functions
6. Willin/FRMD6 Interaction Partners and AD
7. Conclusions
- Do changes in Willin/FRMD6 transcripts in AD translate to protein level changes?
- Does Willin/FRMD6 function upstream of MST1, LATS1, and YAP/TAZ in the induction of oxidative stress and mitochondrial dysfunction in AD?
- What factors are involved in the control of Willin/FRMD6 signaling function in neuronal cells? Does the subcellular localization of Willin/FRMD6 act as a functional switch between different signaling roles?
- How does Willin/FRMD6 influence Hippo signaling in a neuronal context?
- How does Willin/FRMD6 affect the actin cytoskeleton in neurons and does this contribute to cytoskeleton-dependent functions such as axonal trafficking and synaptic vesicle cycling?
- Are AD-associated decreases in Willin/FRMD6 transcripts associated with a specific subpopulation of neurons? Do these decreases also occur in glial cells?
- Does restoring Willin/FRMD6 expression ameliorate AD-induced perturbations?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gunn-Moore, F.J.; Welsh, G.I.; Herron, L.R.; Brannigan, F.; Venkateswarlu, K.; Gillespie, S.; Brandwein-Gensler, M.; Madan, R.; Tavare, J.M.; Brophy, P.J.; et al. A novel 4.1 ezrin radixin moesin (FERM)-containing protein, ‘Willin’. FEBS Lett. 2005, 579, 5089–5094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Ishiuchi, T.; Takeichi, M. Willin and Par3 cooperatively regulate epithelial apical constriction through aPKC-mediated ROCK phosphorylation. Nat. Cell Biol. 2011, 13, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Hamada, K.; Shimizu, T.; Matsui, T.; Tsukita, S.; Hakoshima, T. Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J. 2000, 19, 4449–4462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, M.A.; Reczek, D.; Bretscher, A.; Karplus, P.A. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 2000, 101, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.D.; Keep, N.H. The 2.7 A crystal structure of the activated FERM domain of moesin: An analysis of structural changes on activation. Biochemistry 2001, 40, 7061–7068. [Google Scholar] [CrossRef] [PubMed]
- Frame, M.C.; Patel, H.; Serrels, B.; Lietha, D.; Eck, M.J. The FERM domain: Organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 2010, 11, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Moleirinho, S.; Tilston-Lunel, A.; Angus, L.; Gunn-Moore, F.; Reynolds, P.A. The expanding family of FERM proteins. Biochem. J. 2013, 452, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Baines, A.J.; Lu, H.C.; Bennett, P.M. The Protein 4.1 family: Hub proteins in animals for organizing membrane proteins. Biochim. Biophys. Acta 2014, 1838, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Gunn-Moore, F.J.; Tilston-Lunel, A.M.; Reynolds, P.A. Willing to Be Involved in Cancer. Genes 2016, 7, 37. [Google Scholar] [CrossRef]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteom. 2014, 96, 253–262. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.G.; Reynolds, C.A.; Feldman, A.L.; Kallin, M.; Lambert, J.C.; Amouyel, P.; Ingelsson, E.; Pedersen, N.L.; Prince, J.A. Genome-wide and gene-based association implicates FRMD6 in Alzheimer disease. Hum. Mutat. 2012, 33, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Furney, S.J.; Simmons, A.; Breen, G.; Pedroso, I.; Lunnon, K.; Proitsi, P.; Hodges, A.; Powell, J.; Wahlund, L.O.; Kloszewska, I.; et al. Genome-wide association with MRI atrophy measures as a quantitative trait locus for Alzheimer’s disease. Mol. Psychiatry 2011, 16, 1130–1138. [Google Scholar] [CrossRef] [Green Version]
- Potkin, S.G.; Guffanti, G.; Lakatos, A.; Turner, J.A.; Kruggel, F.; Fallon, J.H.; Saykin, A.J.; Orro, A.; Lupoli, S.; Salvi, E.; et al. Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer’s disease. PLoS ONE 2009, 4, e6501. [Google Scholar] [CrossRef]
- Stein, J.L.; Hua, X.; Lee, S.; Ho, A.J.; Leow, A.D.; Toga, A.W.; Saykin, A.J.; Shen, L.; Foroud, T.; Pankratz, N.; et al. Voxelwise genome-wide association study (vGWAS). Neuroimage 2010, 53, 1160–1174. [Google Scholar] [CrossRef] [Green Version]
- Madan, R.; Brandwein-Gensler, M.; Schlecht, N.F.; Elias, K.; Gorbovitsky, E.; Belbin, T.J.; Mahmood, R.; Breining, D.; Qian, H.; Childs, G.; et al. Differential tissue and subcellular expressionof ERM proteins in normal and malignant tissues: Cytoplasmic ezrin expression has prognostic signficance for head and neck squamous cell carcinoma. Head Neck 2006, 28, 1018–1027. [Google Scholar] [CrossRef]
- Moleirinho, S.; Chang, N.; Sims, A.H.; Tilston-Lunel, A.M.; Angus, L.; Steele, A.; Boswell, V.; Barnett, S.C.; Ormandy, C.; Faratian, D.; et al. KIBRA exhibits MST-independent functional regulation of the Hippo signaling pathway in mammals. Oncogene 2013, 32, 1821–1830. [Google Scholar] [CrossRef] [Green Version]
- Moleirinho, S.; Patrick, C.; Tilston-Lunel, A.M.; Higginson, J.R.; Angus, L.; Antkowiak, M.; Barnett, S.C.; Prystowsky, M.B.; Reynolds, P.A.; Gunn-Moore, F.J. Willin, an upstream component of the hippo signaling pathway, orchestrates mammalian peripheral nerve fibroblasts. PLoS ONE 2013, 8, e60028. [Google Scholar] [CrossRef] [Green Version]
- Beck, J.; Kressel, M. FERM domain-containing protein 6 identifies a subpopulation of varicose nerve fibers in different vertebrate species. Cell Tissue Res. 2020, 381, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Lyu, C.; Lyu, G.W.; Mulder, J.; Uhlen, M.; Cai, X.H.; Hokfelt, T.; Sten Shi, T.J. Expression and regulation of FRMD6 in mouse DRG neurons and spinal cord after nerve injury. Sci. Rep. 2020, 10, 1880. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.; Chang, Z.; Gu, X.; Liu, R. MTA2 promotes HCC progression through repressing FRMD6, a key upstream component of hippo signaling pathway. Biochem. Biophys. Res. Commun. 2019, 515, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, J.; Zhi, L.; Cai, F. Linc00887 suppresses tumorigenesis of cervical cancer through regulating the miR-454-3p/FRMD6-Hippo axis. Cancer Cell Int. 2021, 21, 33. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Z.; Wang, X.; Ding, Y.; Li, N. The tumor suppressive effect of long non-coding RNA FRMD6-AS2 in uteri corpus endometrial carcinoma. Life Sci. 2020, 243, 117254. [Google Scholar] [CrossRef]
- Gandini, S.; Guerrieri-Gonzaga, A.; Puntoni, M.; Decensi, A. Metformin and breast cancer risk. J. Clin. Oncol. 2013, 31, 973–974. [Google Scholar] [CrossRef]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, J.; Chen, H.; Wang, R.; Li, P.; Miao, Y.; Liu, P. Metformin suppresses proliferation and invasion of drug-resistant breast cancer cells by activation of the Hippo pathway. J. Cell Mol. Med. 2020, 24, 5786–5796. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, Y.; Chen, X.; Zheng, X.; Xu, G.; Yuan, Z.; Zhao, H.; Chen, W.; Li, L.; Zheng, N.; et al. The TrkB-T1 receptor mediates BDNF-induced migration of aged cardiac microvascular endothelial cells by recruiting Willin. Aging Cell 2019, 18, e12881. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Li, P.; Wang, Y.; Liang, Z.; Liu, J.; Li, J.; Jiang, Y.; Bao, G.; Li, L.; Zhu, B.; et al. CRB3 regulates contact inhibition by activating the Hippo pathway in mammary epithelial cells. Cell Death Dis. 2017, 8, e2546. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Feng, W.; Xin, H.; Tian, Z.; Zhang, Y.; Zhang, L. 14-3-3 Proteins interact with FRMD6 and regulate its subcellular localization in breast cancer cells. Chem. Res. Chin. Univ. 2015, 31, 558–563. [Google Scholar] [CrossRef]
- Ishiuchi, T.; Takeichi, M. Nectins localize Willin to cell-cell junctions. Genes Cells 2012, 17, 387–397. [Google Scholar] [CrossRef]
- Bretscher, A.; Edwards, K.; Fehon, R.G. ERM proteins and merlin: Integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 2002, 3, 586–599. [Google Scholar] [CrossRef]
- Ramesh, V. Merlin and the ERM proteins in Schwann cells, neurons and growth cones. Nat. Rev. Neurosci. 2004, 5, 462–470. [Google Scholar] [CrossRef]
- Angus, L.; Moleirinho, S.; Herron, L.; Sinha, A.; Zhang, X.; Niestrata, M.; Dholakia, K.; Prystowsky, M.B.; Harvey, K.F.; Reynolds, P.A.; et al. Willin/FRMD6 expression activates the Hippo signaling pathway kinases in mammals and antagonizes oncogenic YAP. Oncogene 2012, 31, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Fernando, R.N.; Cotter, L.; Perrin-Tricaud, C.; Berthelot, J.; Bartolami, S.; Pereira, J.A.; Gonzalez, S.; Suter, U.; Tricaud, N. Optimal myelin elongation relies on YAP activation by axonal growth and inhibition by Crb3/Hippo pathway. Nat. Commun. 2016, 7, 12186. [Google Scholar] [CrossRef]
- Flegel, C.; Schobel, N.; Altmuller, J.; Becker, C.; Tannapfel, A.; Hatt, H.; Gisselmann, G. RNA-Seq Analysis of Human Trigeminal and Dorsal Root Ganglia with a Focus on Chemoreceptors. PLoS ONE 2015, 10, e0128951. [Google Scholar] [CrossRef] [Green Version]
- Manteniotis, S.; Lehmann, R.; Flegel, C.; Vogel, F.; Hofreuter, A.; Schreiner, B.S.; Altmuller, J.; Becker, C.; Schobel, N.; Hatt, H.; et al. Comprehensive RNA-Seq expression analysis of sensory ganglia with a focus on ion channels and GPCRs in Trigeminal ganglia. PLoS ONE 2013, 8, e79523. [Google Scholar] [CrossRef]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lonnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggstrom, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef]
- Uttam, S.; Wong, C.; Amorim, I.S.; Jafarnejad, S.M.; Tansley, S.N.; Yang, J.; Prager-Khoutorsky, M.; Mogil, J.S.; Gkogkas, C.G.; Khoutorsky, A. Translational profiling of dorsal root ganglia and spinal cord in a mouse model of neuropathic pain. Neurobiol. Pain 2018, 4, 35–44. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Buzsaki, G. Theta oscillations in the hippocampus. Neuron 2002, 33, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Landgraf, R.; Frank, E.; Aldag, J.M.; Neumann, I.D.; Sharer, C.A.; Ren, X.; Terwilliger, E.F.; Niwa, M.; Wigger, A.; Young, L.J. Viral vector-mediated gene transfer of the vole V1a vasopressin receptor in the rat septum: Improved social discrimination and active social behaviour. Eur. J. Neurosci. 2003, 18, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Stoop, R. Neuromodulation by oxytocin and vasopressin. Neuron 2012, 76, 142–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borbely, E.; Scheich, B.; Helyes, Z. Neuropeptides in learning and memory. Neuropeptides 2013, 47, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, D.; Van Dijck, A.; Janssen, L.; De Deyn, P.P. Neuropeptides in Alzheimer’s disease: From pathophysiological mechanisms to therapeutic opportunities. Curr. Alzheimer Res. 2013, 10, 449–468. [Google Scholar] [CrossRef]
- Li, W.; Cooper, J.; Karajannis, M.A.; Giancotti, F.G. Merlin: A tumour suppressor with functions at the cell cortex and in the nucleus. EMBO Rep. 2012, 13, 204–215. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Wu, M.Z.; Deng, X.T.; Ma, P.C.; Li, Z.H.; Liang, L.; Xia, M.F.; Cui, D.; He, D.D.; Zong, Y.; et al. Inhibition of YAP/TAZ Activity in Spinal Cord Suppresses Neuropathic Pain. J. Neurosci. 2016, 36, 10128–10140. [Google Scholar] [CrossRef]
- Chen, B.P.; Liang, G.; Whelan, J.; Hai, T. ATF3 and ATF3 delta Zip. Transcriptional repression versus activation by alternatively spliced isoforms. J. Biol. Chem. 1994, 269, 15819–15826. [Google Scholar] [CrossRef]
- Chu, H.M.; Tan, Y.; Kobierski, L.A.; Balsam, L.B.; Comb, M.J. Activating transcription factor-3 stimulates 3′,5′-cyclic adenosine monophosphate-dependent gene expression. Mol. Endocrinol. 1994, 8, 59–68. [Google Scholar] [CrossRef]
- Hai, T.; Curran, T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. USA 1991, 88, 3720–3724. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Kim, K.T. Short-term plasticity of small synaptic vesicle (SSV) and large dense-core vesicle (LDCV) exocytosis. Cell Signal. 2009, 21, 1465–1470. [Google Scholar] [CrossRef]
- Ramamoorthy, P.; Wang, Q.; Whim, M.D. Cell type-dependent trafficking of neuropeptide Y-containing dense core granules in CNS neurons. J. Neurosci. 2011, 31, 14783–14788. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Franco, J.J.; Munoz-Cuevas, F.J.; Lujan, R.; Jurado, S. Excitatory and Inhibitory Neurons in the Hippocampus Exhibit Molecularly Distinct Large Dense Core Vesicles. Front. Cell Neurosci. 2016, 10, 202. [Google Scholar] [CrossRef] [Green Version]
- Chirivino, D.; Del Maestro, L.; Formstecher, E.; Hupe, P.; Raposo, G.; Louvard, D.; Arpin, M. The ERM proteins interact with the HOPS complex to regulate the maturation of endosomes. Mol. Biol. Cell 2011, 22, 375–385. [Google Scholar] [CrossRef]
- Erwig, L.P.; McPhilips, K.A.; Wynes, M.W.; Ivetic, A.; Ridley, A.J.; Henson, P.M. Differential regulation of phagosome maturation in macrophages and dendritic cells mediated by Rho GTPases and ezrin-radixin-moesin (ERM) proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 12825–12830. [Google Scholar] [CrossRef] [Green Version]
- Muriel, O.; Tomas, A.; Scott, C.C.; Gruenberg, J. Moesin and cortactin control actin-dependent multivesicular endosome biogenesis. Mol. Biol. Cell 2016, 27, 3305–3316. [Google Scholar] [CrossRef] [Green Version]
- Schwander, M.; Lopes, V.; Sczaniecka, A.; Gibbs, D.; Lillo, C.; Delano, D.; Tarantino, L.M.; Wiltshire, T.; Williams, D.S.; Muller, U. A novel allele of myosin VIIa reveals a critical function for the C-terminal FERM domain for melanosome transport in retinal pigment epithelial cells. J. Neurosci. 2009, 29, 15810–15818. [Google Scholar] [CrossRef] [Green Version]
- Semenova, I.; Ikeda, K.; Ivanov, P.; Rodionov, V. The protein kinase A-anchoring protein moesin is bound to pigment granules in melanophores. Traffic 2009, 10, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Lou, X. Sensing Exocytosis and Triggering Endocytosis at Synapses: Synaptic Vesicle Exocytosis-Endocytosis Coupling. Front. Cell Neurosci. 2018, 12, 66. [Google Scholar] [CrossRef]
- Meunier, F.A.; Gutierrez, L.M. Captivating New Roles of F-Actin Cortex in Exocytosis and Bulk Endocytosis in Neurosecretory Cells. Trends Neurosci. 2016, 39, 605–613. [Google Scholar] [CrossRef]
- Zhao, B.; Tumaneng, K.; Guan, K.L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Mazack, V.; Sudol, M. Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J. Biol. Chem. 2008, 283, 27534–27546. [Google Scholar] [CrossRef] [Green Version]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Wang, S.; Dong, Y.; Yuan, Z. The Role and Regulatory Mechanism of Hippo Signaling Components in the Neuronal System. Front. Immunol. 2020, 11, 281. [Google Scholar] [CrossRef] [Green Version]
- Sahu, M.R.; Mondal, A.C. The emerging role of Hippo signaling in neurodegeneration. J. Neurosci. Res. 2020, 98, 796–814. [Google Scholar] [CrossRef]
- Seo, J.; Kim, J. Regulation of Hippo signaling by actin remodeling. BMB Rep. 2018, 51, 151–156. [Google Scholar] [CrossRef]
- Bossuyt, W.; Chen, C.L.; Chen, Q.; Sudol, M.; McNeill, H.; Pan, D.; Kopp, A.; Halder, G. An evolutionary shift in the regulation of the Hippo pathway between mice and flies. Oncogene 2014, 33, 1218–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldrup, J.; Strand, S.H.; Cieza-Borrella, C.; Jakobsson, M.E.; Riedel, M.; Norgaard, M.; Hedensted, S.; Dagnaes-Hansen, F.; Ulhoi, B.P.; Eeles, R.; et al. FRMD6 has tumor suppressor functions in prostate cancer. Oncogene 2021, 40, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Visser-Grieve, S.; Hao, Y.; Yang, X. Human homolog of Drosophila expanded, hEx, functions as a putative tumor suppressor in human cancer cell lines independently of the Hippo pathway. Oncogene 2012, 31, 1189–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronenberg, N.M.; Tilston-Lunel, A.; Thompson, F.E.; Chen, D.; Yu, W.; Dholakia, K.; Gather, M.C.; Gunn-Moore, F.J. Willin/FRMD6 Influences Mechanical Phenotype and Neuronal Differentiation in Mammalian Cells by Regulating ERK1/2 Activity. Front. Cell Neurosci. 2020, 14, 552213. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, K.; Yu, Q. FRMD6 inhibits human glioblastoma growth and progression by negatively regulating activity of receptor tyrosine kinases. Oncotarget 2016, 7, 70080–70091. [Google Scholar] [CrossRef] [Green Version]
- Gomes, D.A.; Rodrigues, M.A.; Leite, M.F.; Gomez, M.V.; Varnai, P.; Balla, T.; Bennett, A.M.; Nathanson, M.H. c-Met must translocate to the nucleus to initiate calcium signals. J. Biol. Chem. 2008, 283, 4344–4351. [Google Scholar] [CrossRef] [Green Version]
- Twine, N.A.; Janitz, K.; Wilkins, M.R.; Janitz, M. Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PLoS ONE 2011, 6, e16266. [Google Scholar] [CrossRef]
- Castillo, E.; Leon, J.; Mazzei, G.; Abolhassani, N.; Haruyama, N.; Saito, T.; Saido, T.; Hokama, M.; Iwaki, T.; Ohara, T.; et al. Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep. 2017, 7, 17762. [Google Scholar] [CrossRef] [Green Version]
- Vega, I.E.; Umstead, A.; Wygant, C.M.; Beck, J.S.; Counts, S.E. Ezrin Expression is Increased During Disease Progression in a Tauopathy Mouse Model and Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 1086–1095. [Google Scholar] [CrossRef]
- Dejanovic, B.; Huntley, M.A.; De Maziere, A.; Meilandt, W.J.; Wu, T.; Srinivasan, K.; Jiang, Z.; Gandham, V.; Friedman, B.A.; Ngu, H.; et al. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 2018, 100, 1322–1336.e1327. [Google Scholar] [CrossRef] [Green Version]
- Takousis, P.; Sadlon, A.; Schulz, J.; Wohlers, I.; Dobricic, V.; Middleton, L.; Lill, C.M.; Perneczky, R.; Bertram, L. Differential expression of microRNAs in Alzheimer’s disease brain, blood, and cerebrospinal fluid. Alzheimer’s Dement. 2019, 15, 1468–1477. [Google Scholar] [CrossRef]
- Fumagalli, F.; Racagni, G.; Riva, M.A. The expanding role of BDNF: A therapeutic target for Alzheimer’s disease? Pharm. J. 2006, 6, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C. Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer’s Disease (AD): A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.S.; Dunckley, T.; Beach, T.G.; Grover, A.; Mastroeni, D.; Walker, D.G.; Caselli, R.J.; Kukull, W.A.; McKeel, D.; Morris, J.C.; et al. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol. Genom. 2007, 28, 311–322. [Google Scholar] [CrossRef]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446. [Google Scholar] [CrossRef] [Green Version]
- Stopa, E.G.; Tanis, K.Q.; Miller, M.C.; Nikonova, E.V.; Podtelezhnikov, A.A.; Finney, E.M.; Stone, D.J.; Camargo, L.M.; Parker, L.; Verma, A.; et al. Comparative transcriptomics of choroid plexus in Alzheimer’s disease, frontotemporal dementia and Huntington’s disease: Implications for CSF homeostasis. Fluids Barriers CNS 2018, 15, 18. [Google Scholar] [CrossRef]
- Ross, R.A.; Spengler, B.A.; Biedler, J.L. Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J. Natl Cancer Inst. 1983, 71, 741–747. [Google Scholar]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Forster, J.I.; Koglsberger, S.; Trefois, C.; Boyd, O.; Baumuratov, A.S.; Buck, L.; Balling, R.; Antony, P.M. Characterization of Differentiated SH-SY5Y as Neuronal Screening Model Reveals Increased Oxidative Vulnerability. J. Biomol. Screen 2016, 21, 496–509. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, M.; Qi, M.L.; Yoshimura, N.; Miyashita, T.; Tagawa, K.; Wada, Y.; Enokido, Y.; Marubuchi, S.; Harjes, P.; Arai, N.; et al. Transcriptional repression induces a slowly progressive atypical neuronal death associated with changes of YAP isoforms and p73. J. Cell Biol. 2006, 172, 589–604. [Google Scholar] [CrossRef] [Green Version]
- Mueller, K.A.; Glajch, K.E.; Huizenga, M.N.; Wilson, R.A.; Granucci, E.J.; Dios, A.M.; Tousley, A.R.; Iuliano, M.; Weisman, E.; LaQuaglia, M.J.; et al. Hippo Signaling Pathway Dysregulation in Human Huntington’s Disease Brain and Neuronal Stem Cells. Sci. Rep. 2018, 8, 11355. [Google Scholar] [CrossRef] [Green Version]
- Yamanishi, E.; Hasegawa, K.; Fujita, K.; Ichinose, S.; Yagishita, S.; Murata, M.; Tagawa, K.; Akashi, T.; Eishi, Y.; Okazawa, H. A novel form of necrosis, TRIAD, occurs in human Huntington’s disease. Acta Neuropathol. Commun. 2017, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Shin, J.H.; Hwang, S.G.; Gwag, B.J.; McKee, A.C.; Lee, J.; Kowall, N.W.; Ryu, H.; Lim, D.S.; Choi, E.J. MST1 functions as a key modulator of neurodegeneration in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2013, 110, 12066–12071. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Ji, J.X.; Xu, Y.T.; Ni, H.B.; Rui, Q.; Liu, H.X.; Jiang, F.; Gao, R.; Chen, G. Inhibition of Lats1/p-YAP1 pathway mitigates neuronal apoptosis and neurological deficits in a rat model of traumatic brain injury. CNS Neurosci. Ther. 2018, 24, 906–916. [Google Scholar] [CrossRef]
- Tanaka, H.; Homma, H.; Fujita, K.; Kondo, K.; Yamada, S.; Jin, X.; Waragai, M.; Ohtomo, G.; Iwata, A.; Tagawa, K.; et al. YAP-dependent necrosis occurs in early stages of Alzheimer’s disease and regulates mouse model pathology. Nat. Commun. 2020, 11, 507. [Google Scholar] [CrossRef]
- Yu, L.; Liu, Y.; Jin, Y.; Cao, X.; Chen, J.; Jin, J.; Gu, Y.; Bao, X.; Ren, Z.; Xu, Y.; et al. Lentivirus-Mediated HDAC3 Inhibition Attenuates Oxidative Stress in APPswe/PS1dE9 Mice. J. Alzheimer’s Dis. 2018, 61, 1411–1424. [Google Scholar] [CrossRef]
- Mao, Y.; Chen, X.; Xu, M.; Fujita, K.; Motoki, K.; Sasabe, T.; Homma, H.; Murata, M.; Tagawa, K.; Tamura, T.; et al. Targeting TEAD/YAP-transcription-dependent necrosis, TRIAD, ameliorates Huntington’s disease pathology. Hum. Mol. Genet. 2016, 25, 4749–4770. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, D.F.; Luo, R.; Wu, Y.; Zhou, H.; Kong, L.L.; Bi, R.; Yao, Y.G. A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 215–229. [Google Scholar] [CrossRef]
- Shao, D.; Zhai, P.; Del Re, D.P.; Sciarretta, S.; Yabuta, N.; Nojima, H.; Lim, D.S.; Pan, D.; Sadoshima, J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 2014, 5, 3315. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.; Liu, X.; Zhang, J.; Wang, H.G.; Ye, J.M.; Shi, Y. Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy 2015, 11, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, F.; Cui, Y.; Liu, S.; Sun, H. Mst1 overexpression combined with Yap knockdown augments thyroid carcinoma apoptosis via promoting MIEF1-related mitochondrial fission and activating the JNK pathway. Cancer Cell Int. 2019, 19, 143. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, Y.; Hu, G.; Zhou, J.; Mei, L.; Xiong, W.C. YAP Is a Critical Inducer of SOCS3, Preventing Reactive Astrogliosis. Cereb. Cortex 2016, 26, 2299–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Rutten, B.P.F.; Kim, M.O. MST1 Regulates Neuronal Cell Death via JNK/Casp3 Signaling Pathway in HFD Mouse Brain and HT22 Cells. Int. J. Mol. Sci. 2019, 20, 2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Seo, B.R.; Choi, E.J.; Koh, J.Y. The role of reciprocal activation of cAbl and Mst1 in the oxidative death of cultured astrocytes. Glia 2014, 62, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, M.K.; Yuan, Z.; Boag, P.R.; Yang, Y.; Villen, J.; Becker, E.B.; DiBacco, S.; de la Iglesia, N.; Gygi, S.; Blackwell, T.K.; et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 2006, 125, 987–1001. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Chen, D.; Hu, P.; Wu, J.; Liu, W.; Zhao, Y.; Cao, M.; Fang, Y.; Bi, W.; Zheng, Z.; et al. The c-Abl-MST1 signaling pathway mediates oxidative stress-induced neuronal cell death. J. Neurosci. 2011, 31, 9611–9619. [Google Scholar] [CrossRef]
- Yun, H.J.; Yoon, J.H.; Lee, J.K.; Noh, K.T.; Yoon, K.W.; Oh, S.P.; Oh, H.J.; Chae, J.S.; Hwang, S.G.; Kim, E.H.; et al. Daxx mediates activation-induced cell death in microglia by triggering MST1 signalling. EMBO J. 2011, 30, 2465–2476. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Zhou, Z.; Yang, H.; Jiao, F.; Li, N.; Gao, Y.; Wang, L.; Chen, J.; Quan, M. MST1 Suppresses Pancreatic Cancer Progression via ROS-Induced Pyroptosis. Mol. Cancer Res. 2019, 17, 1316–1325. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Ye, M.; Yang, W.; Wang, M.; Li, M.; Gu, C.; Zhao, L.; Zhang, Z.; Han, W.; Fan, W.; et al. Effect of Mst1 on Endometriosis Apoptosis and Migration: Role of Drp1-Related Mitochondrial Fission and Parkin-Required Mitophagy. Cell Physiol. Biochem. 2018, 45, 1172–1190. [Google Scholar] [CrossRef]
- Ma, C.; Fan, L.; Wang, J.; Hao, L.; He, J. Hippo/Mst1 overexpression induces mitochondrial death in head and neck squamous cell carcinoma via activating β-catenin/Drp1 pathway. Cell Stress Chaperones 2019, 24, 807–816. [Google Scholar] [CrossRef]
- Zhao, S.; Yin, J.; Zhou, L.; Yan, F.; He, Q.; Huang, L.; Peng, S.; Jia, J.; Cheng, J.; Chen, H.; et al. Hippo/MST1 signaling mediates microglial activation following acute cerebral ischemia-reperfusion injury. Brain Behav. Immun. 2016, 55, 236–248. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.P.; Kudo, W.; Zhu, X.; Smith, M.A.; Lee, H.G. Early induction of c-Myc is associated with neuronal cell death. Neurosci. Lett. 2011, 505, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Perluigi, M.; Di Domenico, F.; Barone, E.; Butterfield, D.A. mTOR in Alzheimer disease and its earlier stages: Links to oxidative damage in the progression of this dementing disorder. Free Radic. Biol. Med. 2021, 169, 382–396. [Google Scholar] [CrossRef]
- Arancio, O.; Zhang, H.P.; Chen, X.; Lin, C.; Trinchese, F.; Puzzo, D.; Liu, S.; Hegde, A.; Yan, S.F.; Stern, A.; et al. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004, 23, 4096–4105. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.B.; Yang, C.T.; Zheng, D.D.; Mo, L.Q.; Wang, X.Y.; Lan, A.P.; Hu, F.; Chen, P.X.; Feng, J.Q.; Zhang, M.F.; et al. Inhibition of ROS-activated ERK1/2 pathway contributes to the protection of H2S against chemical hypoxia-induced injury in H9c2 cells. Mol. Cell Biochem. 2012, 362, 149–157. [Google Scholar] [CrossRef]
- Rasola, A.; Sciacovelli, M.; Chiara, F.; Pantic, B.; Brusilow, W.S.; Bernardi, P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. USA 2010, 107, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Gan, X.; Huang, S.; Wu, L.; Wang, Y.; Hu, G.; Li, G.; Zhang, H.; Yu, H.; Swerdlow, R.H.; Chen, J.X.; et al. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim. Biophys. Acta 2014, 1842, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.M.; Moeendarbary, E.; Sheridan, G.K. Mechanobiology of the brain in ageing and Alzheimer’s disease. Eur. J. Neurosci. 2021, 53, 3851–3878. [Google Scholar] [CrossRef]
- Lepelletier, F.X.; Mann, D.M.; Robinson, A.C.; Pinteaux, E.; Boutin, H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol. Appl Neurobiol. 2017, 43, 167–182. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Chow, B.W.; Gu, C. The molecular constituents of the blood-brain barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Betz, A.L.; Goldstein, G.W. Polarity of the blood-brain barrier: Neutral amino acid transport into isolated brain capillaries. Science 1978, 202, 225–227. [Google Scholar] [CrossRef]
- Wan, W.; Cao, L.; Liu, L.; Zhang, C.; Kalionis, B.; Tai, X.; Li, Y.; Xia, S. Abeta(1-42) oligomer-induced leakage in an in vitro blood-brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J. Neurochem. 2015, 134, 382–393. [Google Scholar] [CrossRef]
- Kook, S.Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. Abeta(1)(-)(4)(2)-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca(2)(+)-calcineurin signaling. J. Neurosci. 2012, 32, 8845–8854. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Park, J.C.; Baik, S.H.; Han, S.H.; Cho, H.J.; Choi, H.; Kim, H.J.; Choi, H.; Lee, W.; Kim, D.K.; Mook-Jung, I. Annexin A1 restores Abeta1-42 -induced blood-brain barrier disruption through the inhibition of RhoA-ROCK signaling pathway. Aging Cell 2017, 16, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Vanleeuwen, J.E. Impaired regulation of synaptic actin cytoskeleton in Alzheimer’s disease. Brain Res. Rev. 2011, 67, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Rush, T.; Martinez-Hernandez, J.; Dollmeyer, M.; Frandemiche, M.L.; Borel, E.; Boisseau, S.; Jacquier-Sarlin, M.; Buisson, A. Synaptotoxicity in Alzheimer’s Disease Involved a Dysregulation of Actin Cytoskeleton Dynamics through Cofilin 1 Phosphorylation. J. Neurosci. 2018, 38, 10349–10361. [Google Scholar] [CrossRef] [Green Version]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef]
- Swanson, E.; Breckenridge, L.; McMahon, L.; Som, S.; McConnell, I.; Bloom, G.S. Extracellular Tau Oligomers Induce Invasion of Endogenous Tau into the Somatodendritic Compartment and Axonal Transport Dysfunction. J. Alzheimer’s Dis. 2017, 58, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Vos, M.; Lauwers, E.; Verstreken, P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci. 2010, 2, 139. [Google Scholar] [CrossRef] [Green Version]
- van Bergeijk, P.; Hoogenraad, C.C.; Kapitein, L.C. Right Time, Right Place: Probing the Functions of Organelle Positioning. Trends Cell Biol. 2016, 26, 121–134. [Google Scholar] [CrossRef]
- Venkatesh, K.; Mathew, A.; Koushika, S.P. Role of actin in organelle trafficking in neurons. Cytoskeleton 2020, 77, 97–109. [Google Scholar] [CrossRef]
- Xiao, L.; Chen, Y.; Ji, M.; Dong, J. KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases. J. Biol. Chem. 2011, 286, 7788–7796. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.J.; Xue, W.; Peng, J.; Wang, Y.; Wei, L.; Yang, Z.; Zhu, H.H.; Fang, Y.X.; Gao, W.Q. Elevated expression of Par3 promotes prostate cancer metastasis by forming a Par3/aPKC/KIBRA complex and inactivating the hippo pathway. J. Exp. Clin. Cancer Res. 2017, 36, 139. [Google Scholar] [CrossRef]
- Papassotiropoulos, A.; Fountoulakis, M.; Dunckley, T.; Stephan, D.A.; Reiman, E.M. Genetics, transcriptomics, and proteomics of Alzheimer’s disease. J. Clin. Psychiatry 2006, 67, 652–670. [Google Scholar] [CrossRef]
- Bates, T.C.; Price, J.F.; Harris, S.E.; Marioni, R.E.; Fowkes, F.G.; Stewart, M.C.; Murray, G.D.; Whalley, L.J.; Starr, J.M.; Deary, I.J. Association of KIBRA and memory. Neurosci. Lett. 2009, 458, 140–143. [Google Scholar] [CrossRef]
- Schaper, K.; Kolsch, H.; Popp, J.; Wagner, M.; Jessen, F. KIBRA gene variants are associated with episodic memory in healthy elderly. Neurobiol. Aging 2008, 29, 1123–1125. [Google Scholar] [CrossRef]
- Corneveaux, J.J.; Liang, W.S.; Reiman, E.M.; Webster, J.A.; Myers, A.J.; Zismann, V.L.; Joshipura, K.D.; Pearson, J.V.; Hu-Lince, D.; Craig, D.W.; et al. Evidence for an association between KIBRA and late-onset Alzheimer’s disease. Neurobiol. Aging 2010, 31, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Rodriguez, E.; Infante, J.; Llorca, J.; Mateo, I.; Sanchez-Quintana, C.; Garcia-Gorostiaga, I.; Sanchez-Juan, P.; Berciano, J.; Combarros, O. Age-dependent association of KIBRA genetic variation and Alzheimer’s disease risk. Neurobiol. Aging 2009, 30, 322–324. [Google Scholar] [CrossRef]
- Wang, H.F.; Tan, L.; Yu, J.T.; Ma, X.Y.; Liu, Q.Y.; Wang, W. Age-dependent association of KIBRA gene polymorphism with Alzheimer’s disease in Han Chinese. Mol. Biol. Rep. 2013, 40, 7077–7082. [Google Scholar] [CrossRef]
- Papassotiropoulos, A.; Stephan, D.A.; Huentelman, M.J.; Hoerndli, F.J.; Craig, D.W.; Pearson, J.V.; Huynh, K.D.; Brunner, F.; Corneveaux, J.; Osborne, D.; et al. Common Kibra alleles are associated with human memory performance. Science 2006, 314, 475–478. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Tang, S.; Dong, L.; Han, X.; Cong, L.; Dong, J.; Han, X.; Zhang, Q.; Wang, Y.; Du, Y. The Neuroprotection of KIBRA in Promoting Neuron Survival and Against Amyloid β-Induced Apoptosis. Front. Cell Neurosci. 2019, 13, 137. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Zhang, H. Par3 and aPKC regulate BACE1 endosome-to-TGN trafficking through PACS1. Neurobiol. Aging 2017, 60, 129–140. [Google Scholar] [CrossRef]
- Sun, M.; Huang, C.; Wang, H.; Zhang, H. Par3 regulates polarized convergence between APP and BACE1 in hippocampal neurons. Neurobiol. Aging 2019, 77, 87–93. [Google Scholar] [CrossRef]
- Sun, M.; Asghar, S.Z.; Zhang, H. The polarity protein Par3 regulates APP trafficking and processing through the endocytic adaptor protein Numb. Neurobiol. Dis. 2016, 93, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; Marin, C.; Rey, M.J.; Ribalta, T.; Goutan, E.; Blanco, R.; Tolosa, E.; Marti, E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol. 1999, 58, 729–739. [Google Scholar] [CrossRef]
- Jeronimo-Santos, A.; Vaz, S.H.; Parreira, S.; Rapaz-Lerias, S.; Caetano, A.P.; Buee-Scherrer, V.; Castren, E.; Valente, C.A.; Blum, D.; Sebastiao, A.M.; et al. Dysregulation of TrkB Receptors and BDNF Function by Amyloid-β Peptide is Mediated by Calpain. Cereb Cortex 2015, 25, 3107–3121. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Liu, W.; Naumovski, L.; Neve, R.L. ASPP2 inhibits APP-BP1-mediated NEDD8 conjugation to cullin-1 and decreases APP-BP1-induced cell proliferation and neuronal apoptosis. J. Neurochem. 2003, 85, 801–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenkhuis, B.; Somarakis, A.; de Haan, L.; Dzyubachyk, O.; ME, I.J.; de Miranda, N.; Lelieveldt, B.P.F.; Dijkstra, J.; van Roon-Mom, W.M.C.; Hollt, T.; et al. Iron loading is a prominent feature of activated microglia in Alzheimer’s disease patients. Acta Neuropathol. Commun. 2021, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Sanphui, P.; Biswas, S.C. FoxO3a is activated and executes neuron death via Bim in response to β-amyloid. Cell Death Dis. 2013, 4, e625. [Google Scholar] [CrossRef]
- Rogelj, B.; Mitchell, J.C.; Miller, C.C.; McLoughlin, D.M. The X11/Mint family of adaptor proteins. Brain Res. Rev. 2006, 52, 305–315. [Google Scholar] [CrossRef]
- Swistowski, A.; Zhang, Q.; Orcholski, M.E.; Crippen, D.; Vitelli, C.; Kurakin, A.; Bredesen, D.E. Novel mediators of amyloid precursor protein signaling. J. Neurosci. 2009, 29, 15703–15712. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, T.; Hong, Y.; Yang, W.; Zhang, X.; Luo, H.; Xu, H.; Wang, X. The Retromer Complex and Sorting Nexins in Neurodegenerative Diseases. Front. Aging Neurosci. 2018, 10, 79. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Cell Type | Effect of Willin Overexpression | Effect of Willin Knockdown/Knockout | Reference |
|---|---|---|---|---|
| Hippo | Primary fibroblasts, MCF10A, HeLa | Activation (increased phosphorylation of MST1/2, LATS1/2, YAP/TAZ; increased cytoplasmic YAP) | Inhibition | [19,23,34] |
| PC | Unknown | Inhibition (decreased phosphorylation of MOB1A, LATS1, YAP) | [71] | |
| Aged CMECs | Unknown | Activation (increased MST1/2, LATS 1/2, YAP transcripts) | [30] | |
| SH-SY5Y | Possible inhibition (increased nuclear TAZ) | Possible activation (decreased nuclear TAZ, decreased YAP) | [73] | |
| c-MYC | PC | Unknown | Activation | [71] |
| mTOR | PC | Unknown | Modulation | [71] |
| ERK | GBM | Inhibition | Unknown | [74] |
| SH-SY5Y | Inhibition | Activation | [73] |
| SNP | Position | MAF | p-Value | SNV | Location | Study |
|---|---|---|---|---|---|---|
| rs7140150 | 52010799 | 0.4566 | 4.77 × 10−7 | C > T | Intron | [16] |
| rs11626056 | 52233276 | 0.3295 | 1.18 × 10−6 | C > A, T | Intron | [15] |
| rs7153703 | 51919822 | 0.213 | 3.38 × 10−6 | A > G, T | Promoter | [14] |
| rs11626565 | 52075152 | 0.0599 | 2.45 × 10−5 | A > C | Intron | [13] |
| rs17586545 | 52035018 | 0.0334 | 4.18 × 10−5 | C > T | Intron | [13] |
| rs17123958 | 51942124 | 0.104 | 7.59 × 10−5 | C > G, T | Promoter | [13] |
| rs12885443 | 52075653 | 0.1769 | 5.34 × 10−4 | A > C | Intron | [13] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.; Yu, W.; Aitken, L.; Gunn-Moore, F. Willin/FRMD6: A Multi-Functional Neuronal Protein Associated with Alzheimer’s Disease. Cells 2021, 10, 3024. https://doi.org/10.3390/cells10113024
Chen D, Yu W, Aitken L, Gunn-Moore F. Willin/FRMD6: A Multi-Functional Neuronal Protein Associated with Alzheimer’s Disease. Cells. 2021; 10(11):3024. https://doi.org/10.3390/cells10113024
Chicago/Turabian StyleChen, Doris, Wanjia Yu, Laura Aitken, and Frank Gunn-Moore. 2021. "Willin/FRMD6: A Multi-Functional Neuronal Protein Associated with Alzheimer’s Disease" Cells 10, no. 11: 3024. https://doi.org/10.3390/cells10113024
APA StyleChen, D., Yu, W., Aitken, L., & Gunn-Moore, F. (2021). Willin/FRMD6: A Multi-Functional Neuronal Protein Associated with Alzheimer’s Disease. Cells, 10(11), 3024. https://doi.org/10.3390/cells10113024