
A Unique Topoisomerase II Inhibitor with Dose-Affected Anticancer Mechanisms and Less Cardiotoxicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Cultures
2.3. Cell Viability Assay
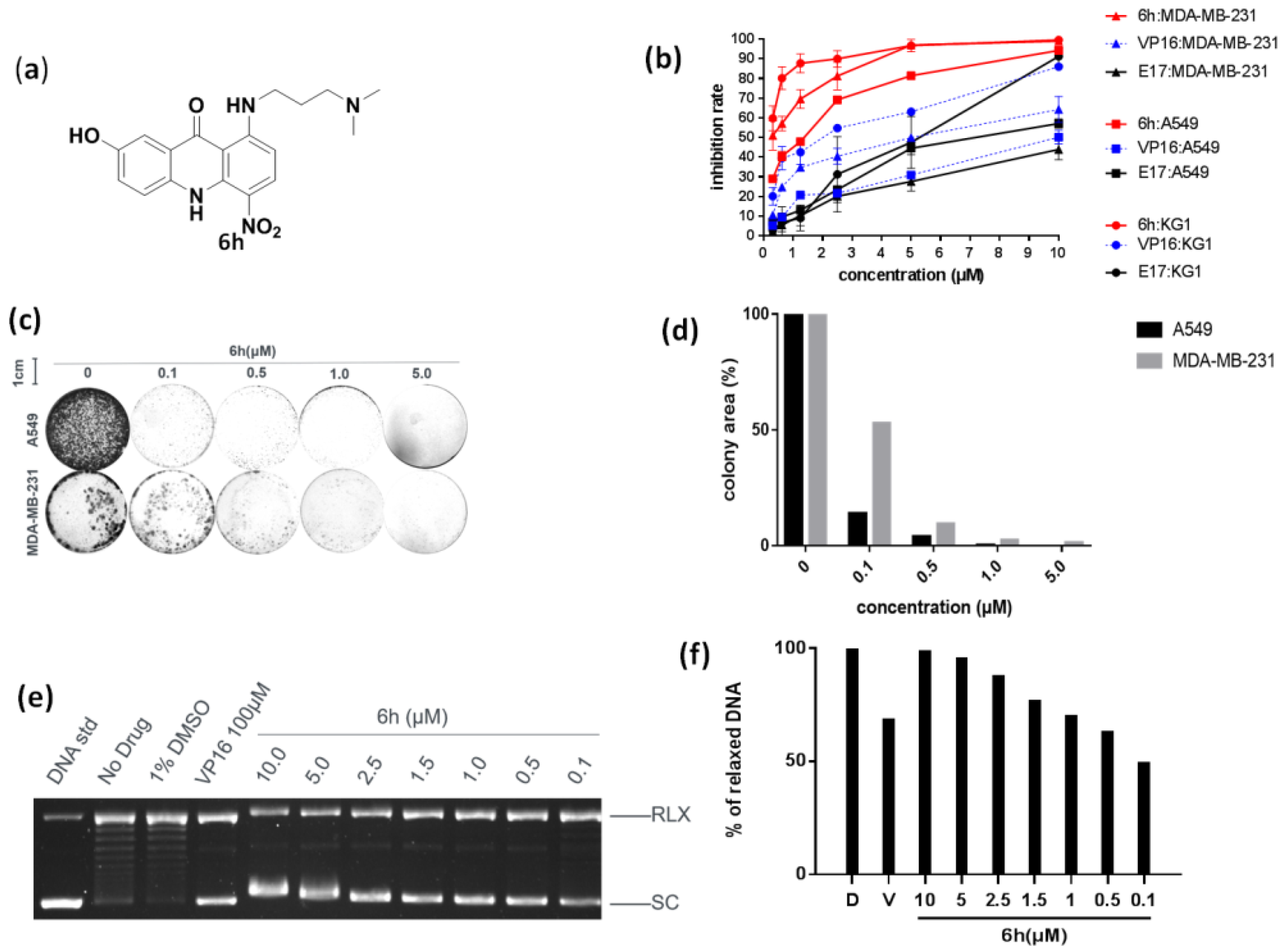
2.4. Colony Formation Assay
2.5. Topo IIα-Mediated pBR322 DNA Relaxation Assay
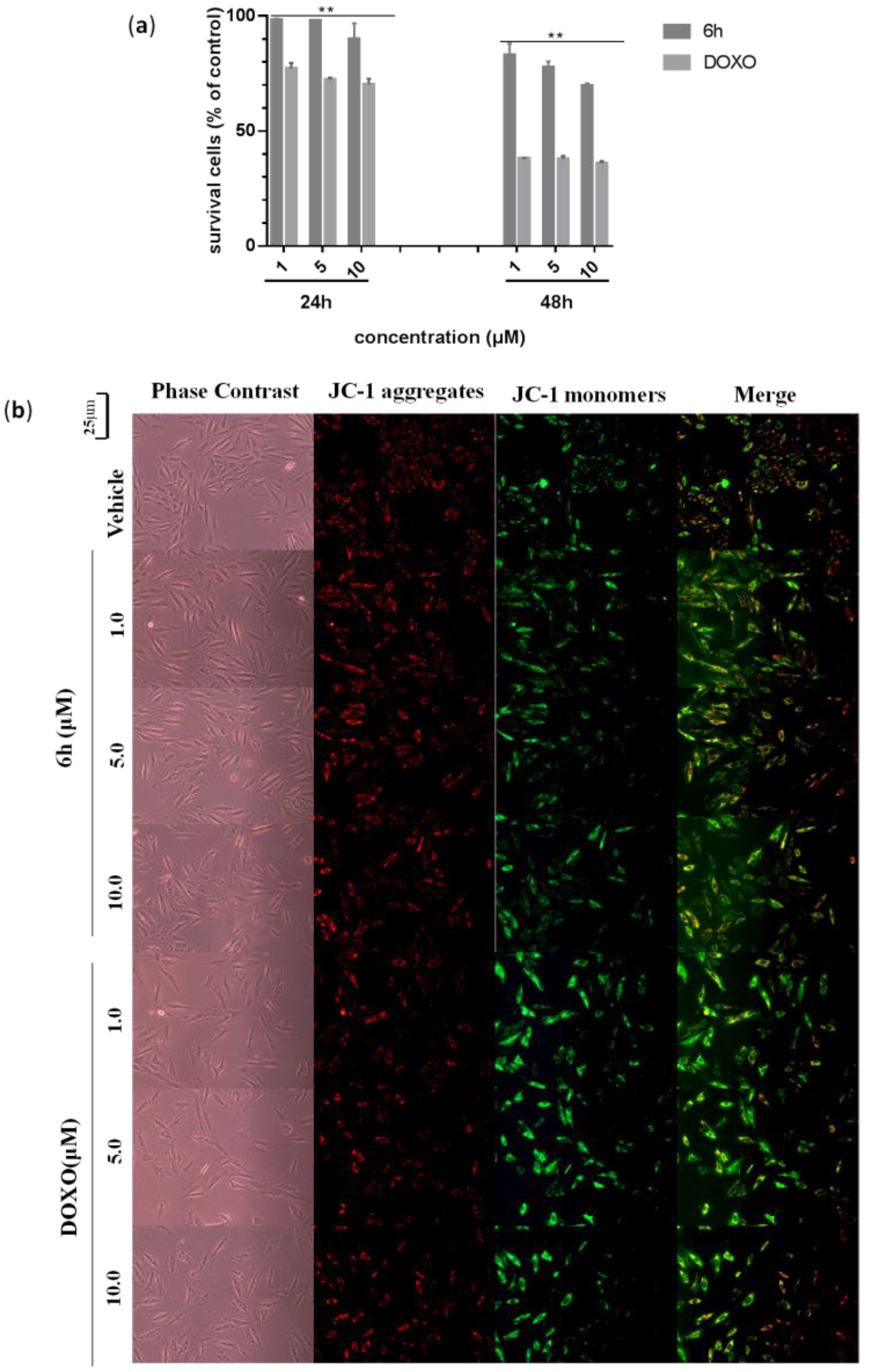
2.6. Measurement of Mitochondrial Membrane Potential (∆ψm)
2.7. Cell Cycle Analysis
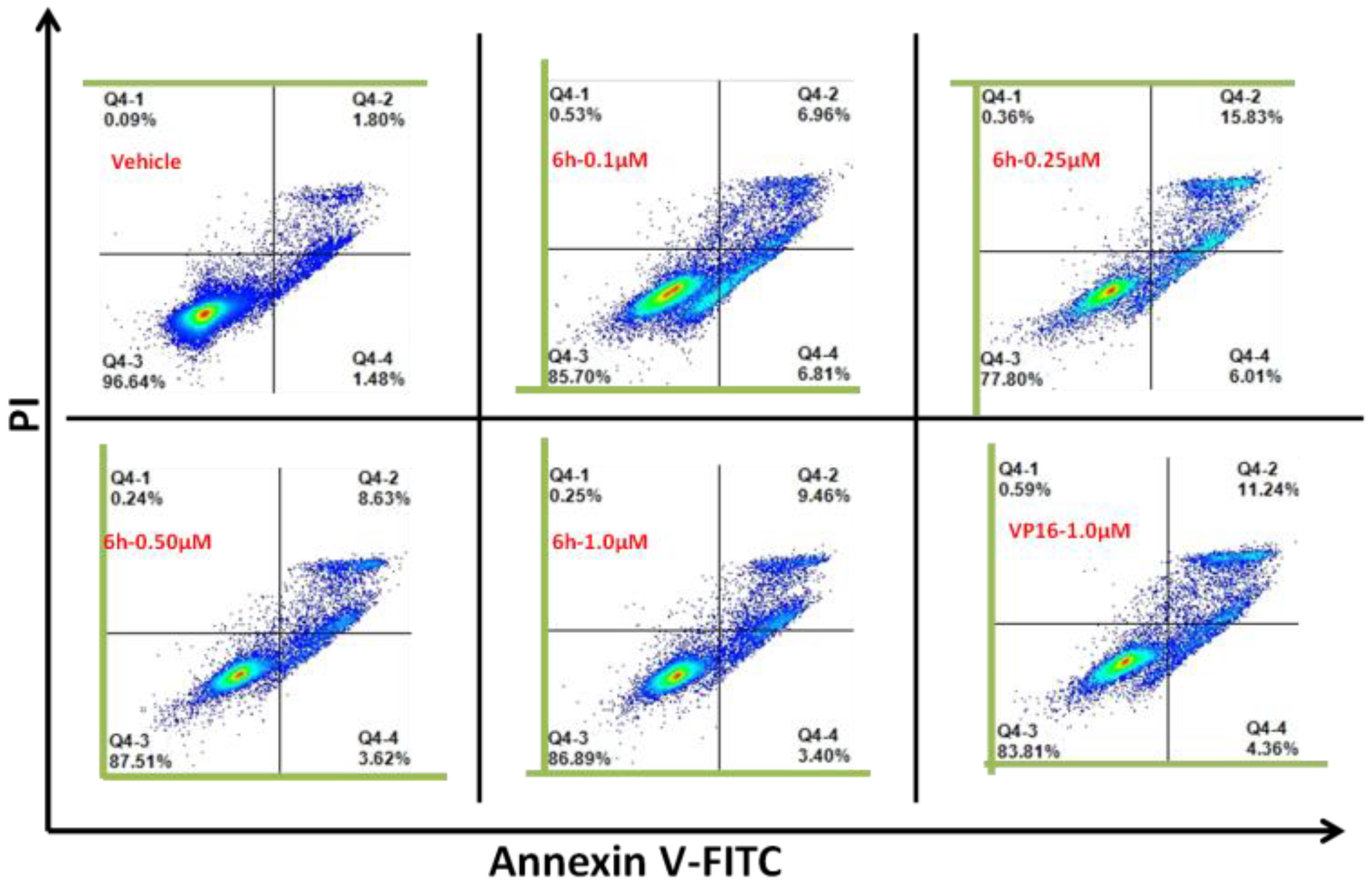
2.8. Annexin V-FITC/PI Apoptosis Detection Assay
2.9. Data Analysis
3. Results
3.1. 6h Exhibited Significant Anti-Proliferative Activity by Inhibiting Topo IIα
3.2. 6h Induced Different Cell Cycle Arrests in Cancer Cells
3.3. 6h Induced Obvious Apoptosis in KG1 Cells
3.4. 6h Induced Less Cardiotoxicity in H9c2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell. Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Huang, X.; Wu, J.; Yang, L.; Zheng, Y.; Shen, Y.; Li, Z.; Li, X. Discovery of novel topoisomerase II inhibitors by medicinal chemistry approaches. J. Med. Chem. 2018, 61, 8947–8980. [Google Scholar] [CrossRef]
- Champoux, J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Farh, L.; Lin, L.; Lin, T.; Yu, Y.; Yen, T.; Chiang, C.; Chan, N. Structural basis of Type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [Green Version]
- Azarova, A.; Lin, C.; Tsai, Y.; Lau, J.; Wang, J.; Liu, L. Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc. Natl. Acad. Sci. USA 2007, 104, 11014–11019. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wu, X.; Lu, C.; Li, X. Structure-activity relationship of novel acridone derivatives as antiproliferative agents. Bioorg. Med. Chem. 2021, 29, 115868. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Hu, W.; Li, C.; Wu, J.; Yang, L.; Han, X.; Shen, Y.; Li, Z.; Li, X. Design, synthesis, biological evaluation and molecular docking study on peptidomimetic analogues of XK469. Eur. J. Med. Chem. 2016, 124, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Liu, Z.; Li, X. Design, synthesis, bioevaluation of LFC- and PA-tethered anthraquinone analogues of mitoxantrone. Bioorg. Chem. 2020, 101, 104005. [Google Scholar] [CrossRef]
- Chen, J.; Wu, X.; Lu, C.; Li, X. E17 exerts anti-tumor activity through inhibiting topo II-mediated chromosomes condensation in CRC cells. Biochem. Biophys. Res. Commun. 2019, 513, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, B.; Zhang, W.; Yang, T.; Wang, N.; Gao, C.; Tan, C.; Liu, H.; Jiang, Y. Synthesis and antiproliferative activity of 9-benzylamino-6-chloro-2-methoxy-acridine derivatives as potent DNA-binding ligands and topoisomerase II inhibitors. Eur. J. Med. Chem. 2016, 116, 59–70. [Google Scholar] [CrossRef]
- Prasher, P.; Sharma, M. Medicinal chemistry of acridine and its analogues. Med. Chem. Commun. 2018, 9, 1589–1618. [Google Scholar] [CrossRef]
- Hong, D.; Ding, J.; Li, O.; He, Q.; Ke, M.; Zhu, M.; Liu, L.; Ou, W.; He, Y.; Wu, Y. Human-induced pluripotent stem cell-derived macrophages and their immunological function in response to tuberculosis infection. Stem. Cell. Res. Ther. 2018, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Braselmann, H.; Michna, A.; Hess, J.; Unger, K. CFAssay: Statistical analysis of the colony formation assay. Radiat. Oncol. 2015, 10, 223. [Google Scholar] [CrossRef] [Green Version]
- Franken, N.; Rodermond, H.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Vejpongsa, P.; Yeh, E. Topoisomerase 2beta: A promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin. Pharmacol. Ther. 2014, 95, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Lacopo, F.; Cipolla, C. Cardiotoxicity of anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.; Ait-Oudhia, S. Insights into doxorubicin-induced cardiotoxicity: Molecular mechanisms, preventive strategies, and early monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef] [Green Version]
- Gorini, S.; Angelis, A.; Berrino, D.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic drugs and mitochondrial dysfunction: Focus on doxorubicin, trastuzumab, and sunitinib. Oxid. Med. Cell Longev. 2018, 2018, 7582730. [Google Scholar] [CrossRef] [Green Version]
- Marinello, J.; Delcuratolo, M.; Capranico, G. Anthracyclines as topoisomerase II poisons: From early studies to new perspectives. Int. J. Mol. Sci. 2018, 19, 3480. [Google Scholar] [CrossRef] [Green Version]
- Bax, B.; Murshudov, G.; Maxwell, A.; Germe, T. DNA Topoisomerase Inhibitors: Trapping a DNA-cleaving machine in motion. J. Mol. Biol. 2019, 431, 3427–3449. [Google Scholar] [CrossRef] [PubMed]
- Tewey, K.; Chen, G.; Nelson, E.; Liu, L. Intercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J. Biol. Chem. 1984, 259, 9182–9187. [Google Scholar] [CrossRef]
- Tewey, K.; Rowe, T.; Yang, L.; Halligan, B.; Liu, L. Adriamycin-Induced DNA Damage Mediated by Mammalian DNA Topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.-Y.; Xu, G.-S.; Li, X. A Unique Topoisomerase II Inhibitor with Dose-Affected Anticancer Mechanisms and Less Cardiotoxicity. Cells 2021, 10, 3138. https://doi.org/10.3390/cells10113138
Li Z-Y, Xu G-S, Li X. A Unique Topoisomerase II Inhibitor with Dose-Affected Anticancer Mechanisms and Less Cardiotoxicity. Cells. 2021; 10(11):3138. https://doi.org/10.3390/cells10113138
Chicago/Turabian StyleLi, Zhi-Ying, Guang-Sen Xu, and Xun Li. 2021. "A Unique Topoisomerase II Inhibitor with Dose-Affected Anticancer Mechanisms and Less Cardiotoxicity" Cells 10, no. 11: 3138. https://doi.org/10.3390/cells10113138