
Drosophila Heart as a Model for Cardiac Development and Diseases

Abstract
1. Introduction
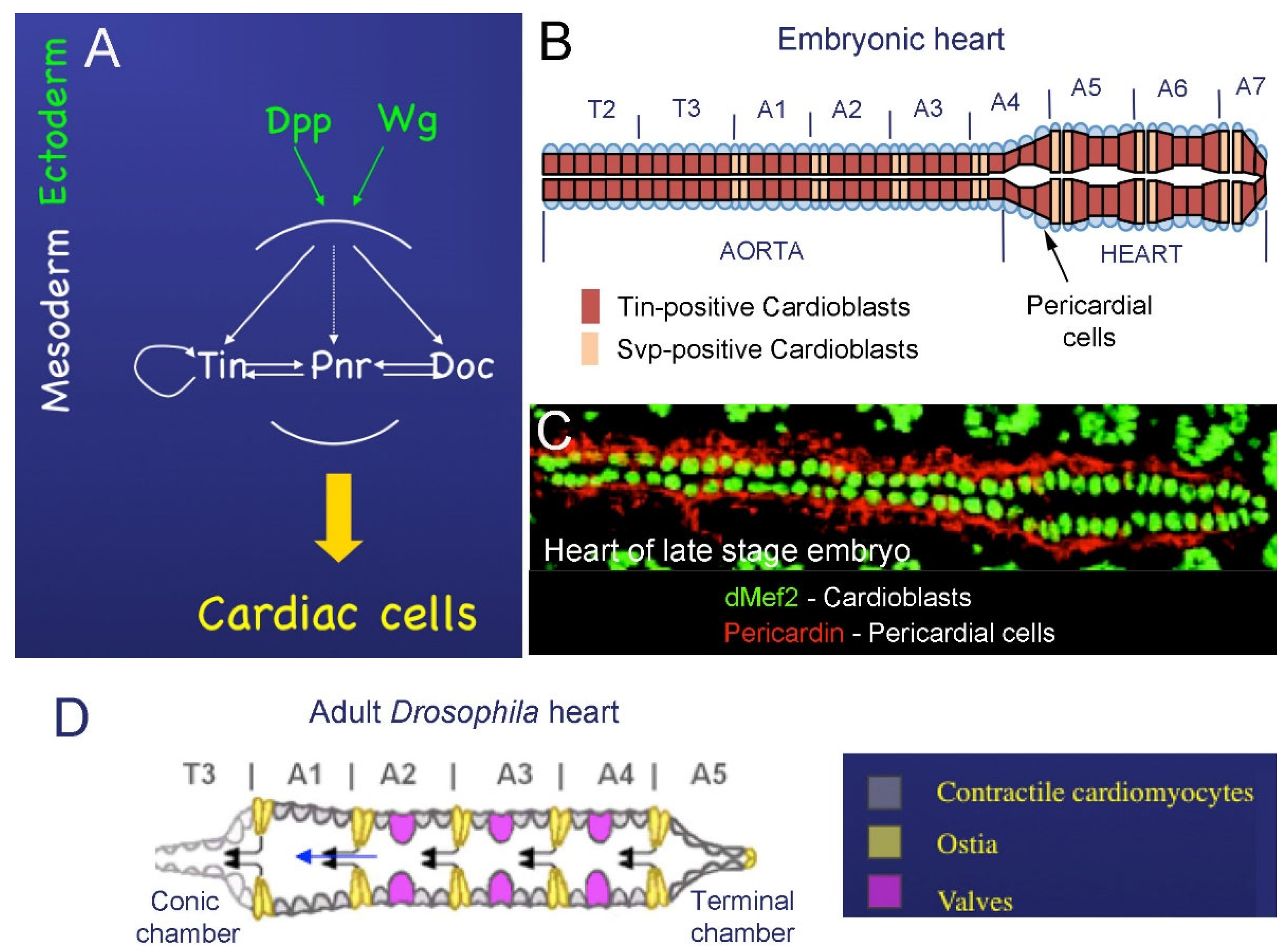
2. The Drosophila Heart
2.1. Cardiac Development in Embryos
2.2. Development of the Adult Fly Heart—Hox-Dependent Remodeling of the Cardiac Tube
3. Vertebrate Heart and Its Similarity with the Drosophila Cardiac Tube

{kind=link}
{kind=link}
| Fly Gene | Expression | Mutants | Vertebrate Ortholog | Expression | Mutants |
|---|---|---|---|---|---|
| Tinman | Expressed early during development uniformly in mesoderm After gastrulation: restricted to the dorsal portion of the mesoderm Later: transiently in visceral mesoderm and permanently in heart (in cardiac and pericardial cells) [8,28] | Lack of visceral and cardiac mesoderm. The somatic mesoderm shows moderate patterning defects later in development [28,33] | Nkx2–5 | Initially expressed in the bilateral cardiac progenitors of the anterior lateral plate mesoderm and in part of the pharyngeal endoderm | Lack of ventricular-specific myosin light-chain gene expression Heart tube fails to undergo normal looping [34] |
| Dmef2 | Expressed myogenic precursor lineages and their descendants [8] | Heart differentiation affected [35] | Mef2 | Expressed in cardiac, skeletal, and smooth muscle precursor lineages [8] | Defects in heart looping and Hand2 downregulation [8] |
| HAND | Heart, lymph glands, circular visceral musculature, and a subset of CNS cells | In embryos: lack of lymph glands. In adult: disorganized myofibrillar structure, reduced systolic and diastolic diameter, abnormal heartbeat contractions, midguts highly deformed, and premature lethality [36] | Hand2/3 | Heart neural crest derivatives [37] | Aortic sac defects Heart looping defects [37] |
| Dpp | Dorsal ectoderm | Lack of heart and visceral mesoderm [8] | BMPs | Expressed in endoderm and ectoderm | Affected heart development Down regulation of Nkx2–5 [38] |
| Wg | Ectoderm—adjacent to cardiac mesoderm [39] | Loss of repeated clusters of even-skipped expressing cells in mesoderm [39] Loss of heart precursors [10] | Wnt | Wnt5a and Wnt11 expressed in second heart field [40] | Defective right ventricle development [41] |
4. Identifying Cardiac Aging Genes and Modeling Human Heart Diseases in Drosophila
4.1. Studying Cardiac Aging and Heart Failure in Drosophila
4.2. Identifying Genes Involved in Congenital Heart Defects
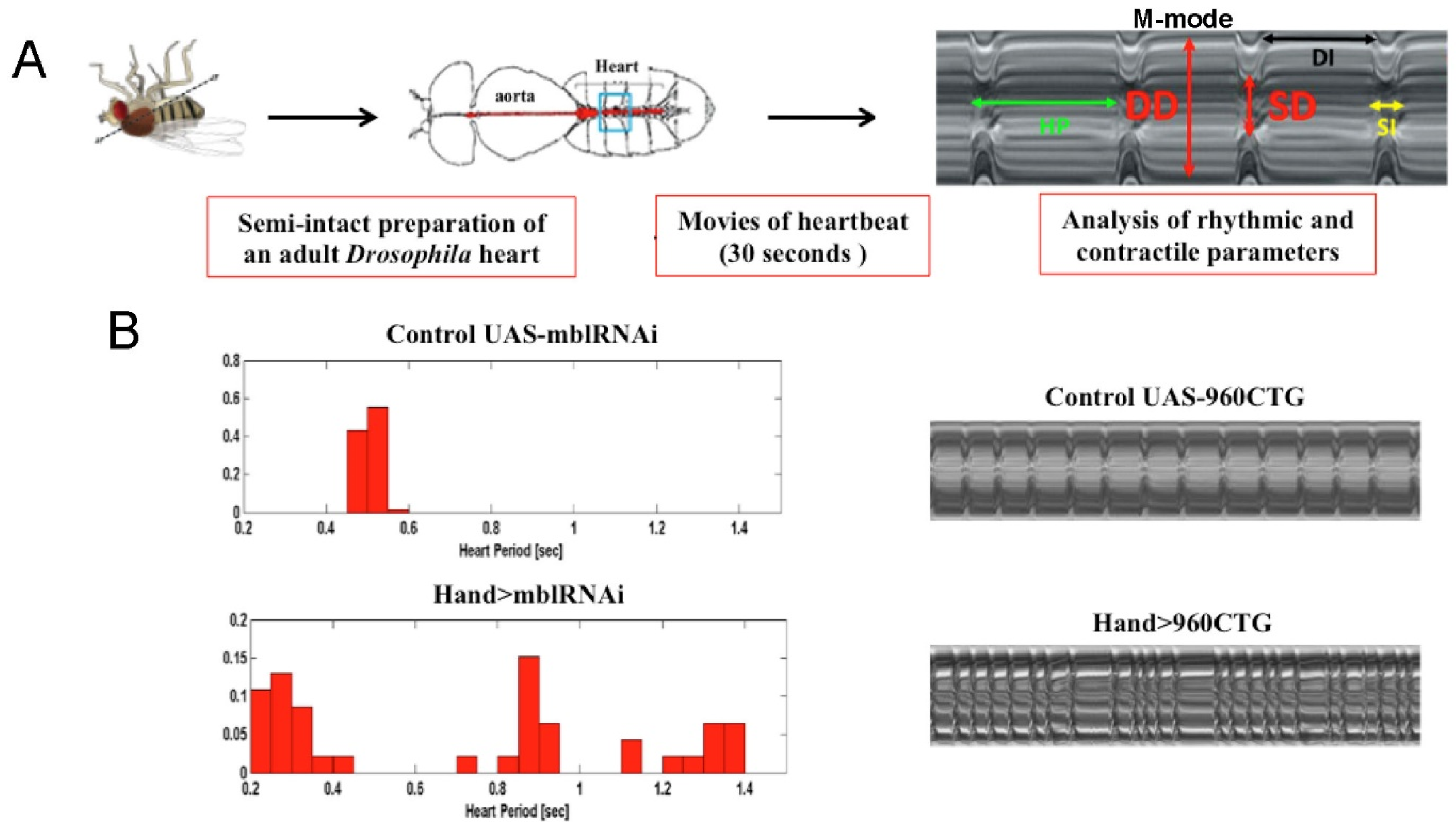
4.3. Modeling Myotonic Dystrophy Type 1 Heart Defects
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brand, A.H.; Perrimon, N. Targeted Gene Expression as a Means of Altering Cell Fates and Generating Dominant Phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Gratz, S.J.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. Precise Genome Editing of Drosophila with CRISPR RNA-Guided Cas9. Methods Mol. Biol. 2015, 1311, 335–348. [Google Scholar] [CrossRef]
- Housden, B.E.; Hu, Y.; Perrimon, N. Design and Generation of Drosophila Single Guide RNA Expression Constructs. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef]
- Port, F.; Strein, C.; Stricker, M.; Rauscher, B.; Heigwer, F.; Zhou, J.; Beyersdörffer, C.; Frei, J.; Hess, A.; Kern, K.; et al. A Large-Scale Resource for Tissue-Specific CRISPR Mutagenesis in Drosophila. eLife 2020, 9, e53865. [Google Scholar] [CrossRef]
- Hu, Y.; Comjean, A.; Rodiger, J.; Liu, Y.; Gao, Y.; Chung, V.; Zirin, J.; Perrimon, N.; Mohr, S.E. FlyRNAi.Org—the Database of the Drosophila RNAi Screening Center and Transgenic RNAi Project: 2021 Update. Nucleic Acids Res. 2021, 49, D908–D915. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The Genome Sequence of Drosophila Melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A Systematic Analysis of Human Disease-Associated Gene Sequences in Drosophila Melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef]
- Bodmer, R.; Venkatesh, T.V. Heart Development in Drosophila and Vertebrates: Conservation of Molecular Mechanisms. Dev. Gen. 1998, 22, 181–186. [Google Scholar] [CrossRef]
- Rugendorff, A.; Younossi-Hartenstein, A.; Hartenstein, V. Embryonic Origin and Differentiation of the Drosophila Heart. Roux’s Arch. Dev. Bio.l 1994, 203, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Golden, K.; Bodmer, R. Heart Development in Drosophila Requires the Segment Polarity Gene Wingless. Dev. Biol. 1995, 169, 619–628. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zmojdzian, M.; Ponte, J.P.D.; Jagla, K. Cellular Components and Signals Required for the Cardiac Outflow Tract Assembly in Drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 2475–2480. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, W.K.; Bodmer, R. The Patterns of Wingless, Decapentaplegic, and Tinman Position the Drosophila Heart. Mech. Dev. 2002, 114, 13–26. [Google Scholar] [CrossRef]
- Klinedinst, S.L.; Bodmer, R. Gata Factor Pannier Is Required to Establish Competence for Heart Progenitor Formation. Development 2003, 130, 3027–3038. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.; Schneider, M.; Schwendele, B.; Renault, A.D. Drosophila Heart Cell Movement to the Midline Occurs through Both Cell Autonomous Migration and Dorsal Closure. Dev. Biol. 2014, 396, 169–182. [Google Scholar] [CrossRef]
- Zaffran, S.; Frasch, M. Early Signals in Cardiac Development. Circ. Res. 2002, 91, 457–469. [Google Scholar] [CrossRef]
- Jagla, K.; Frasch, M.; Jagla, T.; Dretzen, G.; Bellard, F.; Bellard, M. Ladybird, a New Component of the Cardiogenic Pathway in Drosophila Required for Diversification of Heart Precursors. Development 1997, 124, 3471–3479. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, K. D-Mef2 Is a Target for Tinman Activation during Drosophila Heart Development. EMBO J. 1997, 16, 515–522. [Google Scholar] [CrossRef]
- Monier, B.; Astier, M.; Sémériva, M.; Perrin, L. Steroid-Dependent Modification of Hox Function Drives Myocyte Reprogramming in the Drosophila Heart. Development 2005, 132, 5283–5293. [Google Scholar] [CrossRef] [PubMed]
- Perrin, L.; Monier, B.; Ponzielli, R.; Astier, M.; Semeriva, M. Drosophila Cardiac Tube Organogenesis Requires Multiple Phases of Hox Activity. Dev. Biol. 2004, 272, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M.; Hoshizaki, D.K.; Cripps, R.M. Homeotic Selector Genes Control the Patterning of Seven-up Expressing Cells in the Drosophila Dorsal Vessel. Mech. Dev. 2005, 122, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Lovato, T.L.; Nguyen, T.P.; Molina, M.R.; Cripps, R.M. The Hox Gene Abdominal-A Specifies Heart Cell Fate in TheDrosophila Dorsal Vessel. Development 2002, 129, 5019–5027. [Google Scholar] [CrossRef]
- Shah, A.P.; Nongthomba, U.; Kelly Tanaka, K.K.; Denton, M.L.B.; Meadows, S.M.; Bancroft, N.; Molina, M.R.; Cripps, R.M. Cardiac Remodeling in Drosophila Arises from Changes in Actin Gene Expression and from a Contribution of Lymph Gland-like Cells to the Heart Musculature. Mech. Dev. 2011, 128, 222–233. [Google Scholar] [CrossRef]
- Curtis, N.J.; Ringo, J.M.; Dowse, H.B. Morphology of the Pupal Heart, Adult Heart, and Associated Tissues in the Fruit Fly, Drosophila Melanogaster. J. Morphol. 1999, 240, 225–235. [Google Scholar] [CrossRef]
- Frasch, M. Genome-Wide Approaches to Drosophila Heart Development. J. Cardiovasc. Dev. Dis. 2016, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Bulatovic, I.; Månsson-Broberg, A.; Sylvén, C.; Grinnemo, K.-H. Human Fetal Cardiac Progenitors: The Role of Stem Cells and Progenitors in the Fetal and Adult Heart. Best Pract. Res. Clin. Obst. Gynaecol. 2016, 31, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.; Webb, S.; Brown, N.A.; Lamers, W.; Anderson, R.H. Development of the Heart: (1) Formation of the Cardiac Chambers and Arterial Trunks. Heart 2003, 89, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Lamers, W.H.; Moorman, A.F.M. Cardiac Septation. Circ. Res. 2002, 91, 93–103. [Google Scholar] [CrossRef]
- Bodmer, R. Heart Development in Drosophila and Its Relationship to Vertebrates. Trends Cardiovasc. Med. 1995, 5, 21–28. [Google Scholar] [CrossRef]
- Ahmad, S.M. Conserved Signaling Mechanisms in Drosophila Heart Development: Signaling Mechanisms in Drosophila CARDIOGENESIS. Dev. Dyn. 2017, 246, 641–656. [Google Scholar] [CrossRef]
- Zikova, M.; Ponte, J.-P.D.; Dastugue, B.; Jagla, K. Patterning of the Cardiac Outflow Region in Drosophila. Proc. Natl. Acad. Sci. USA 2003, 100, 12189–12194. [Google Scholar] [CrossRef]
- Sieber-Blum, M. Cardiac Neural Crest Stem Cells. Anatom. Record Part A Discov. Mol. Cell. Evolut. Biol. 2004, 276A, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Rotstein, B.; Paululat, A. On the Morphology of the Drosophila Heart. J. Cardiovasc. Dev. Disease 2016, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, R. The Gene Tinman Is Required for Specification of the Heart and Visceral Muscles in Drosophila. Development 1993, 118, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Lyons, I.; Parsons, L.M.; Hartley, L.; Li, R.; Andrews, J.E.; Robb, L.; Harvey, R.P. Myogenic and Morphogenetic Defects in the Heart Tubes of Murine Embryos Lacking the Homeo Box Gene Nkx2-5. Genes Dev. 1995, 9, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Lilly, B.; Zhao, B.; Ranganayakulu, G.; Paterson, B.M.; Schulz, R.A.; Olson, E.N. Requirement of MADS Domain Transcription Factor D-MEF2 for Muscle Formation in Drosophila. Science 1995, 267, 688–693. [Google Scholar] [CrossRef]
- Lo, P.C.H.; Zaffran, S.; Sénatore, S.; Frasch, M. The Drosophila Hand Gene Is Required for Remodeling of the Developing Adult Heart and Midgut during Metamorphosis. Dev. Biol. 2007, 311, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Firulli, A.B.; McFadden, D.G.; Lin, Q.; Srivastava, D.; Olson, E.N. Heart and Extra-Embryonic Mesodermal Defects in Mouse Embryos Lacking the BHLH Transcription Factor Hand1. Nat. Genet. 1998, 18, 266–270. [Google Scholar] [CrossRef]
- Shi, Y.; Katsev, S.; Cai, C.; Evans, S. BMP Signaling Is Required for Heart Formation in Vertebrates. Dev. Biol. 2000, 224, 226–237. [Google Scholar] [CrossRef]
- Lawrence, P.A.; Bodmer, R.; Vincent, J.-P. Segmental Patterning of Heart Precursors in Drosophila. Development 1996, 121, 4303–4308. [Google Scholar] [CrossRef]
- Tian, Y.; Cohen, E.D.; Morrisey, E.E. The Importance of Wnt Signaling in Cardiovascular Development. Pediatr Cardiol 2010, 31, 342–348. [Google Scholar] [CrossRef]
- Ai, D.; Fu, X.; Wang, J.; Lu, M.-F.; Chen, L.; Baldini, A.; Klein, W.H.; Martin, J.F. Canonical Wnt Signaling Functions in Second Heart Field to Promote Right Ventricular Growth. Proc. Natl. Acad. Sci. USA 2007, 104, 9319–9324. [Google Scholar] [CrossRef] [PubMed]
- Blice-Baum, A.C.; Guida, M.C.; Hartley, P.S.; Adams, P.D.; Bodmer, R.; Cammarato, A. As Time Flies by: Investigating Cardiac Aging in the Short-Lived Drosophila Model. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 1831–1844. [Google Scholar] [CrossRef]
- Zhu, S.; Han, Z.; Luo, Y.; Chen, Y.; Zeng, Q.; Wu, X.; Yuan, W. Molecular Mechanisms of Heart Failure: Insights from Drosophila. Heart Fail. Rev. 2017, 22, 91–98. [Google Scholar] [CrossRef]
- Paternostro, G.; Vignola, C.; Bartsch, D.-U.; Omens, J.H.; McCulloch, A.D.; Reed, J.C. Age-Associated Cardiac Dysfunction in Drosophila Melanogaster. Circ. Res. 2001, 88, 1053–1058. [Google Scholar] [CrossRef]
- Fleg, J.L.; O’Connor, F.; Gerstenblith, G.; Becker, L.C.; Clulow, J.; Schulman, S.P.; Lakatta, E.G. Impact of Age on the Cardiovascular Response to Dynamic Upright Exercise in Healthy Men and Women. J. Appl. Physiol. (1985) 1995, 78, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Cannon, L.; Zambon, A.C.; Cammarato, A.; Zhang, Z.; Vogler, G.; Munoz, M.; Taylor, E.; Cartry, J.; Bernstein, S.I.; Melov, S.; et al. Expression Patterns of Cardiac Aging in Drosophila. Aging Cell 2017, 16, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Ocorr, K.; Reeves, N.L.; Wessells, R.J.; Fink, M.; Chen, H.-S.V.; Akasaka, T.; Yasuda, S.; Metzger, J.M.; Giles, W.; Posakony, J.W.; et al. KCNQ Potassium Channel Mutations Cause Cardiac Arrhythmias in Drosophila That Mimic the Effects of Aging. Proc. Natl. Acad. Sci. USA 2007, 104, 3943–3948. [Google Scholar] [CrossRef] [PubMed]
- Wessells, R.; Fitzgerald, E.; Piazza, N.; Ocorr, K.; Morley, S.; Davies, C.; Lim, H.-Y.; Mitchell, L.; Hayes, M.; Oldham, S.; et al. D4eBP Acts Downstream of Both DTOR and DFoxo to Modulate Cardiac Functional Aging in Drosophila. Aging Cell 2009, 8, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Wessells, R.J.; Bodmer, R. Screening Assays for Heart Function Mutants in Drosophila. BioTechniques 2004, 37, 58–66. [Google Scholar] [CrossRef]
- Santalla, M.; Valverde, C.A.; Harnichar, E.; Lacunza, E.; Aguilar-Fuentes, J.; Mattiazzi, A.; Ferrero, P. Aging and CaMKII Alter Intracellular Ca2+ Transients and Heart Rhythm in Drosophila Melanogaster. PLoS ONE 2014, 9, e101871. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Ocorr, K.; Bodmer, R.; Cartry, J. Drosophila as a Model to Study Cardiac Aging. Exp. Gerontol. 2011, 46, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Illenberger, S.; Kishimoto, N.Y.; Huttelmaier, S.; Keating, M.T.; Jockusch, B.M. Metavinculin Mutations Alter Actin Interaction in Dilated Cardiomyopathy. Circulation 2002, 105, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, G.; Spenlehauer, A.; Sessions, A.O.; Trujillo, A.S.; Fuhrmann, A.; Fu, Z.; Venkatraman, V.; Pohl, D.; Tuler, J.; Wang, M.; et al. Vinculin Network-Mediated Cytoskeletal Remodeling Regulates Contractile Function in the Aging Heart. Sci. Transl. Med. 2015, 7, 292ra99. [Google Scholar] [CrossRef] [PubMed]
- Albert, T.K.; Lemaire, M.; van Berkum, N.L.; Gentz, R.; Collart, M.A.; Timmers, H.T. Isolation and Characterization of Human Orthologs of Yeast CCR4-NOT Complex Subunits. Nucleic Acids Res. 2000, 28, 809–817. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Neely, G.G.; Kuba, K.; Cammarato, A.; Isobe, K.; Amann, S.; Zhang, L.; Murata, M.; Elmén, L.; Gupta, V.; Arora, S.; et al. A Global in Vivo Drosophila RNAi Screen Identifies NOT3 as a Conserved Regulator of Heart Function. Cell 2010, 141, 142–153. [Google Scholar] [CrossRef]
- Reller, M.D.; Strickland, M.J.; Riehle-Colarusso, T.; Mahle, W.T.; Correa, A. Prevalence of Congenital Heart Defects in Metropolitan Atlanta, 1998–2005. J. Pediatr. 2008, 153, 807–813. [Google Scholar] [CrossRef]
- Cowan, J.R.; Ware, S.M. Genetics and Genetic Testing in Congenital Heart Disease. Clin. Perinatol. 2015, 42, 373–393. [Google Scholar] [CrossRef]
- Gelb, B.; Brueckner, M.; Chung, W.; Goldmuntz, E.; Kaltman, J.; Kaski, J.P.; Kim, R.; Kline, J.; Mercer-Rosa, L.; Pediatric Cardiac Genomics Consortium, Writing Committee; et al. The Congenital Heart Disease Genetic Network Study: Rationale, Design, and Early Results. Circ. Res. 2013, 112, 698–706. [Google Scholar] [CrossRef]
- Rufaihah, A.J.; Chen, C.K.; Yap, C.H.; Mattar, C.N.Z. Mending a Broken Heart: In Vitro, in Vivo and in Silico Models of Congenital Heart Disease. Dis. Model Mech. 2021, 14, dmm047522. [Google Scholar] [CrossRef]
- Zhu, J.; Fu, Y.; Nettleton, M.; Richman, A.; Han, Z. High Throughput in Vivo Functional Validation of Candidate Congenital Heart Disease Genes in Drosophila. eLife 2017, 6, e22617. [Google Scholar] [CrossRef]
- Schroeder, A.M.; Allahyari, M.; Vogler, G.; Missinato, M.A.; Nielsen, T.; Yu, M.S.; Theis, J.L.; Larsen, L.A.; Goyal, P.; Rosenfeld, J.A.; et al. Model System Identification of Novel Congenital Heart Disease Gene Candidates: Focus on RPL13. Hum. Mol. Genet. 2019, 28, 3954–3969. [Google Scholar] [CrossRef] [PubMed]
- Theadom, A.; Rodrigues, M.; Roxburgh, R.; Balalla, S.; Higgins, C.; Bhattacharjee, R.; Jones, K.; Krishnamurthi, R.; Feigin, V. Prevalence of Muscular Dystrophies: A Systematic Literature Review. NED 2014, 43, 259–268. [Google Scholar] [CrossRef]
- Meola, G.; Cardani, R. Myotonic Dystrophies: An Update on Clinical Aspects, Genetic, Pathology, and Molecular Pathomechanisms. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Allard, P.; Potvin, L.; Prévost, C.; Bégin, P. A 10-Year Study of Mortality in a Cohort of Patients with Myotonic Dystrophy. Neurology 1999, 52, 1658–1662. [Google Scholar] [CrossRef] [PubMed]
- Buxton, J.; Shelbourne, P.; Davies, J.; Jones, C.; Tongeren, T.V.; Aslanidis, C.; de Jong, P.; Jansen, G.; Anvret, M.; Riley, B.; et al. Detection of an Unstable Fragment of DNA Specific to Individuals with Myotonic Dystrophy. Nature 1992, 355, 547–548. [Google Scholar] [CrossRef]
- Fardaei, M.; Rogers, M.T.; Thorpe, H.M.; Larkin, K.; Hamshere, M.G.; Harper, P.S.; Brook, J.D. Three Proteins, MBNL, MBLL and MBXL, Co-Localize in Vivo with Nuclear Foci of Expanded-Repeat Transcripts in DM1 and DM2 Cells. Hum. Mol. Genet. 2002, 11, 805–814. [Google Scholar] [CrossRef]
- Fardaei, M.; Larkin, K.; Brook, J.D.; Hamshere, M.G. In Vivo Co-Localisation of MBNL Protein with DMPK Expanded-Repeat Transcripts. Nucleic Acids Res. 2001, 29, 2766–2771. [Google Scholar] [CrossRef]
- Kuyumcu-Martinez, N.M.; Wang, G.-S.; Cooper, T.A. Increased Steady State Levels of CUGBP1 in Myotonic Dystrophy 1 Are Due to PKC-Mediated Hyper-Phosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Cooper, T.A. Pathogenic Mechanisms of Myotonic Dystrophy. Biochem. Soc. Trans. 2009, 37, 1281–1286. [Google Scholar] [CrossRef]
- Lund, M.; Diaz, L.J.; Ranthe, M.F.; Petri, H.; Duno, M.; Juncker, I.; Eiberg, H.; Vissing, J.; Bundgaard, H.; Wohlfahrt, J.; et al. Cardiac Involvement in Myotonic Dystrophy: A Nationwide Cohort Study. Eur. Heart J. 2014, 35, 2158–2164. [Google Scholar] [CrossRef]
- Benhayon, D.; Lugo, R.; Patel, R.; Carballeira, L.; Elman, L.; Cooper, J.M. Long-Term Arrhythmia Follow-up of Patients with Myotonic Dystrophy. J. Cardiovasc. Electrophysiol. 2015, 26, 305–310. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Wolfe, J.T.; Holmes, D.R.; Edwards, W.D. Pathology of the Cardiac Conduction System in Myotonic Dystrophy: A Study of 12 Cases. J. Am. Coll. Cardiol. 1988, 11, 662–671. [Google Scholar] [CrossRef]
- Plantié, E.; Migocka-Patrzałek, M.; Daczewska, M.; Jagla, K. Model Organisms in the Fight against Muscular Dystrophy: Lessons from Drosophila and Zebrafish. Molecules 2015, 20, 6237–6253. [Google Scholar] [CrossRef] [PubMed]
- Souidi, A.; Zmojdzian, M.; Jagla, K. Dissecting Pathogenetic Mechanisms and Therapeutic Strategies in Drosophila Models of Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2018, 19, 4104. [Google Scholar] [CrossRef]
- Ocorr, K.; Fink, M.; Cammarato, A.; Bernstein, S.I.; Bodmer, R. Semi-Automated Optical Heartbeat Analysis of Small Hearts. J. Vis. Exp. 2009, 31, 1435. [Google Scholar] [CrossRef] [PubMed]
- Auxerre-Plantié, E.; Nakamori, M.; Renaud, Y.; Huguet, A.; Choquet, C.; Dondi, C.; Miquerol, L.; Takahashi, M.P.; Gourdon, G.; Junion, G.; et al. Straightjacket/A2δ3 Deregulation Is Associated with Cardiac Conduction Defects in Myotonic Dystrophy Type 1. eLife 2019, 8, e51114. [Google Scholar] [CrossRef]
- Picchio, L.; Plantie, E.; Renaud, Y.; Poovthumkadavil, P.; Jagla, K. Novel Drosophila Model of Myotonic Dystrophy Type 1: Phenotypic Characterization and Genome-Wide View of Altered Gene Expression. Hum. Mol. Genet. 2013, 22, 2795–2810. [Google Scholar] [CrossRef]
- Chakraborty, M.; Selma-Soriano, E.; Magny, E.; Couso, J.P.; Pérez-Alonso, M.; Charlet-Berguerand, N.; Artero, R.; Llamusi, B. Pentamidine Rescues Contractility and Rhythmicity in a Drosophila Model of Myotonic Dystrophy Heart Dysfunction. Dis. Models Mech. 2015, 8, 1569–1578. [Google Scholar] [CrossRef]
- Chakraborty, M.; Sellier, C.; Ney, M.; Pascal, V.; Charlet-Berguerand, N.; Artero, R.; Llamusi, B. Daunorubicin Reduces MBNL1 Sequestration Caused by CUG-Repeat Expansion and Rescues Cardiac Dysfunctions in a Drosophila Model of Myotonic Dystrophy. Dis. Models Mech. 2018, 11, dmm032557. [Google Scholar] [CrossRef]
- Zhang, D.; Ke, L.; Mackovicova, K.; Van Der Want, J.J.L.; Sibon, O.C.M.; Tanguay, R.M.; Morrow, G.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J.J.M. Effects of Different Small HSPB Members on Contractile Dysfunction and Structural Changes in a Drosophila Melanogaster Model for Atrial Fibrillation. J. Mol. Cell Cardiol. 2011, 51, 381–389. [Google Scholar] [CrossRef]
- Taghli-Lamallem, O.; Plantié, E.; Jagla, K. Drosophila in the Heart of Understanding Cardiac Diseases: Modeling Channelopathies and Cardiomyopathies in the Fruitfly. J. Cardiovasc. Dev. Dis. 2016, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Bloemink, M.J.; Melkani, G.C.; Dambacher, C.M.; Bernstein, S.I.; Geeves, M.A. Two Drosophila Myosin Transducer Mutants with Distinct Cardiomyopathies Have Divergent ADP and Actin Affinities. J. Biol. Chem. 2011, 286, 28435–28443. [Google Scholar] [CrossRef]
- Cozhimuttam Viswanathan, M.; Kaushik, G.; Engler, A.J.; Lehman, W.; Cammarato, A. A Drosophila Melanogaster Model of Diastolic Dysfunction and Cardiomyopathy Based on Impaired Troponin-T Function. Circ. Res. 2014, 114, e6–e17. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.-M.; Wolf, M.J. Serial Examination of an Inducible and Reversible Dilated Cardiomyopathy in Individual Adult Drosophila. PLoS ONE 2009, 4, e7132. [Google Scholar] [CrossRef][Green Version]
- Selma-Soriano, E.; Casillas-Serra, C.; Artero, R.; Llamusi, B.; Navarro, J.A.; Redón, J. Rabphilin Silencing Causes Dilated Cardiomyopathy in a Drosophila Model of Nephrocyte Damage. Sci. Rep. 2021, 11, 15287. [Google Scholar] [CrossRef] [PubMed]
- Taghli-Lamallem, O.; Akasaka, T.; Hogg, G.; Nudel, U.; Yaffe, D.; Chamberlain, J.S.; Ocorr, K.; Bodmer, R. Dystrophin Deficiency in Drosophila Reduces Lifespan and Causes a Dilated Cardiomyopathy Phenotype. Aging Cell 2008, 7, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J. Modeling Dilated Cardiomyopathies in Drosophila. Trends Cardiovasc. Med. 2012, 22, 55–61. [Google Scholar] [CrossRef]
- Bell, K.M.; Kronert, W.A.; Huang, A.; Bernstein, S.I.; Swank, D.M. The R249Q Hypertrophic Cardiomyopathy Myosin Mutation Decreases Contractility in Drosophila by Impeding Force Production. J. Physiol. 2019, 597, 2403–2420. [Google Scholar] [CrossRef]
- Kronert, W.A.; Bell, K.M.; Viswanathan, M.C.; Melkani, G.C.; Trujillo, A.S.; Huang, A.; Melkani, A.; Cammarato, A.; Swank, D.M.; Bernstein, S.I. Prolonged Cross-Bridge Binding Triggers Muscle Dysfunction in a Drosophila Model of Myosin-Based Hypertrophic Cardiomyopathy. eLife 2018, 7, e38064. [Google Scholar] [CrossRef]
- Tallo, C.A.; Duncan, L.H.; Yamamoto, A.H.; Slaydon, J.D.; Arya, G.H.; Turlapati, L.; Mackay, T.F.C.; Carbone, M.A. Heat Shock Proteins and Small Nucleolar RNAs Are Dysregulated in a Drosophila Model for Feline Hypertrophic Cardiomyopathy. G3 Genes Genom. Gen. 2021, 11. [Google Scholar] [CrossRef]
- Achal, M.; Trujillo, A.S.; Melkani, G.C.; Farman, G.P.; Ocorr, K.; Viswanathan, M.C.; Kaushik, G.; Newhard, C.S.; Glasheen, B.M.; Melkani, A.; et al. A Restrictive Cardiomyopathy Mutation in an Invariant Proline at the Myosin Head/Rod Junction Enhances Head Flexibility and Function, Yielding Muscle Defects in Drosophila. J. Mol. Biol. 2016, 428, 2446–2461. [Google Scholar] [CrossRef] [PubMed]
- Cammarato, A.; Alayari, N.N.; Gucek, M.; Reedy, M.C.; Rucker, J.; Eyk, J.E.V.; Cole, R.N.; O’Rourke, B.; Bodmer, R.; Bernstein, S.I.; et al. A Systems Biology Approach to Restrictive Cardiomyopathy in Drosophila. Biophys. J. 2010, 98, 717a–718a. [Google Scholar] [CrossRef][Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souidi, A.; Jagla, K. Drosophila Heart as a Model for Cardiac Development and Diseases. Cells 2021, 10, 3078. https://doi.org/10.3390/cells10113078
Souidi A, Jagla K. Drosophila Heart as a Model for Cardiac Development and Diseases. Cells. 2021; 10(11):3078. https://doi.org/10.3390/cells10113078
Chicago/Turabian StyleSouidi, Anissa, and Krzysztof Jagla. 2021. "Drosophila Heart as a Model for Cardiac Development and Diseases" Cells 10, no. 11: 3078. https://doi.org/10.3390/cells10113078
APA StyleSouidi, A., & Jagla, K. (2021). Drosophila Heart as a Model for Cardiac Development and Diseases. Cells, 10(11), 3078. https://doi.org/10.3390/cells10113078