
TRPV4 Mechanotransduction in Fibrosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
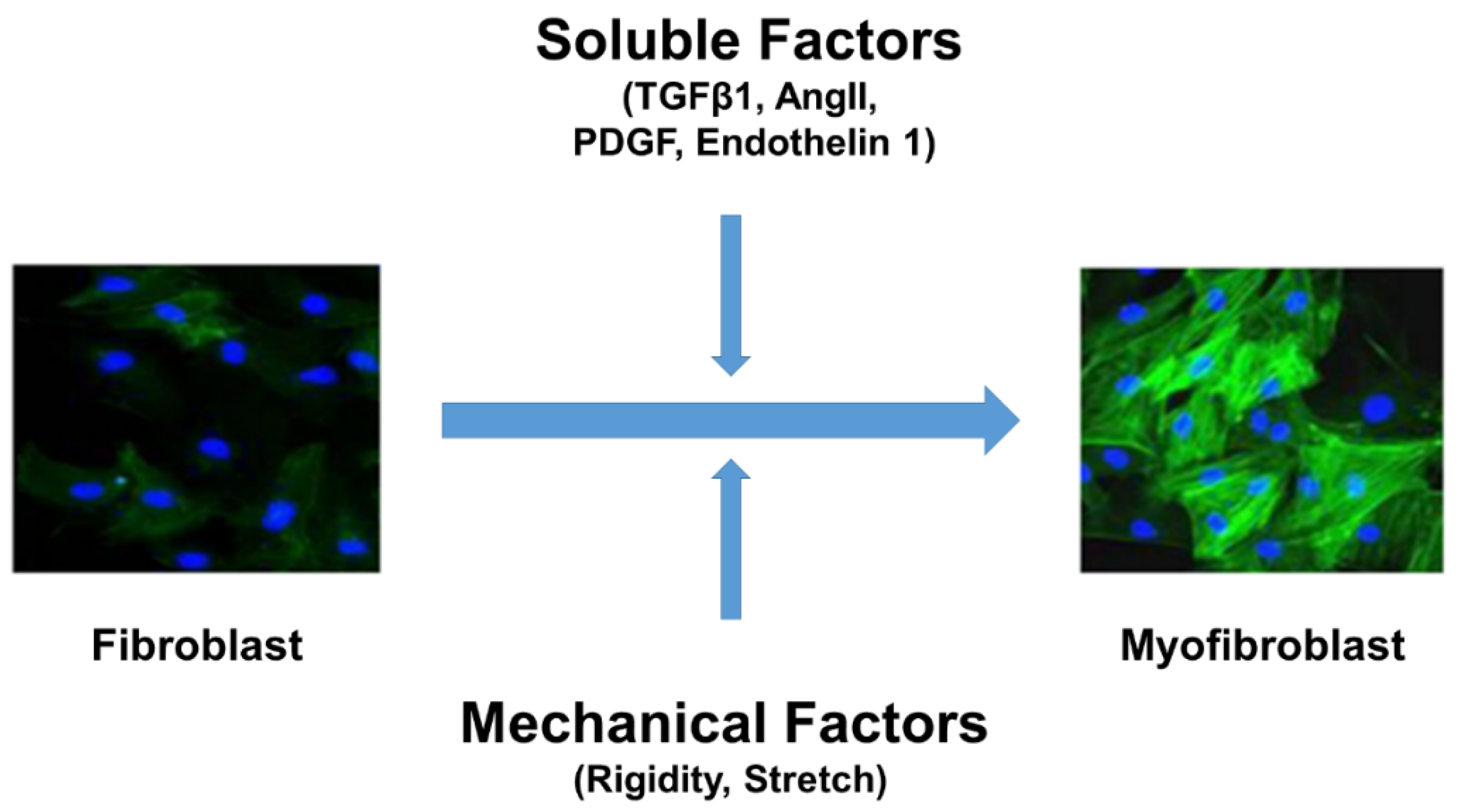
2. Fibroblast Differentiation and Signaling Mechanism
3. TRP Channels in Fibroblasts
4. TRPV4 Structure and Mechanotransduction
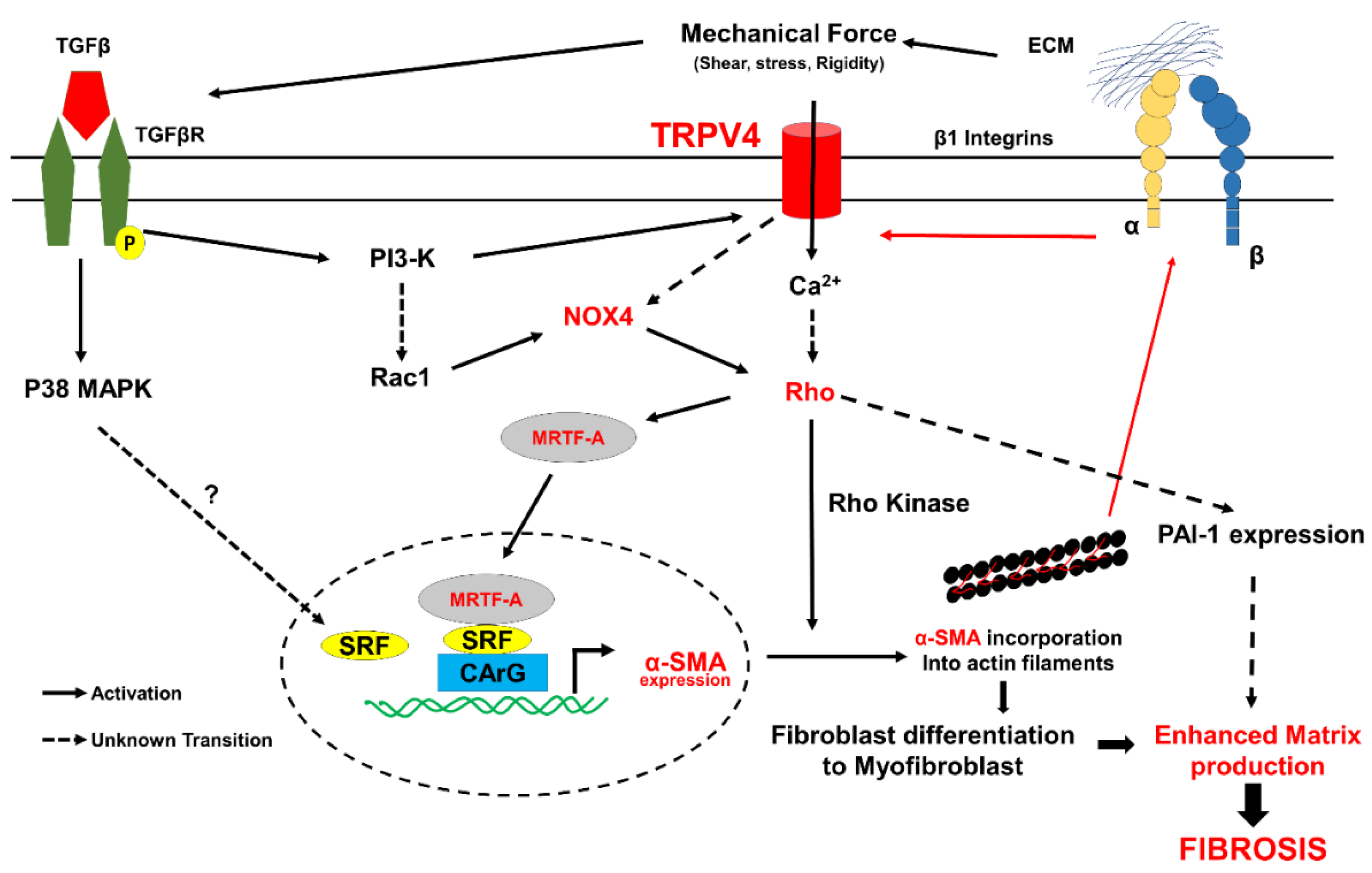
5. TRPV4 Mechanotransduction in Cardiac Fibrosis
6. Role of TRPV4 Mechanotransduction in Pulmonary Fibrosis
7. Summary and Clinical Implications
Supplementary Materials
Funding
Conflicts of Interest
References
- Horn, M.; Trafford, A.W. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J. Mol. Cell. Cardiol. 2016, 93, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [Green Version]
- DeLeon-Pennell, K.Y.; Meschiari, C.A.; Jung, M.; Lindsey, M.L. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Prog. Mol. Biol. Transl. Sci. 2017, 147, 75–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Guan, J. Antifibrotic therapies to control cardiac fibrosis. Biomater. Res. 2016, 20, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leask, A. Potential therapeutic targets for cardiac fibrosis: TGFβ, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, R.; Hartog, A.W.D.; Radonic, T.; Micha, D.; Maugeri, A.; Van Dijk, F.S.; Meijers-Heijboer, H.E.; Timmermans, J.; Scholte, A.J.; Berg, M.V.D.; et al. Beneficial Outcome of Losartan Therapy Depends on Type of FBN1 Mutation in Marfan Syndrome. Circ. Cardiovasc. Genet. 2015, 8, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Shibasaki, Y.; Nishiue, T.; Masaki, H.; Tamura, K.; Matsumoto, N.; Mori, Y.; Nishikawa, M.; Matsubara, H.; Iwasaka, T. Impact of the Angiotensin II Receptor Antagonist, Losartan, on Myocardial Fibrosis in Patients with End-Stage Renal Disease: Assessment by Ultrasonic Integrated Backscatter and Biochemical Markers. Hypertens. Res. 2005, 28, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Burghardt, I.; Tritschler, F.; Opitz, C.; Frank, B.; Weller, M.; Wick, W. Pirfenidone inhibits TGF-β expression in malignant glioma cells. Biochem. Biophys. Res. Commun. 2007, 354, 542–547. [Google Scholar] [CrossRef]
- Halevy, O.; Nagler, A.; Levi-Schaffer, F.; Genina, O.; Pines, M. Inhibition of collagen type I synthesis by skin fibroblasts of graft versus host disease and scleroderma patients: Effect of halofuginone. Biochem. Pharmacol. 1996, 52, 1057–1063. [Google Scholar] [CrossRef]
- Nagler, A.; Firman, N.; Feferman, R.; Cotev, S.; Pines, M.; Shoshan, S. Reduction in pulmonary fibrosis in vivo by halofuginone. Am. J. Respir. Crit. Care Med. 1996, 154, 1082–1086. [Google Scholar] [CrossRef]
- Fang, L.; Murphy, A.; Dart, A.M. A Clinical Perspective of Anti-Fibrotic Therapies for Cardiovascular Disease. Front. Pharmacol. 2017, 8, 186. [Google Scholar] [CrossRef]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133, 133. [Google Scholar] [CrossRef] [PubMed]
- van Nieuwenhoven, F.; Turner, N. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vasc. Pharmacol. 2013, 58, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Shinde, A.V.; Frangogiannis, N.G. Mechanisms of Fibroblast Activation in the Remodeling Myocardium. Curr. Pathobiol. Rep. 2017, 5, 145–152. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Platelet-derived growth factor (PDGF) therapy in myocardial infarction: Challenges and opportunities. Int. J. Cardiol. 2021, 341, 74–75. [Google Scholar] [CrossRef]
- Budi, E.H.; Schaub, J.R.; Decaris, M.; Turner, S.; Derynck, R. TGF-β as a driver of fibrosis: Physiological roles and therapeutic opportunities. J. Pathol. 2021, 254, 358–373. [Google Scholar] [CrossRef]
- Adapala, R.K.; Kanugula, A.K.; Paruchuri, S.; Chilian, W.M.; Thodeti, C.K. TRPV4 deletion protects heart from myocardial infarction-induced adverse remodeling via modulation of cardiac fibroblast differentiation. Basic Res. Cardiol. 2020, 115, 14. [Google Scholar] [CrossRef]
- Adapala, R.K.; Thoppil, R.J.; Luther, D.J.; Paruchuri, S.; Meszaros, J.G.; Chilian, W.M.; Thodeti, C.K. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol. 2013, 54, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Rahaman, S.O.; Grove, L.M.; Paruchuri, S.; Southern, B.; Abraham, S.; Niese, K.A.; Scheraga, R.; Ghosh, S.; Thodeti, C.K.; Zhang, D.X.; et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J. Clin. Investig. 2014, 124, 5225–5238. [Google Scholar] [CrossRef]
- Thodeti, C.K.; Matthews, B.; Ravi, A.; Mammoto, A.; Ghosh, K.; Bracha, A.L.; Ingber, D.E. TRPV4 Channels Mediate Cyclic Strain–Induced Endothelial Cell Reorientation Through Integrin-to-Integrin Signaling. Circ. Res. 2009, 104, 1123–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gombedza, F.; Kondeti, V.; Al-Azzam, N.; Koppes, S.; Duah, E.; Patil, P.; Hexter, M.; Phillips, D.; Thodeti, C.K.; Paruchuri, S. Mechanosensitive transient receptor potential vanilloid 4 regulates Dermatophagoides farinae–induced airway remodeling via 2 distinct pathways modulating matrix synthesis and degradation. FASEB J. 2016, 31, 1556–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herum, K.M.; Choppe, J.; Kumar, A.; Engler, A.J.; McCulloch, A.D. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol. Biol. Cell 2017, 28, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, G.S.; Pavesi, A.; Rasponi, M.; Fiore, G.B.; Kamm, R.D.; Soncini, M. Human cardiac fibroblasts adaptive responses to controlled combined mechanical strain and oxygen changes in vitro. eLife 2017, 6, e22847. [Google Scholar] [CrossRef] [Green Version]
- Stewart, L.; Turner, N. Channelling the Force to Reprogram the Matrix: Mechanosensitive Ion Channels in Cardiac Fibroblasts. Cells 2021, 10, 990. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef] [Green Version]
- Piersma, B.; de Rond, S.; Werker, P.M.; Boo, S.; Hinz, B.; van Beuge, M.M.; Bank, R.A. YAP1 Is a Driver of Myofibroblast Differentiation in Normal and Diseased Fibroblasts. Am. J. Pathol. 2015, 185, 3326–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nat. Cell Biol. 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Janssen, L.J.; Mukherjee, S.; Ask, K. Calcium Homeostasis and Ionic Mechanisms in Pulmonary Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2015, 53, 135–148. [Google Scholar] [CrossRef]
- Feng, J.; Armillei, M.K.; Yu, A.S.; Liang, B.T.; Runnels, L.W.; Yue, L. Ca2+ Signaling in Cardiac Fibroblasts and Fibrosis-Associated Heart Diseases. J. Cardiovasc. Dev. Dis. 2019, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPHIE, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Ayaub, E.A.; Murphy, J.; Lu, C.; Kolb, M.; Ask, K.; Janssen, L.J. Disruption of Calcium Signaling in Fibroblasts and Attenuation of Bleomycin-Induced Fibrosis by Nifedipine. Am. J. Respir. Cell Mol. Biol. 2015, 53, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Meszaros, J.G.; Gonzalez, A.M.; Endo-Mochizuki, Y.; Villegas, S.; Villarreal, F.; Brunton, L.L. Identification of G protein-coupled signaling pathways in cardiac fibroblasts: Cross talk between Gq and Gs. Am. J. Physiol. Physiol. 2000, 278, C154–C162. [Google Scholar] [CrossRef] [Green Version]
- Thodeti, C.K.; Paruchuri, S.; Meszaros, J.G. A TRP to cardiac fibroblast differentiation. Channels 2013, 7, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-Dependent Pathway for Myofibroblast Transdifferentiation and Wound Healing In Vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Nishida, M.; Onohara, N.; Sato, Y.; Suda, R.; Ogushi, M.; Tanabe, S.; Inoue, R.; Mori, Y.; Kurose, H. Gα12/13-mediated Up-regulation of TRPC6 Negatively Regulates Endothelin-1-induced Cardiac Myofibroblast Formation and Collagen Synthesis through Nuclear Factor of Activated T Cells Activation. J. Biol. Chem. 2007, 282, 23117–23128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, M.; Luo, X.; Qi, X.Y.; Tadevosyan, A.; Maguy, A.; Ordog, B.; Ledoux, J.; Kato, T.; Naud, P.; Voigt, N.; et al. Transient Receptor Potential Canonical-3 Channel–Dependent Fibroblast Regulation in Atrial Fibrillation. Circulation 2012, 126, 2051–2064. [Google Scholar] [CrossRef]
- Hu, F.; Li, M.; Han, F.; Zhang, Q.; Zeng, Y.; Zhang, W.; Cheng, X. Role of TRPM7 in cardiac fibrosis: A potential therapeutic target (Review). Exp. Ther. Med. 2021, 21, 173. [Google Scholar] [CrossRef]
- Du, J.; Xie, J.; Zhang, Z.; Tsujikawa, H.; Fusco, D.; Silverman, D.; Liang, B.; Yue, L. TRPM7-Mediated Ca2+ Signals Confer Fibrogenesis in Human Atrial Fibrillation. Circ. Res. 2010, 106, 992–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Ying, S.; Zhao, X.; Liu, J.; Wang, Y. Increased expression of lung TRPV1/TRPA1 in a cough model of bleomycin-induced pulmonary fibrosis in Guinea pigs. BMC Pulm. Med. 2019, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, N.-X.; Gibbs, A.; Tonks, A.; Hope-Gill, B.D. Airway expression of Transient Receptor Potential (TRP) Vanniloid-1 and Ankyrin-1 channels is not increased in patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2017, 12, e0187847. [Google Scholar] [CrossRef] [Green Version]
- Becker, D.; Bereiter-Hahn, J.; Jendrach, M. Functional interaction of the cation channel transient receptor potential vanilloid 4 (TRPV4) and actin in volume regulation. Eur. J. Cell Biol. 2009, 88, 141–152. [Google Scholar] [CrossRef]
- Toft-Bertelsen, T.; MacAulay, N. TRPing to the Point of Clarity: Understanding the Function of the Complex TRPV4 Ion Channel. Cells 2021, 10, 165. [Google Scholar] [CrossRef]
- Hatano, N.; Itoh, Y.; Muraki, K. Cardiac fibroblasts have functional TRPV4 activated by 4α-phorbol 12,13-didecanoate. Life Sci. 2009, 85, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Mukherjee, S.; Sheng, W.; Nilius, B.; Janssen, L.J. Electrophysiological characterization of voltage-dependent calcium currents and TRPV4 currents in human pulmonary fibroblasts. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L603–L614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earley, S.; Pauyo, T.; Drapp, R.; Tavares, M.J.; Liedtke, W.; Brayden, J.E. TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am. J. Physiol. Circ. Physiol. 2009, 297, H1096–H1102. [Google Scholar] [CrossRef] [Green Version]
- Sianati, S.; Schroeter, L.; Richardson, J.; Tay, A.; Lamandé, S.R.; Poole, K. Modulating the Mechanical Activation of TRPV4 at the Cell-Substrate Interface. Front. Bioeng. Biotechnol. 2020, 8, 608951. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Cho, T.-J.; Unger, S.; Lausch, E.; Nishimura, G.; Kim, O.-H.; Superti-Furga, A.; Ikegawa, S. TRPV4-pathy, a novel channelopathy affecting diverse systems. J. Hum. Genet. 2010, 55, 400–402. [Google Scholar] [CrossRef]
- Liedtke, W.; Friedman, J.M. Abnormal osmotic regulation in trpv4-/- mice. Proc. Natl. Acad. Sci. USA 2003, 100, 13698–13703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, B.D.; Thodeti, C.K.; Tytell, J.; Mammoto, A.; Overby, D.; Ingber, D.E. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface β1 integrins. Integr. Biol. 2010, 2, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Adapala, R.K.; Thoppil, R.J.; Ghosh, K.; Cappelli, H.; Dudley, A.C.; Paruchuri, S.; Keshamouni, V.; Klagsbrun, M.; Meszaros, J.G.; Chilian, W.M.; et al. Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene 2016, 35, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Colbert, H.A.; Smith, T.L.; Bargmann, C.I. OSM-9, a novel protein with structural sim-ilarity to channels, is required for olfaction, mechanosensation, and olfactory adaptation in Caenorhabditis elegans. J. Neurosci. 1997, 17, 8259–8269. [Google Scholar] [CrossRef] [Green Version]
- A Colbert, H.; Bargmann, C. Odorant-specific adaptation pathways generate olfactory plasticity in C. elegans. Neuron 1995, 14, 803–812. [Google Scholar] [CrossRef] [Green Version]
- KoÖhler, R.; Heyken, W.-T.; Heinau, P.; Schubert, R.; Si, H.; Kacik, M.; Busch, C.; Grgic, I.; Maier, T.; Hoyer, J. Evidence for a Functional Role of Endothelial Transient Receptor Potential V4 in Shear Stress–Induced Vasodilatation. Arter. Thromb. Vasc. Biol. 2006, 26, 1495–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmannsgruber, V.; Heyken, W.-T.; Kacik, M.; Kaistha, A.; Grgic, I.; Harteneck, C.; Liedtke, W.; Hoyer, J.; Kohler, R. Arterial Response to Shear Stress Critically Depends on Endothelial TRPV4 Expression. PLoS ONE 2007, 2, e827. [Google Scholar] [CrossRef] [PubMed]
- Willette, R.N.; Bao, W.; Nerurkar, S.; Yue, T.-L.; Doe, C.P.; Stankus, G.; Turner, G.H.; Ju, H.; Thomas, H.; Fishman, C.E.; et al. Systemic Activation of the Transient Receptor Potential Vanilloid Subtype 4 Channel Causes Endothelial Failure and Circulatory Collapse: Part 2. J. Pharmacol. Exp. Ther. 2008, 326, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Baylie, R.L.; Brayden, J.E. TRPV channels and vascular function. Acta Physiol. 2011, 203, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Filosa, J.A.; Yao, X.; Rath, G. TRPV4 and the Regulation of Vascular Tone. J. Cardiovasc. Pharmacol. 2013, 61, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heathcote, H.R.; Lee, M.D.; Zhang, X.; Saunter, C.D.; Wilson, C.; McCarron, J.G. Endothelial TRPV4 channels modulate vascular tone by Ca2+ -induced Ca2+ release at inositol 1,4,5-trisphosphate receptors. Br. J. Pharmacol. 2019, 176, 3297–3317. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.X.; Gutterman, D.D. Transient Receptor Potential Channel Activation and Endothelium-dependent Dilation in the Systemic Circulation. J. Cardiovasc. Pharmacol. 2011, 57, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baratchi, S.; Knoerzer, M.; Khoshmanesh, K.; Mitchell, A.; McIntyre, P. Shear Stress Regulates TRPV4 Channel Clustering and Translocation from Adherens Junctions to the Basal Membrane. Sci. Rep. 2017, 7, 15942. [Google Scholar] [CrossRef] [Green Version]
- Thoppil, R.J.; Adapala, R.K.; Cappelli, H.C.; Kondeti, V.; Dudley, A.C.; Gary Meszaros, J.; Paruchuri, S.; Thodeti, C.K. TRPV4 channel activation selectively inhibits tumor endothelial cell proliferation. Sci. Rep. 2015, 5, 14257. [Google Scholar] [CrossRef] [Green Version]
- Kanugula, A.K.; Adapala, R.K.; Jamaiyar, A.; Lenkey, N.; Guarino, B.D.; Liedtke, W.; Yin, L.; Paruchuri, S.; Thodeti, C.K. Endothelial TRPV4 channels prevent tumor growth and metastasis via modulation of tumor angiogenesis and vascular integrity. Angiogenesis 2021, 24, 647–656. [Google Scholar] [CrossRef]
- Kanugula, A.K.; Adapala, R.K.; Midha, P.; Cappelli, H.C.; Meszaros, J.G.; Paruchuri, S.; Chilian, W.M.; Thodeti, C.K. Novel noncanonical regulation of soluble VEGF/VEGFR2 signaling by mechanosensitive ion channel TRPV4. FASEB J. 2019, 33, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.L.; Peana, D.; Veteto, A.; Lambert, M.D.; Nourian, Z.; Karasseva, N.G.; Hill, M.; Lindman, B.; Baines, C.; Krenz, M.; et al. TRPV4 increases cardiomyocyte calcium cycling and contractility yet contributes to damage in the aged heart following hypoosmotic stress. Cardiovasc. Res. 2019, 115, 46–56. [Google Scholar] [CrossRef]
- Peana, D.; Polo-Parada, L.; Domeier, T.L. Arrhythmogenesis in the aged heart following ischaemia–reperfusion: Role of transient receptor potential vanilloid 4. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chaigne, S.; Cardouat, G.; Louradour, J.; Vaillant, F.; Charron, S.; Sacher, F.; Ducret, T.; Guinamard, R.; Vigmond, E.; Hof, T. Transient receptor potential vanilloid 4 channel participates in mouse ventricular electrical activity. Am. J. Physiol. Circ. Physiol. 2021, 320, H1156–H1169. [Google Scholar] [CrossRef]
- Zhou, R.; Hang, P.; Zhu, W.; Su, Z.; Liang, H.; Du, Z. Whole Genome Network Analysis of Ion Channels and Connexins in Myocardial Infarction. Cell. Physiol. Biochem. 2011, 27, 299–304. [Google Scholar] [CrossRef]
- Ahn, M.-S.; Eom, Y.W.; Oh, J.-E.; Cha, S.-K.; Park, K.S.; Son, J.-W.; Lee, J.-W.; Youn, Y.J.; Ahn, S.G.; Kim, J.-Y.; et al. Transient receptor potential channel TRPV4 mediates TGF-β1-induced differentiation of human ventricular fibroblasts. Cardiol. J. 2020, 27, 162–170. [Google Scholar] [CrossRef]
- Jia, X.; Xiao, C.; Sheng, D.; Yang, M.; Cheng, Q.; Wu, J.; Zhang, S. TRPV4 Mediates Cardiac Fibrosis via the TGF-β1/Smad3 Signaling Pathway in Diabetic Rats. Cardiovasc. Toxicol. 2020, 20, 492–499. [Google Scholar] [CrossRef]
- Sgalla, G.; Biffi, A.; Richeldi, L. Idiopathic pulmonary fibrosis: Diagnosis, epidemiology and natural history. Respirology 2016, 21, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Hoffmann, J.; Kaestle, S.M.; Neye, N.; Wang, L.; Baeurle, J.; Liedtke, W.; Wu, S.; Kuppe, H.; Pries, A.R.; et al. Negative-Feedback Loop Attenuates Hydrostatic Lung Edema via a cGMP-Dependent Regulation of Transient Receptor Potential Vanilloid 4. Circ. Res. 2008, 102, 966–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Wang, X.; Varty, L.; Rizzo, C.A.; Yang, R.; Correll, C.C.; Phelps, P.T.; Egan, R.W.; Hey, J.A. Functional TRPV4 channels are expressed in human airway smooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 2004, 287, L272–L278. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, D.; King, J.A.; Weber, D.S.; Addison, E.; Liedtke, W.; Townsley, M.I. Transient Receptor Potential Vanilloid 4–Mediated Disruption of the Alveolar Septal Barrier. Circ. Res. 2006, 99, 988–995. [Google Scholar] [CrossRef]
- Sidhaye, V.; Güler, A.D.; Schweitzer, K.S.; D’Alessio, F.; Caterina, M.J.; King, L.S. Transient receptor potential vanilloid 4 regulates aquaporin-5 abundance under hypotonic conditions. Proc. Natl. Acad. Sci. USA 2006, 103, 4747–4752. [Google Scholar] [CrossRef] [Green Version]
- Grove, L.M.; Mohan, M.L.; Abraham, S.; Scheraga, R.G.; Southern, B.D.; Crish, J.F.; Prasad, S.V.N.; Olman, M.A. Translocation of TRPV4-PI3Kγ complexes to the plasma membrane drives myofibroblast transdifferentiation. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Al-Azzam, N.; Teegala, L.R.; Pokhrel, S.; Ghebreigziabher, S.; Chachkovskyy, T.; Thodeti, S.; Gavilanes, I.; Covington, K.; Thodeti, C.K.; Paruchuri, S. Transient Receptor Potential Vanilloid channel regulates fibroblast differentiation and airway remodeling by modulating redox signals through NADPH Oxidase 4. Sci. Rep. 2020, 10, 9827. [Google Scholar] [CrossRef]
- Pini, A.; Viappiani, S.; Bolla, M.; Masini, E.; Bani, D. Prevention of Bleomycin-Induced Lung Fibrosis in Mice by a Novel Approach of Parallel Inhibition of Cyclooxygenase and Nitric-Oxide Donation Using NCX 466, a Prototype Cyclooxygenase Inhibitor and Nitric-Oxide Donor. J. Pharmacol. Exp. Ther. 2012, 341, 493–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuyama, H.; Tsuruda, T.; Sekita, Y.; Hatakeyama, K.; Imamura, T.; Kato, J.; Asada, Y.; Stasch, J.-P.; Kitamura, K. Pressure-independent effects of pharmacological stimulation of soluble guanylate cyclase on fibrosis in pressure-overloaded rat heart. Hypertens. Res. 2009, 32, 597–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-H.; Kim, M.; Yim, B.; Park, C.Y. Nitric oxide attenuated transforming growth factor-β induced myofibroblast differentiation of human keratocytes. Sci. Rep. 2021, 11, 8183. [Google Scholar] [CrossRef] [PubMed]
- Sampson, N.; Berger, P.; Zenzmaier, C. Redox Signaling as a Therapeutic Target to Inhibit Myofibroblast Activation in Degenerative Fibrotic Disease. BioMed Res. Int. 2014, 2014, 131737. [Google Scholar] [CrossRef] [Green Version]
- Marziano, C.; Hong, K.; Cope, E.L.; Kotlikoff, M.I.; Isakson, B.E.; Sonkusare, S.K. Nitric Oxide–Dependent Feedback Loop Regulates Transient Receptor Potential Vanilloid 4 (TRPV4) Channel Cooperativity and Endothelial Function in Small Pulmonary Arteries. J. Am. Hear. Assoc. 2017, 6, e007157. [Google Scholar] [CrossRef] [Green Version]
- Adapala, R.K.; Talasila, P.K.; Bratz, I.N.; Zhang, D.X.; Suzuki, M.; Meszaros, J.G.; Thodeti, C.K. PKCα mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. Am. J. Physiol. Circ. Physiol. 2011, 301, H757–H765. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adapala, R.K.; Katari, V.; Teegala, L.R.; Thodeti, S.; Paruchuri, S.; Thodeti, C.K. TRPV4 Mechanotransduction in Fibrosis. Cells 2021, 10, 3053. https://doi.org/10.3390/cells10113053
Adapala RK, Katari V, Teegala LR, Thodeti S, Paruchuri S, Thodeti CK. TRPV4 Mechanotransduction in Fibrosis. Cells. 2021; 10(11):3053. https://doi.org/10.3390/cells10113053
Chicago/Turabian StyleAdapala, Ravi K., Venkatesh Katari, Lakshminarayan Reddy Teegala, Sathwika Thodeti, Sailaja Paruchuri, and Charles K. Thodeti. 2021. "TRPV4 Mechanotransduction in Fibrosis" Cells 10, no. 11: 3053. https://doi.org/10.3390/cells10113053
APA StyleAdapala, R. K., Katari, V., Teegala, L. R., Thodeti, S., Paruchuri, S., & Thodeti, C. K. (2021). TRPV4 Mechanotransduction in Fibrosis. Cells, 10(11), 3053. https://doi.org/10.3390/cells10113053