
Adaptation to Chronic-Cycling Hypoxia Renders Cancer Cells Resistant to MTH1-Inhibitor Treatment Which Can Be Counteracted by Glutathione Depletion

 , , and
, , and 
Abstract
: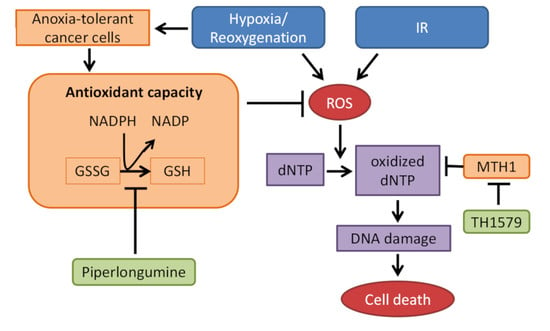
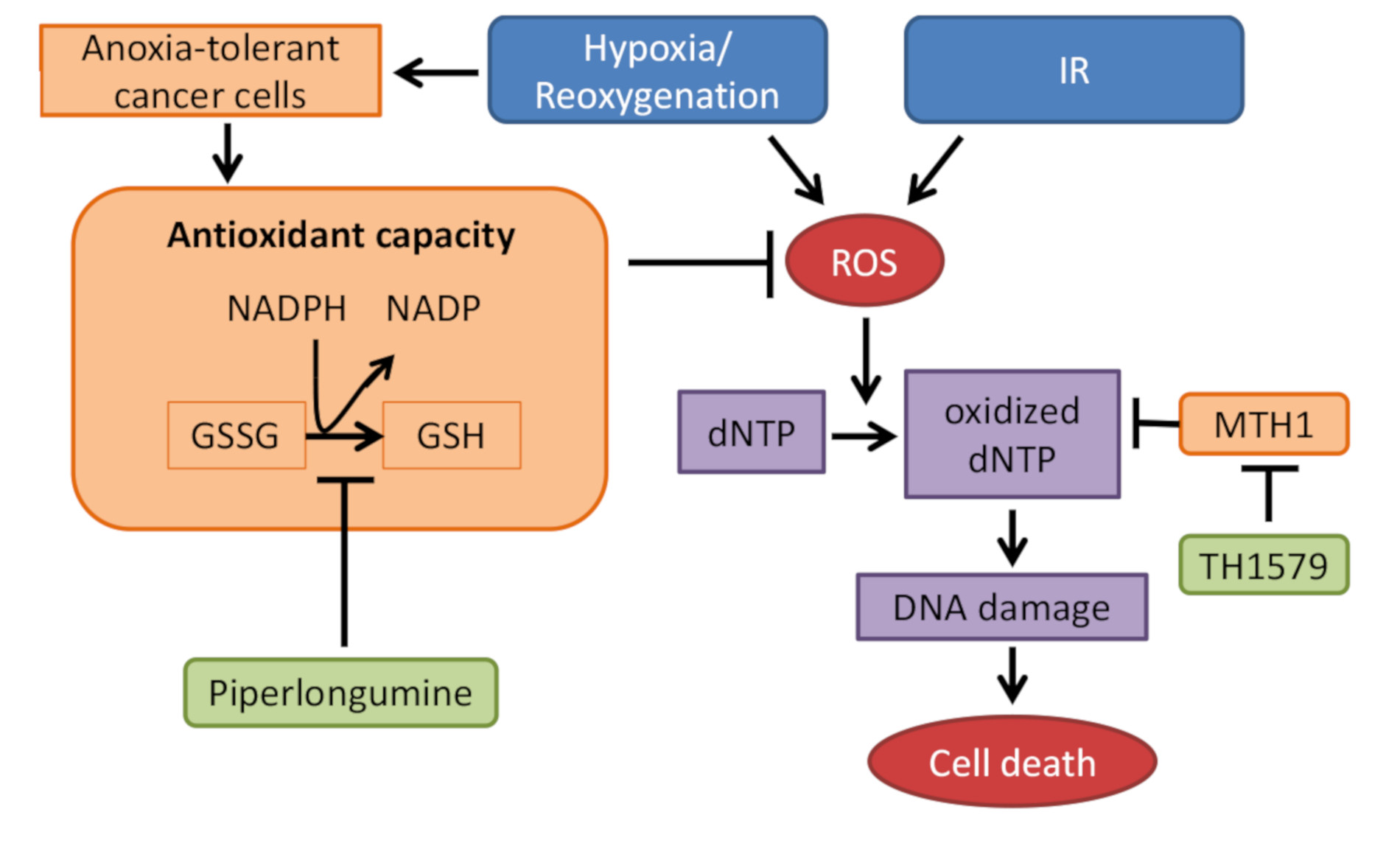{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents, Cell Lines and Culturing Conditions
2.2. Irradiation and Treatment
2.3. qRT-PCR Analysis
2.4. Assessment of Oxidative DNA Damage
2.5. Determination of GSH Levels
2.6. Flow Cytometry Analysis
2.7. Colony Formation Assays
2.8. Statistics
3. Results
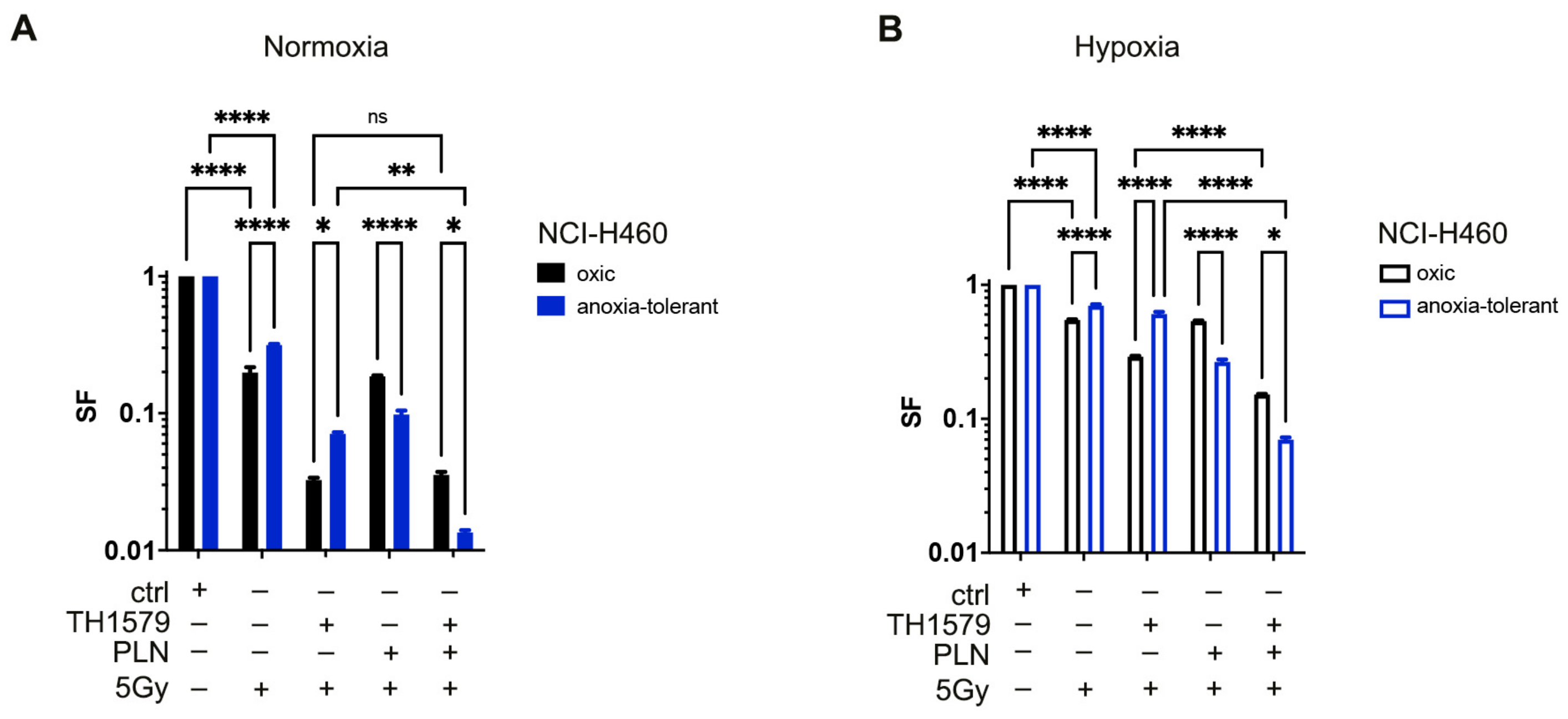
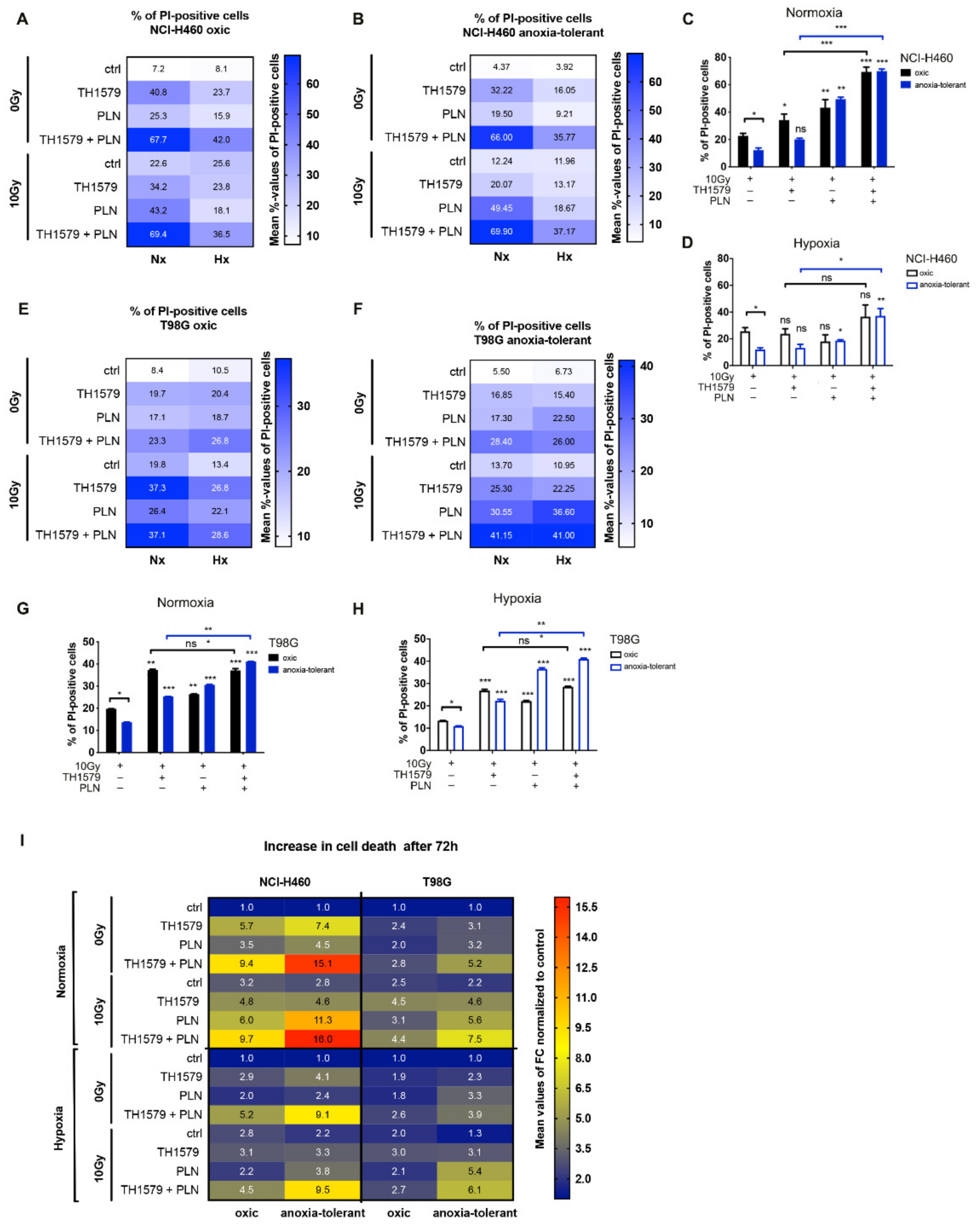
3.1. Inhibition of Glutathione System Overcomes Acute and Chronic-Cycling Hypoxia-Induced Reduction of Cell Death upon MTH1-Inhibition in Combination with IR
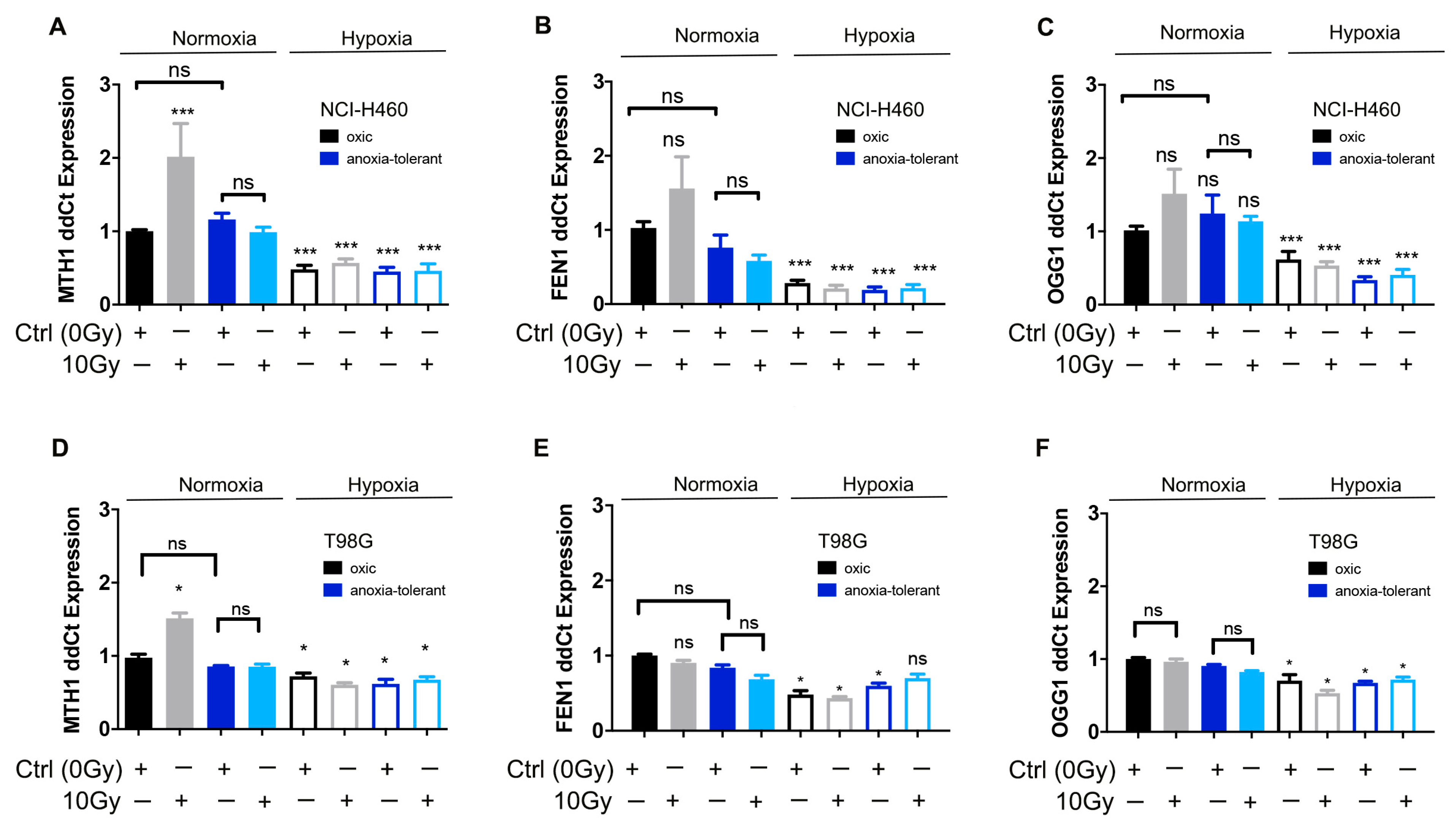
3.2. Ionizing Radiation Increases Expression of MTH1 in Normoxia While Acute and Chronic Cycling Hypoxia Decreases Their Expression
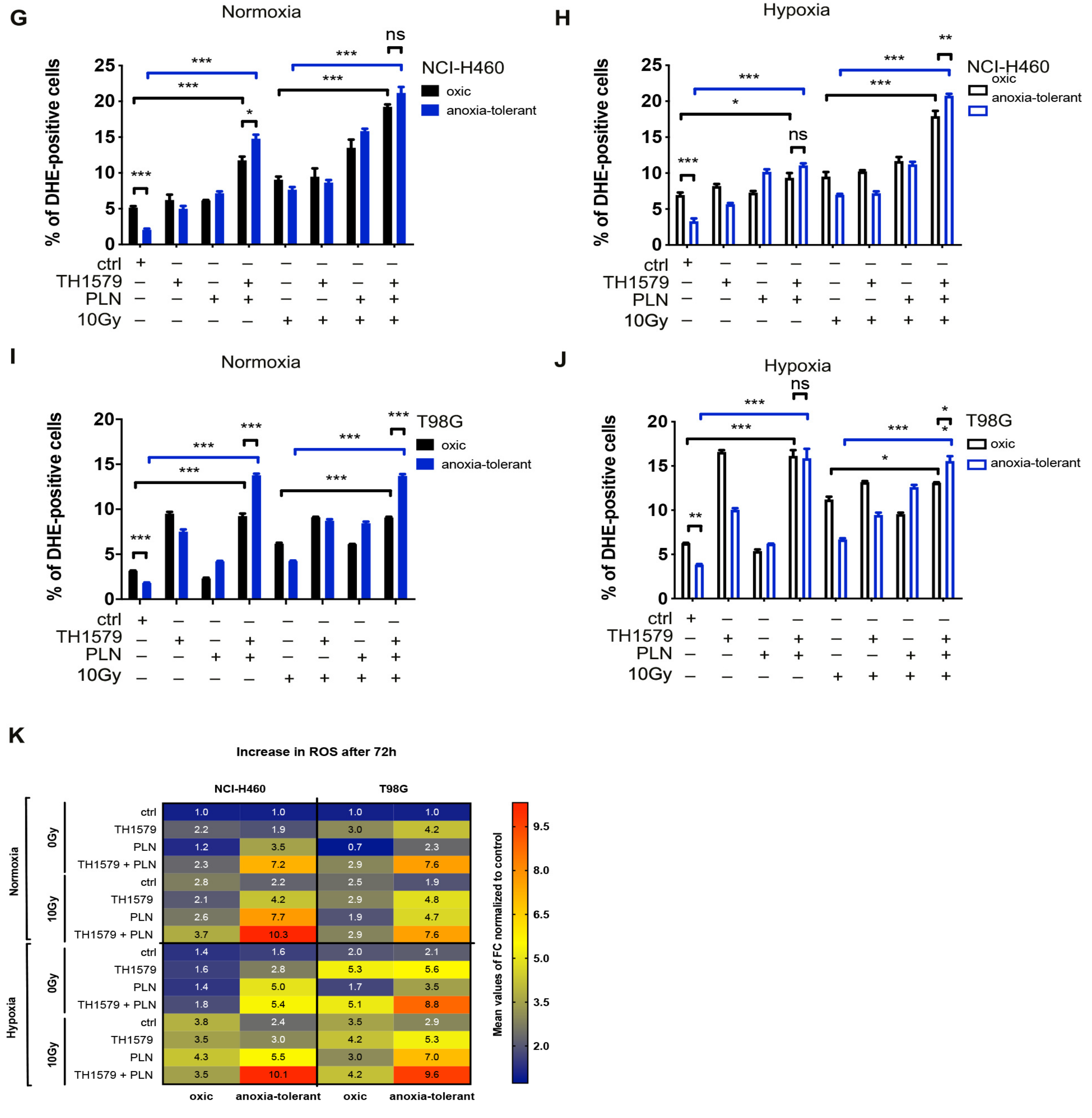
3.3. Exposure to IR or Acute Hypoxia Triggers Antioxidant Defense Offering Opportunity for Multimodal Inhibition of MTH1 and Glutathione Regeneration
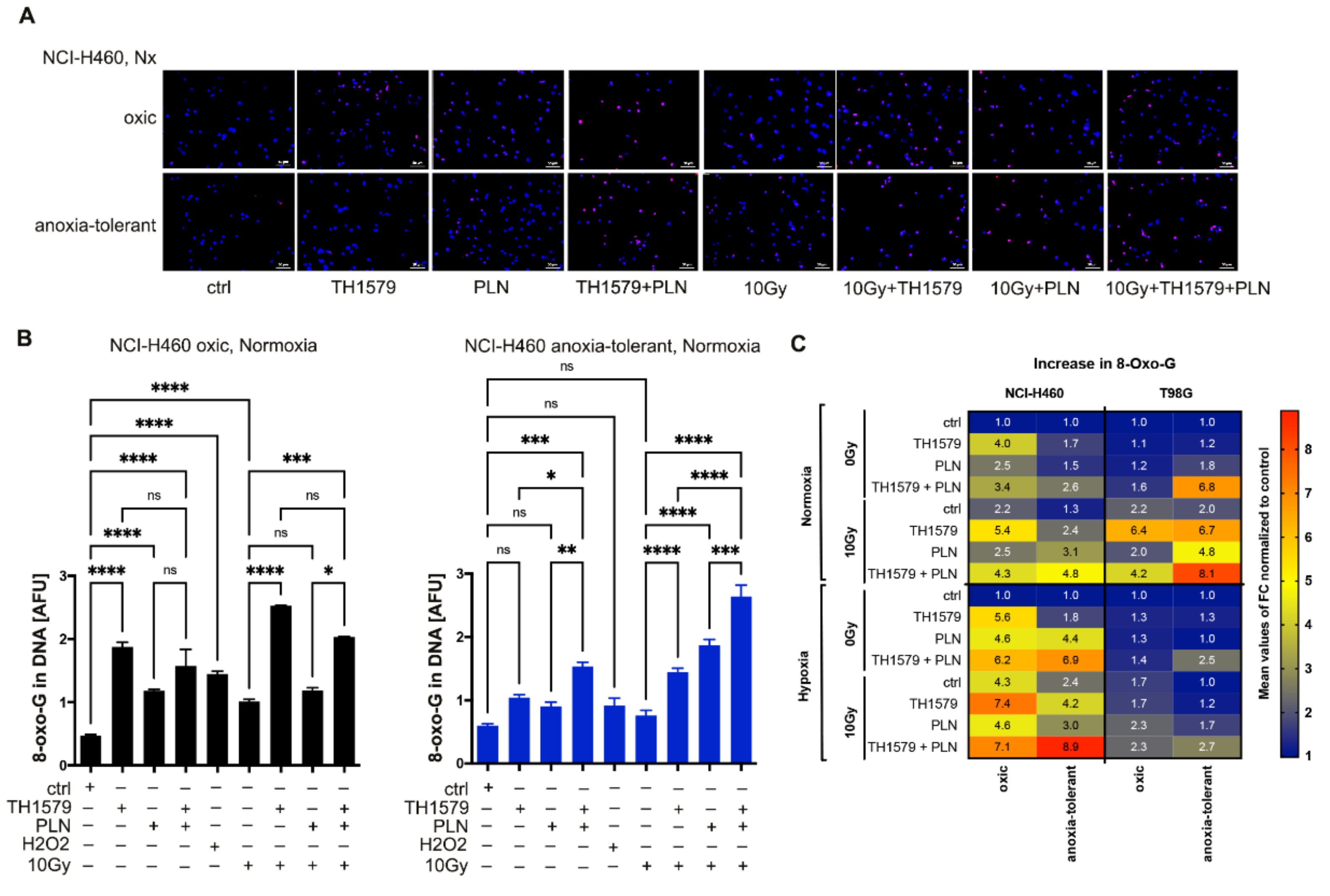
3.4. Radiation Triggers Oxidative DNA Damage and Synergizes with Multimodal Targeting of MTH1 and Glutathione System
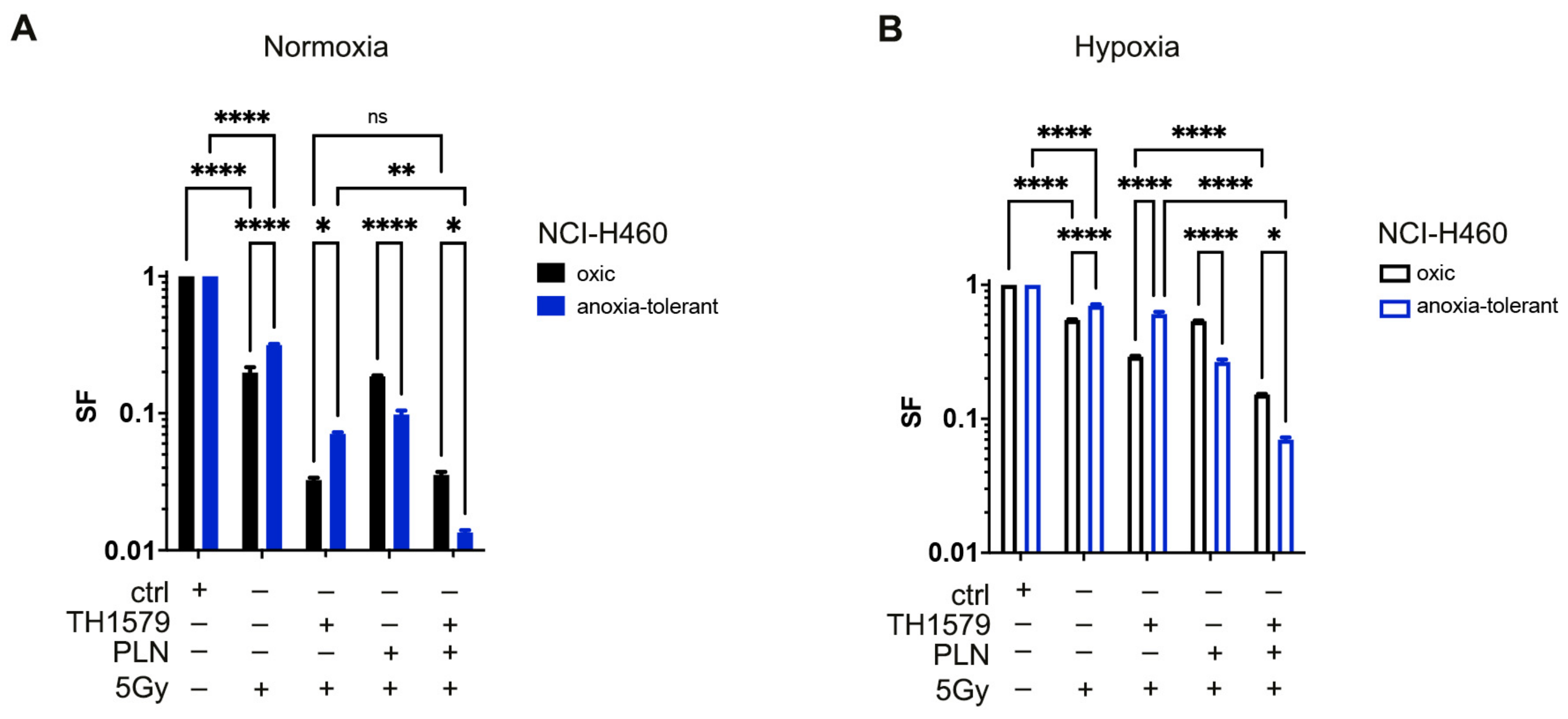
3.5. Inhibition of MTH1 Combined with Inhibition of Glutathione Regeneration Overcomes Radioresistance Induced by Adaptation to Chronic-Cycling Hypoxia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Matschke, J.; Riffkin, H.; Klein, D.; Handrick, R.; Lüdemann, L.; Metzen, E.; Shlomi, T.; Stuschke, M.; Jendrossek, V. Targeted inhibition of glutamine-dependent glutathione metabolism overcomes death resistance induced by chronic cycling hypoxia. Antioxid. Redox Signal. 2016, 25, 89–107. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; Beucken, T.V.D.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. PERK/eIF2 signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc. Natl. Acad. Sci. USA 2013, 110, 4622–4627. [Google Scholar] [CrossRef] [Green Version]
- Hlouschek, J.; Ritter, V.; Wirsdörfer, F.; Klein, D.; Jendrossek, V.; Matschke, J. Targeting SLC25A10 alleviates improved antioxidant capacity and associated radioresistance of cancer cells induced by chronic-cycling hypoxia. Cancer Lett. 2018, 439, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Hlouschek, J.; Hansel, C.; Jendrossek, V.; Matschke, J. The Mitochondrial Citrate Carrier (SLC25A1) sustains redox homeostasis and mitochondrial metabolism supporting radioresistance of cancer cells with tolerance to cycling severe hypoxia. Front. Oncol. 2018, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, R.; Goto, Y.; Koyasu, S.; Kobayashi, M.; Morinibu, A.; Yoshimura, M.; Hiraoka, M.; Hammond, E.M.; Harada, H. UCHL1-HIF-1 axis-mediated antioxidant property of cancer cells as a therapeutic target for radiosensitization. Sci. Rep. 2017, 7, 6879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Wang, H.; De Ridder, M. Targeting antioxidant enzymes as a radiosensitizing strategy. Cancer Lett. 2018, 438, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic radioresistance: Can ROS be the key to overcome it? Cancers 2019, 11, 112. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef]
- Lieber, M. The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair. BioEssays 1997, 19, 233–240. [Google Scholar] [CrossRef]
- Boiteux, S.; Radicella, J.P. The human OGG1 Gene: Structure, functions, and its implication in the process of carcinogenesis. Arch. Biochem. Biophys. 2000, 377, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.N.; Bindra, R.S. Targeting the DNA damage response for cancer therapy. DNA Repair 2009, 8, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA damaging revolution: PARP inhibitors and beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195. [Google Scholar] [CrossRef]
- Gad, H.; Koolmeister, T.; Jemth, A.-S.; Eshtad, S.; Jacques, S.A.; Ström, C.E.; Svensson, L.M.; Schultz, N.; Lundbäck, T.; Einarsdottir, B.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Kettle, J.G.; Alwan, H.; Bista, M.; Breed, J.; Davies, N.L.; Eckersley, K.; Fillery, S.; Foote, K.M.; Goodwin, L.; Jones, D.R.; et al. Potent and selective inhibitors of MTH1 probe its role in cancer cell survival. J. Med. Chem. 2016, 59, 2346–2361. [Google Scholar] [CrossRef]
- Samaranayake, G.J.; Troccoli, C.I.; Zhang, L.; Huynh, M.; Jayaraj, C.J.; Ji, D.; McPherson, L.; Onishi, Y.; Nguyen, D.M.; Robbins, D.J.; et al. The existence of MTH1-independent 8-oxodGTPase activity in cancer cells as a compensatory mechanism against on-target effects of MTH1 inhibitors. Mol. Cancer Ther. 2019, 19, 432–446. [Google Scholar] [CrossRef]
- Bräutigam, L.; Pudelko, L.; Jemth, A.-S.; Gad, H.; Narwal, M.; Gustafsson, R.; Karsten, S.; Puigvert, J.C.; Homan, E.; Berndt, C.; et al. Hypoxic signaling and the cellular redox tumor environment determine sensitivity to MTH1 inhibition. Cancer Res. 2016, 76, 2366–2375. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.; Glazer, P.M. Impact of hypoxia on DNA repair and genome integrity. Mutagenesis 2019, 35, 61–68. [Google Scholar] [CrossRef]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Matschke, J.; Wiebeck, E.; Hurst, S.; Rudner, J.; Jendrossek, V. Role of SGK1 for fatty acid uptake, cell survival and radioresistance of NCI-H460 lung cancer cells exposed to acute or chronic cycling severe hypoxia. Radiat. Oncol. 2016, 11, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef]
- Berglund, U.W.; Sanjiv, K.; Gad, H.; Kalderén, C.; Koolmeister, T.; Pham, T.; Gokturk, C.; Jafari, R.; Maddalo, G.; Seashore-Ludlow, B.; et al. Validation and development of MTH1 inhibitors for treatment of cancer. Ann. Oncol. 2016, 27, 2275–2283. [Google Scholar] [CrossRef] [PubMed]
- Caradec, J.; Sirab, N.; Keumeugni, C.; Moutereau, S.; Chimingqi, M.; Matar, C.; Revaud, D.; Bah, M.; Manivet, P.; Conti, M.; et al. ‘Desperate house genes’: The dramatic example of hypoxia. Br. J. Cancer 2010, 102, 1037–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Thomas, J.P.; Lautermann, J.; Liedert, B.; Seiler, F.; Thomale, J. High accumulation of platinum-DNA adducts in strial marginal cells of the cochlea is an early event in cisplatin but not carboplatin ototoxicity. Mol. Pharmacol. 2006, 70, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Raj, L.; Ide, T.; Gurkar, A.U.; Foley, M.; Schenone, M.; Li, X.; Tolliday, N.J.; Golub, T.R.; Carr, S.A.; Shamji, A.F.; et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, M.; Jendrossek, V.; Güner, D.; Goecke, B.; Belka, C. Cyclic exposure to hypoxia and reoxygenation selects for tumor cells with defects in mitochondrial apoptotic pathways. FASEB J. 2004, 18, 1906–1908. [Google Scholar] [CrossRef]
- Pompsch, M.; Vogel, J.; Classen, F.; Kranz, P.; Iliakis, G.; Riffkin, H.; Brockmeier, U.; Metzen, E. The presumed MTH1-inhibitor TH588 sensitizes colorectal carcinoma cells to ionizing radiation in hypoxia. BMC Cancer 2018, 18, 1190. [Google Scholar] [CrossRef]
- McPherson, L.A.; Troccoli, C.I.; Ji, D.; Bowles, A.E.; Gardiner, M.L.; Mohsen, M.G.; Nagathihalli, N.S.; Nguyen, D.M.; Robbins, D.J.; Merchant, N.B.; et al. Increased MTH1-specific 8-oxodGTPase activity is a hallmark of cancer in colon, lung and pancreatic tissue. DNA Repair 2019, 83, 102644. [Google Scholar] [CrossRef] [PubMed]
- Das, I.; Gad, H.; Bräutigam, L.; Pudelko, L.; Tuominen, R.; Höiom, V.; Almlöf, I.; Rajagopal, V.; Hansson, J.; Helleday, T.; et al. AXL and CAV-1 play a role for MTH1 inhibitor TH1579 sensitivity in cutaneous malignant melanoma. Cell Death Differ. 2020, 27, 2081–2098. [Google Scholar] [CrossRef]
- Toth, R.K.; Warfel, N.A. Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants 2017, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, H.M.; Kansanen, E.; Pölönen, P.; Heinäniemi, M.; Levonen, A.-L. Role of the Keap1–Nrf2 Pathway in Cancer. Adv. Cancer Res. 2014, 122, 281–320. [Google Scholar] [CrossRef]
- Sekhar, K.R.; Freeman, M.L. Nrf2 promotes survival following exposure to ionizing radiation. Free. Radic. Biol. Med. 2015, 88, 268–274. [Google Scholar] [CrossRef] [Green Version]
- Hatem, E.; El Banna, N.; Huang, M.-E. Multifaceted roles of glutathione and glutathione-based systems in carcinogenesis and anticancer drug resistance. Antioxid. Redox Signal. 2017, 27, 1217–1234. [Google Scholar] [CrossRef]
- De Santis, M.C.; Porporato, P.E.; Martini, M.; Morandi, A. Signaling pathways regulating redox balance in cancer metabolism. Front. Oncol. 2018, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, H.R.; Gadre, S.M.; Tan, M.; Graham, G.T.; Mosaoa, R.; Ongkeko, M.S.; Kim, K.A.; Riggins, R.B.; Parasido, E.; Petrini, I.; et al. The mitochondrial citrate carrier, SLC25A1, drives stemness and therapy resistance in non-small cell lung cancer. Cell Death Differ. 2018, 25, 1239–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Zhou, X.; Curbo, S.; Karlsson, A. Metformin downregulates the mitochondrial carrier SLC25A10 in a glucose dependent manner. Biochem. Pharmacol. 2018, 156, 444–450. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansel, C.; Hlouschek, J.; Xiang, K.; Melnikova, M.; Thomale, J.; Helleday, T.; Jendrossek, V.; Matschke, J. Adaptation to Chronic-Cycling Hypoxia Renders Cancer Cells Resistant to MTH1-Inhibitor Treatment Which Can Be Counteracted by Glutathione Depletion. Cells 2021, 10, 3040. https://doi.org/10.3390/cells10113040
Hansel C, Hlouschek J, Xiang K, Melnikova M, Thomale J, Helleday T, Jendrossek V, Matschke J. Adaptation to Chronic-Cycling Hypoxia Renders Cancer Cells Resistant to MTH1-Inhibitor Treatment Which Can Be Counteracted by Glutathione Depletion. Cells. 2021; 10(11):3040. https://doi.org/10.3390/cells10113040
Chicago/Turabian StyleHansel, Christine, Julian Hlouschek, Kexu Xiang, Margarita Melnikova, Juergen Thomale, Thomas Helleday, Verena Jendrossek, and Johann Matschke. 2021. "Adaptation to Chronic-Cycling Hypoxia Renders Cancer Cells Resistant to MTH1-Inhibitor Treatment Which Can Be Counteracted by Glutathione Depletion" Cells 10, no. 11: 3040. https://doi.org/10.3390/cells10113040
APA StyleHansel, C., Hlouschek, J., Xiang, K., Melnikova, M., Thomale, J., Helleday, T., Jendrossek, V., & Matschke, J. (2021). Adaptation to Chronic-Cycling Hypoxia Renders Cancer Cells Resistant to MTH1-Inhibitor Treatment Which Can Be Counteracted by Glutathione Depletion. Cells, 10(11), 3040. https://doi.org/10.3390/cells10113040