
Loss of Polycomb Repressive Complex 2 Function Alters Digestive Organ Homeostasis and Neuronal Differentiation in Zebrafish

 , , ,
, , , 
 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Zebrafish Maintenance, Embryo Preparation, and Animal Ethics Statements
2.2. TALEN Design and Assembly
2.3. mRNA Injection into Zebrafish Embryos
2.4. Genotype Analyses
2.5. Kaplan–Meier Analysis
2.6. Oil Red-O Staining
2.7. Smurf Assay
2.8. Histological Analysis
2.9. Biometric Measurements
2.10. Confocal Microscopy
2.11. Whole-Mount In Situ Hybridization
2.12. Histone Extraction and Western Blot Analysis
2.13. RNA Extraction, RT-PCR and RT-qPCR
2.14. Behavioral Studies
3. Results
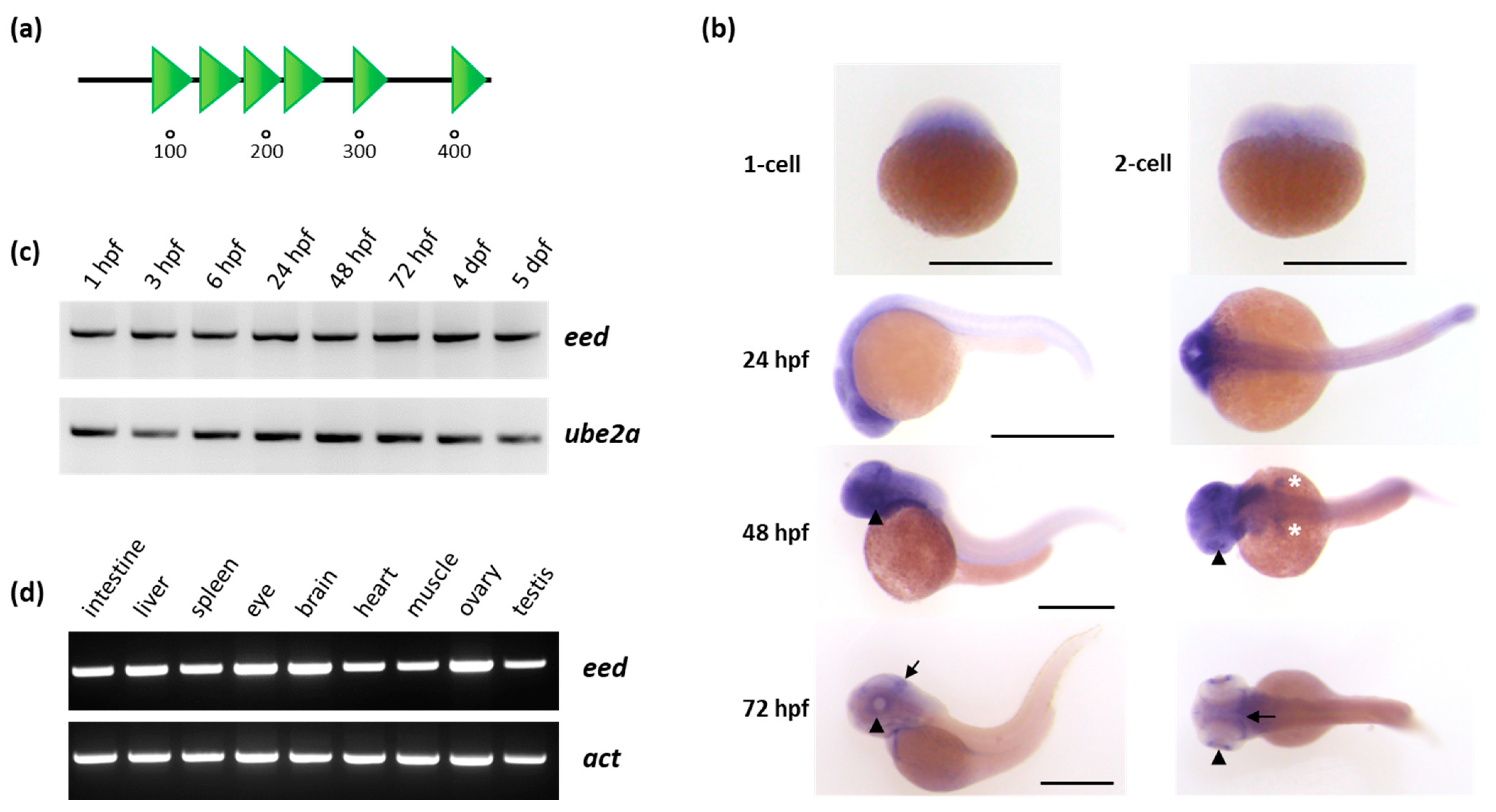
3.1. eed Expression during Zebrafish Development
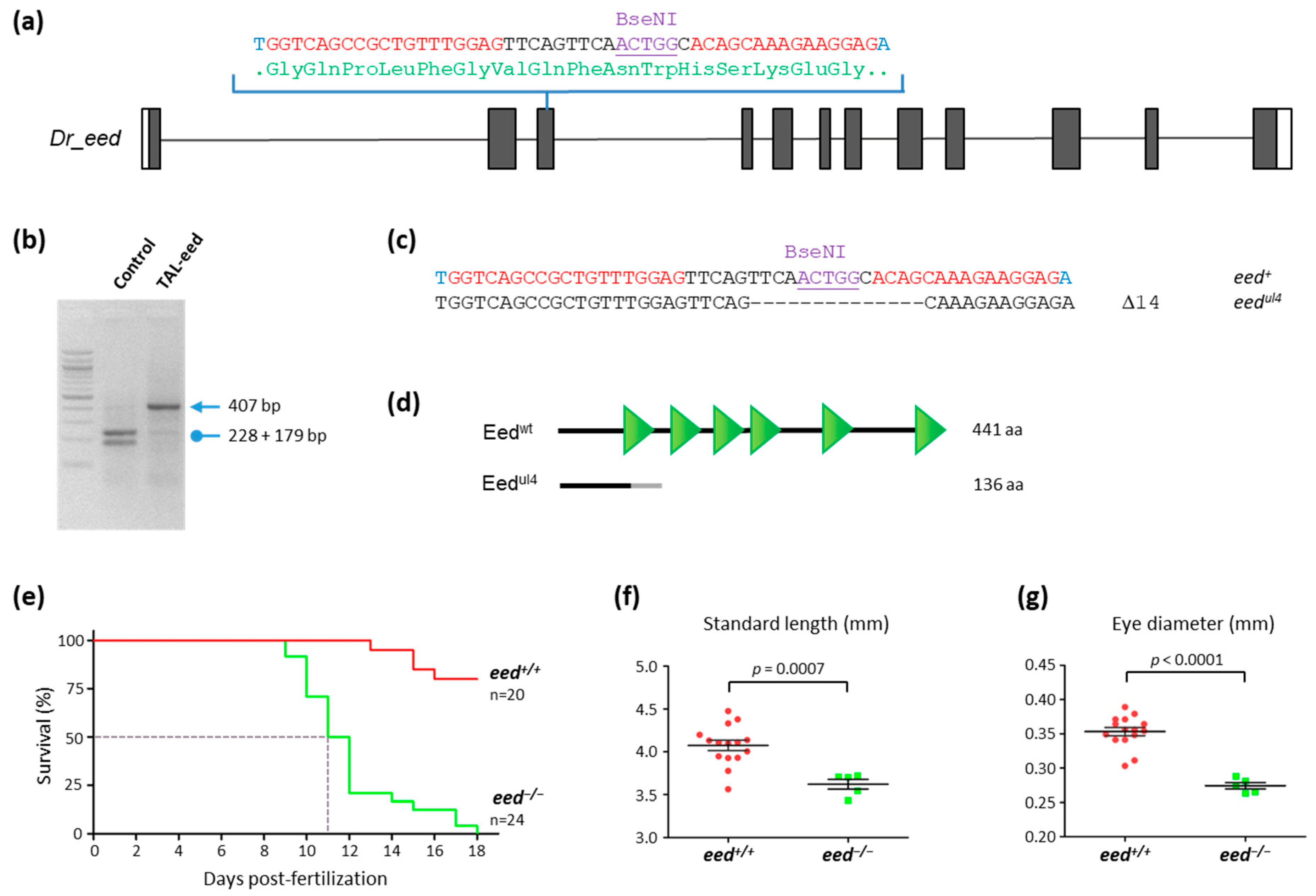
3.2. TALEN-Mediated Inactivation of eed in Zebrafish
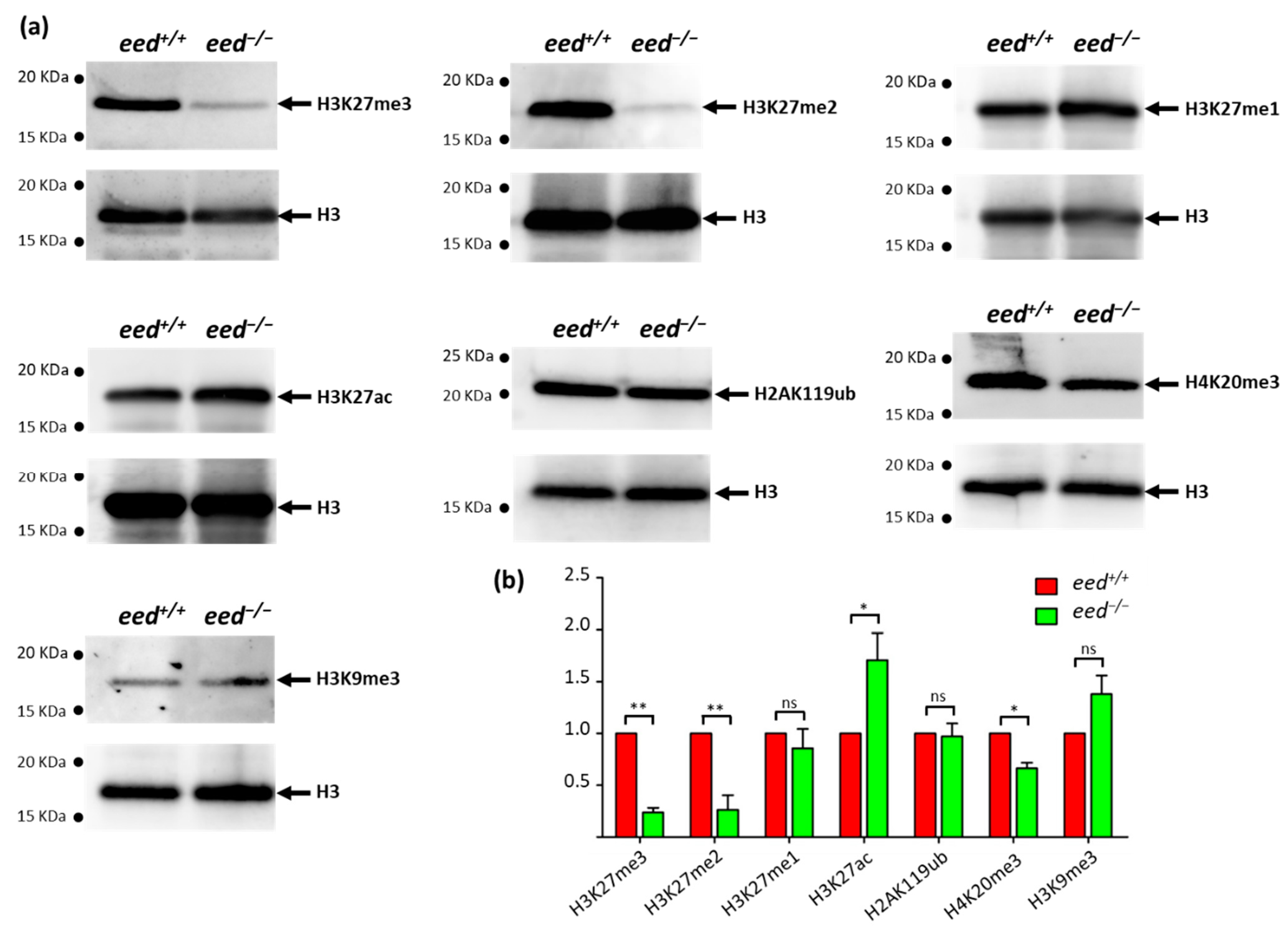
3.3. Loss of eed Function Alters H3K27me2/3 Levels, but Not H3K27me1 nor H2AK119ub
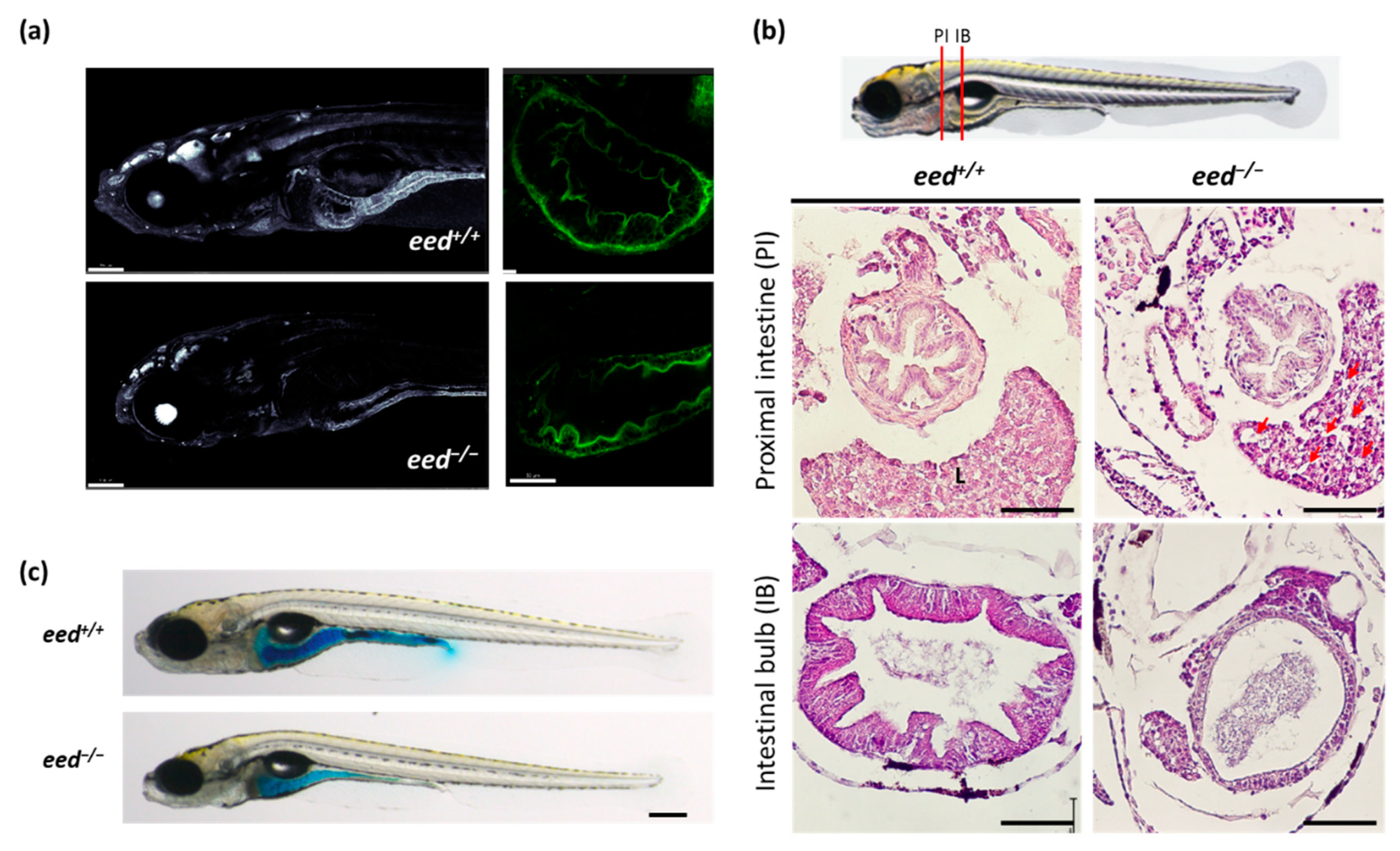
3.4. Loss of eed Function Alters the Homeostasis of Digestive Organs in Zebrafish
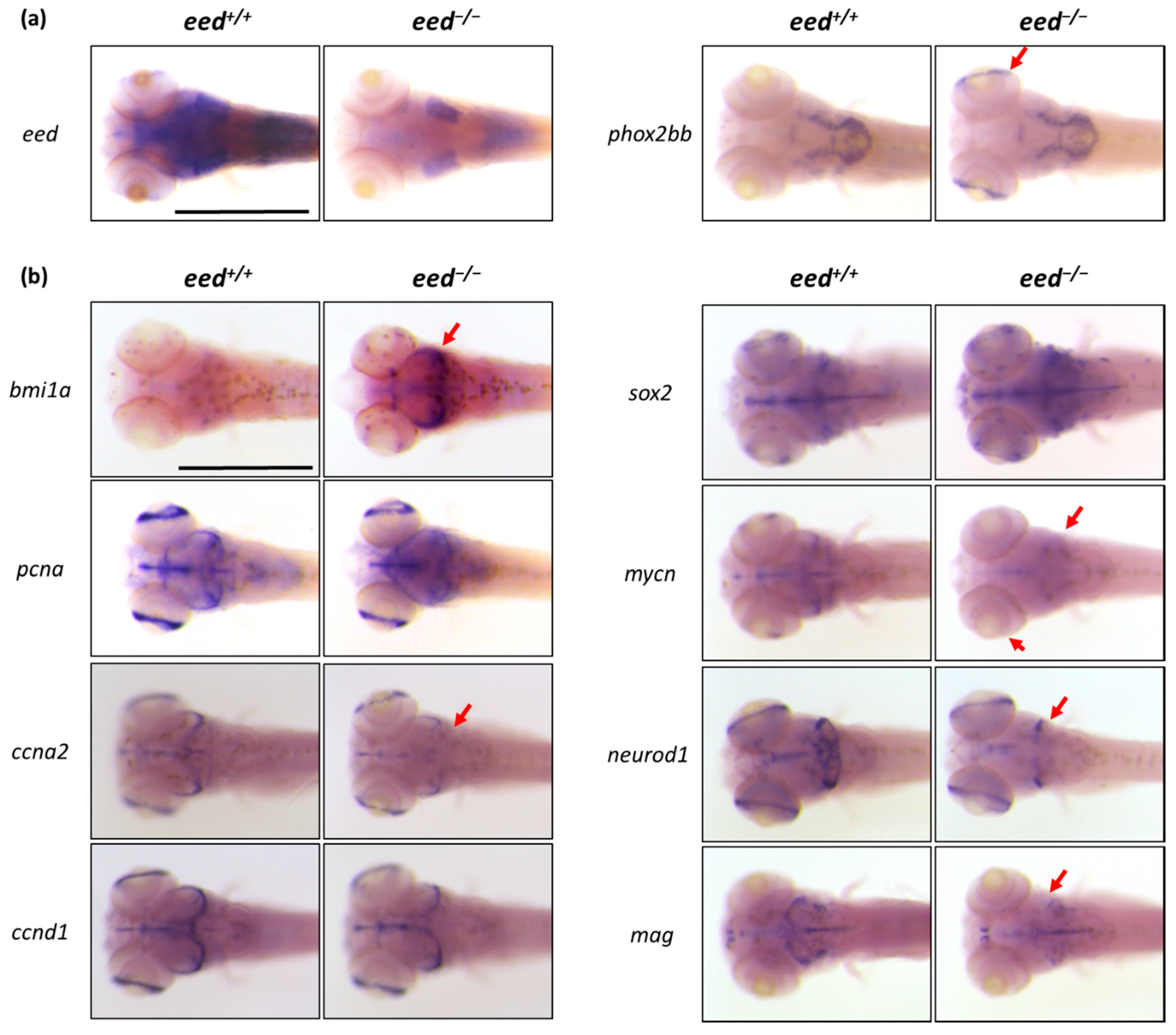
3.5. Loss of eed Function Alters Neuronal Differentiation
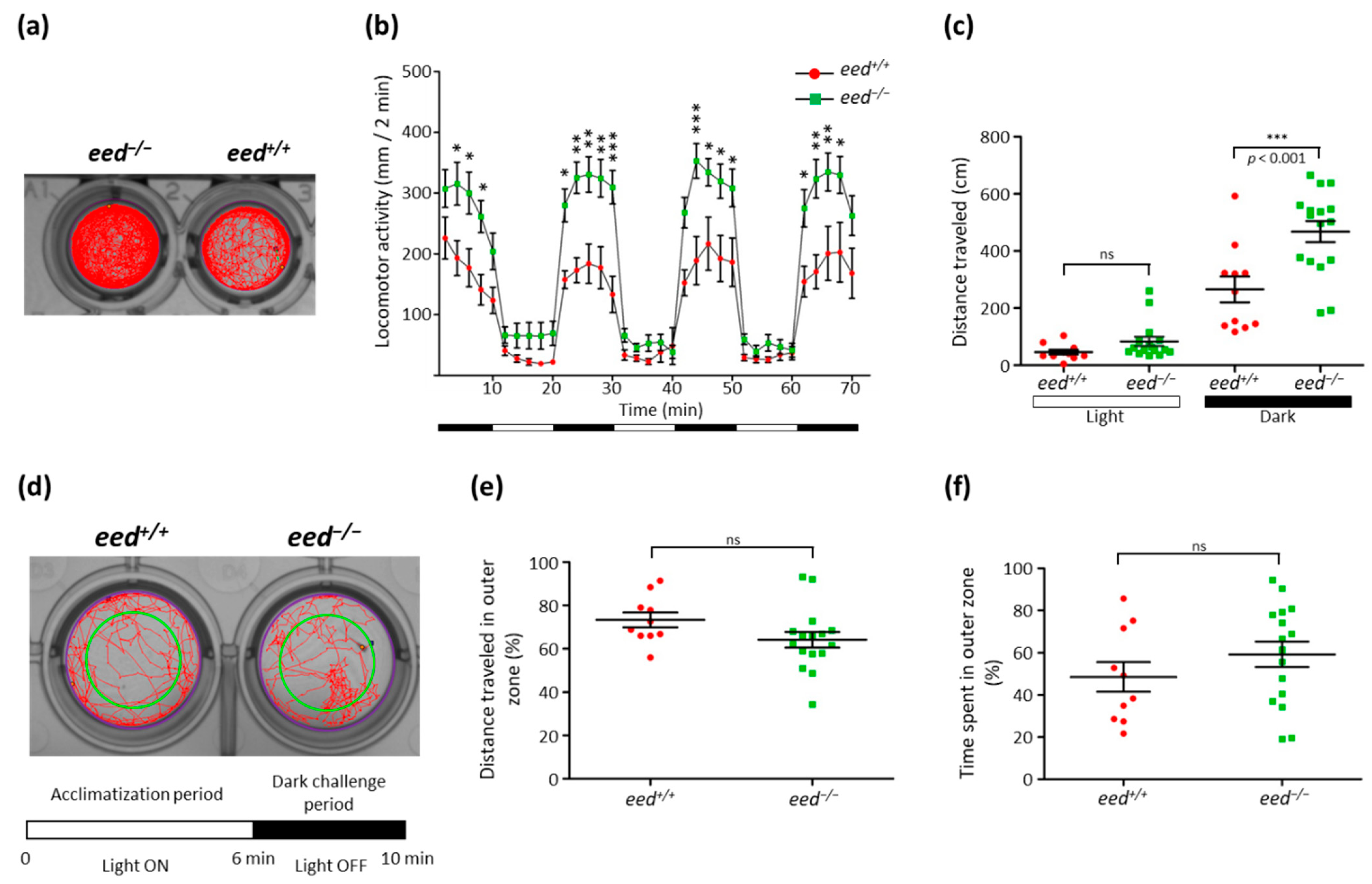
3.6. Loss of eed Function Alters Locomotor Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, M.; Sauvageau, G. Polycomb group proteins: Multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 2010, 7, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Surface, L.E.; Thornton, S.R.; Boyer, L.A. Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell 2010, 7, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Geng, Z.; Gao, Z. Mammalian PRC1 Complexes: Compositional Complexity and Diverse Molecular Mechanisms. Int. J. Mol. Sci. 2020, 21, 8594. [Google Scholar] [CrossRef]
- Glancy, E.; Ciferri, C.; Bracken, A.P. Structural basis for PRC2 engagement with chromatin. Curr. Opin. Struct. Biol. 2021, 67, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Shilatifard, A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nat. Rev. Mol. Cell Biol. 2021, 22, 326–345. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Hart, C.M.; Francis, N.J.; Vargas, M.L.; Sengupta, A.; Wild, B.; Miller, E.L.; O’Connor, M.B.; Kingston, R.E.; Simon, J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 2002, 111, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef]
- de Napoles, M.; Mermoud, J.E.; Wakao, R.; Tang, Y.A.; Endoh, M.; Appanah, R.; Nesterova, T.B.; Silva, J.; Otte, A.P.; Vidal, M.; et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 2004, 7, 663–676. [Google Scholar] [CrossRef]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Azuara, V.; Perry, P.; Sauer, S.; Spivakov, M.; Jørgensen, H.F.; John, R.M.; Gouti, M.; Casanova, M.; Warnes, G.; Merkenschlager, M.; et al. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006, 8, 532–538. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef]
- Pan, G.; Tian, S.; Nie, J.; Yang, C.; Ruotti, V.; Wei, H.; Jonsdottir, G.A.; Stewart, R.; Thomson, J.A. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 2007, 1, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.D.; Han, X.; Chew, J.L.; Liu, J.; Chiu, K.P.; Choo, A.; Orlov, Y.L.; Sung, W.K.; Shahab, A.; Kuznetsov, V.A.; et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 2007, 1, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [Green Version]
- Kouznetsova, V.L.; Tchekanov, A.; Li, X.; Yan, X.; Tsigelny, I.F. Polycomb repressive 2 complex-Molecular mechanisms of function. Protein Sci. 2019, 28, 1387–1399. [Google Scholar] [CrossRef]
- Van Mierlo, G.; Veenstra, G.J.C.; Vermeulen, M.; Marks, H. The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019, 29, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Chammas, P.; Mocavini, I.; Di Croce, L. Engaging chromatin: PRC2 structure meets function. Br. J. Cancer 2020, 122, 315–328. [Google Scholar] [CrossRef]
- Pasini, D.; Bracken, A.P.; Jensen, M.R.; Lazzerini Denchi, E.; Helin, K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004, 23, 4061–4071. [Google Scholar] [CrossRef] [Green Version]
- Faust, C.; Schumacher, A.; Holdener, B.; Magnuson, T. The eed mutation disrupts anterior mesoderm production in mice. Development 1995, 121, 273–285. [Google Scholar] [CrossRef]
- Ueda, T.; Sanada, M.; Matsui, H.; Yamasaki, N.; Honda, Z.I.; Shih, L.Y.; Mori, H.; Inaba, T.; Ogawa, S.; Honda, H. EED mutants impair polycomb repressive complex 2 in myelodysplastic syndrome and related neoplasms. Leukemia 2012, 26, 2557–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erokhin, M.; Chetverina, O.; Győrffy, B.; Tatarskiy, V.V.; Mogila, V.; Shtil, A.A.; Roninson, I.B.; Moreaux, J.; Georgiev, P.; Cavalli, G.; et al. Clinical Correlations of Polycomb Repressive Complex 2 in Different Tumor Types. Cancers 2021, 13, 3155. [Google Scholar] [CrossRef]
- Cohen, A.S.; Tuysuz, B.; Shen, Y.; Bhalla, S.K.; Jones, S.J.; Gibson, W.T. A novel mutation in EED associated with overgrowth. J. Hum. Genet. 2015, 60, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Imagawa, E.; Higashimoto, K.; Sakai, Y.; Numakura, C.; Okamoto, N.; Matsunaga, S.; Ryo, A.; Sato, Y.; Sanefuji, M.; Ihara, K.; et al. Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome. Hum. Mutat. 2017, 38, 637–648. [Google Scholar] [CrossRef]
- Cooney, E.; Bi, W.; Schlesinger, A.E.; Vinson, S.; Potocki, L. Novel EED mutation in patient with Weaver syndrome. Am. J. Med. Genet. A 2017, 173, 541–545. [Google Scholar] [CrossRef]
- Griffiths, S.; Loveday, C.; Zachariou, A.; Behan, L.A.; Chandler, K.; Cole, T.; D’Arrigo, S.; Dieckmann, A.; Foster, A.; Gibney, J.; et al. EED and EZH2 constitutive variants: A study to expand the Cohen-Gibson syndrome phenotype and contrast it with Weaver syndrome. Am. J. Med. Genet. A 2019, 179, 588–594. [Google Scholar] [CrossRef]
- Spellicy, C.J.; Peng, Y.; Olewiler, L.; Cathey, S.S.; Rogers, R.C.; Bartholomew, D.; Johnson, J.; Alexov, E.; Lee, J.A.; Friez, M.J.; et al. Three additional patients with EED-associated overgrowth: Potential mutation hotspots identified? J. Hum. Genet. 2019, 64, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Völkel, P.; Bary, A.; Raby, L.; Chapart, A.; Dupret, B.; Le Bourhis, X.; Angrand, P.O. Ezh1 arises from Ezh2 gene duplication but its function is not required for zebrafish development. Sci. Rep. 2019, 9, 4319. [Google Scholar] [CrossRef]
- Soto, R.A.; Najia, M.A.T.; Hachimi, M.; Frame, J.M.; Yette, G.A.; Lummertz da Rocha, E.; Stankunas, K.; Daley, G.Q.; North, T.E. Sequential regulation of hemogenic fate and hematopoietic stem and progenitor cell formation from arterial endothelium by Ezh1/2. Stem Cell Rep. 2021, 16, 1718–1734. [Google Scholar] [CrossRef]
- San, B.; Chrispijn, N.D.; Wittkopp, N.; van Heeringen, S.J.; Lagendijk, A.K.; Aben, M.; Bakkers, J.; Ketting, R.F.; Kamminga, L.M. Normal formation of a vertebrate body plan and loss of tissue maintenance in the absence of ezh2. Sci. Rep. 2016, 6, 24658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupret, B.; Völkel, P.; Vennin, C.; Toillon, R.A.; Le Bourhis, X.; Angrand, P.O. The histone lysine methyltransferase Ezh2 is required for maintenance of the intestine integrity and for caudal fin regeneration in zebrafish. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 1079–1093. [Google Scholar] [CrossRef]
- Oppel, F.; Ki, D.H.; Zimmerman, M.W.; Ross, K.N.; Tao, T.; Shi, H.; He, S.; Aster, J.C.; Look, A.T. suz12 inactivation in p53- and nf1-deficient zebrafish accelerates the onset of malignant peripheral nerve sheath tumors and expands the spectrum of tumor types. Dis. Model Mech. 2020, 13, dmm042341. [Google Scholar] [CrossRef] [PubMed]
- Maître, J.L.; Berthoumieux, H.; Krens, S.F.; Salbreux, G.; Jülicher, F.; Paluch, E.; Heisenberg, C.P. Adhesion functions in cell sorting by mechanically coupling the cortices of adhering cells. Science 2012, 338, 253–256. [Google Scholar] [CrossRef]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Dupret, B.; Völkel, P.; Le Bourhis, X.; Angrand, P.O. The Polycomb Group Protein Pcgf1 Is Dispensable in Zebrafish but Involved in Early Growth and Aging. PLoS ONE 2016, 11, e0158700. [Google Scholar] [CrossRef] [Green Version]
- Doyle, E.L.; Booher, N.J.; Standage, D.S.; Voytas, D.F.; Brendel, V.P.; Vandyk, J.K.; Bogdanove, A.J. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: Tools for TAL effector design and target prediction. Nucleic Acids Res. 2012, 40, W117–W122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlem, T.J.; Hoshijima, K.; Jurynec, M.J.; Gunther, D.; Starker, C.G.; Locke, A.S.; Weis, A.M.; Voytas, D.F.; Grunwald, D.J. Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 2012, 8, e1002861. [Google Scholar] [CrossRef] [Green Version]
- Dupret, B.; Völkel, P.; Follet, P.; Le Bourhis, X.; Angrand, P.O. Combining genotypic and phenotypic analyses on single mutant zebrafish larvae. MethodsX 2018, 5, 244–256. [Google Scholar] [CrossRef]
- Dambroise, E.; Monnier, L.; Ruisheng, L.; Aguilaniu, H.; Joly, J.S.; Tricoire, H.; Rera, M. Two phases of aging separated by the Smurf transition as a public path to death. Sci. Rep. 2016, 6, 23523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parichy, D.M.; Elizondo, M.R.; Mills, M.G.; Gordon, T.N.; Engeszer, R.E. Normal table of postembryonic zebrafish development: Staging by externally visible anatomy of the living fish. Dev. Dyn. 2009, 238, 2975–3015. [Google Scholar] [CrossRef] [Green Version]
- Thisse, C.; Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008, 3, 59–69. [Google Scholar] [CrossRef]
- Raby, L.; Völkel, P.; Le Bourhis, X.; Angrand, P.O. The Polycomb Orthologues in Teleost Fishes and Their Expression in the Zebrafish Model. Genes 2020, 11, 362. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Xing, X.; Hu, M.; Zhang, Y.; Liu, P.; Chai, J. Structural basis of EZH2 recognition by EED. Structure 2007, 15, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Pelegri, F. Maternal factors in zebrafish development. Dev. Dyn. 2003, 228, 535–554. [Google Scholar] [CrossRef]
- Dosch, R.; Wagner, D.S.; Mintzer, K.A.; Runke, G.; Wiemelt, A.P.; Mullins, M.C. Maternal control of vertebrate development before the midblastula transition: Mutants from the zebrafish I. Dev. Cell 2004, 6, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrispijn, N.D.; Andralojc, K.M.; Castenmiller, C.; Kamminga, L.M. Gene expression profile of a selection of Polycomb Group genes during zebrafish embryonic and germ line development. PLoS ONE 2018, 13, e0200316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.J.; Collins, J.E.; Sealy, I.M.; Wali, N.; Dooley, C.M.; Digby, Z.; Stemple, D.L.; Murphy, D.N.; Billis, K.; Hourlier, T.; et al. A high-resolution mRNA expression time course of embryonic development in zebrafish. eLife 2017, 6, e30860. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Chen, X.; Xiong, J.; Li, Y.; Li, H.; Ding, X.; Liu, S.; Chen, S.; Gao, S.; Zhu, B. Histone methyltransferase G9a contributes to H3K27 methylation in vivo. Cell Res. 2011, 21, 365–367. [Google Scholar] [CrossRef] [Green Version]
- Lavarone, E.; Barbieri, C.M.; Pasini, D. Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat. Commun. 2019, 10, 1679. [Google Scholar] [CrossRef]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.H.; Woodfin, A.R.; et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Tavares, L.; Dimitrova, E.; Oxley, D.; Webster, J.; Poot, R.; Demmers, J.; Bezstarosti, K.; Taylor, S.; Ura, H.; Koide, H.; et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 2012, 148, 664–678. [Google Scholar] [CrossRef] [Green Version]
- Morey, L.; Aloia, L.; Cozzuto, L.; Benitah, S.A.; Di Croce, L. RYBP and Cbx7 define specific biological functions of polycomb complexes in mouse embryonic stem cells. Cell Rep. 2013, 3, 60–69. [Google Scholar] [CrossRef] [Green Version]
- San, B.; Aben, M.; Elurbe, D.M.; Voeltzke, K.; Den Broeder, M.J.; Rougeot, J.; Legler, J.; Kamminga, L.M. Genetic and Epigenetic Regulation of Zebrafish Intestinal Development. Epigenomes 2018, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Behrndt, M.; Salbreux, G.; Campinho, P.; Hauschild, R.; Oswald, F.; Roensch, J.; Grill, S.W.; Heisenberg, C.P. Forces driving epithelial spreading in zebrafish gastrulation. Science 2012, 338, 257–260. [Google Scholar] [CrossRef]
- Araya, C.; Häkkinen, H.M.; Carcamo, L.; Cerda, M.; Savy, T.; Rookyard, C.; Peyriéras, N.; Clarke, J.D.W. Cdh2 coordinates Myosin-II dependent internalisation of the zebrafish neural plate. Sci. Rep. 2019, 9, 1835. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Wang, K.; Zheng, X.; Chen, X.; Zhang, W.; Zhang, Y.; Hou, J.; Liu, L. High fat plus high cholesterol diet lead to hepatic steatosis in zebrafish larvae: A novel model for screening anti-hepatic steatosis drugs. Nutr. Metab. 2015, 12, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Zheng, Y.M.; Zhang, J.P. Comparative Study of Different Diets-Induced NAFLD Models of Zebrafish. Front. Endocrinol. 2018, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- Howarth, D.L.; Lindtner, C.; Vacaru, A.M.; Sachidanandam, R.; Tsedensodnom, O.; Vasilkova, T.; Buettner, C.; Sadler, K.C. Activating transcription factor 6 is necessary and sufficient for alcoholic fatty liver disease in zebrafish. PLoS Genet. 2014, 10, e1004335. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Cao, L.; Du, J.; Jia, R.; Kitazawa, T.; Kubota, A.; Teraoka, H. Dexamethasone-induced hepatomegaly and steatosis in larval zebrafish. J. Toxicol. Sci. 2017, 42, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Katoch, S.; Patial, V. Zebrafish: An emerging model system to study liver diseases and related drug discovery. J. Appl. Toxicol. 2021, 41, 33–51. [Google Scholar] [CrossRef]
- den Broeder, M.J.; Ballangby, J.; Kamminga, L.M.; Aleström, P.; Legler, J.; Lindeman, L.C.; Kamstra, J.H. Inhibition of methyltransferase activity of enhancer of zeste 2 leads to enhanced lipid accumulation and altered chromatin status in zebrafish. Epigenet. Chromatin 2020, 13, 5. [Google Scholar] [CrossRef] [Green Version]
- Alunni, A.; Hermel, J.M.; Heuzé, A.; Bourrat, F.; Jamen, F.; Joly, J.S. Evidence for neural stem cells in the medaka optic tectum proliferation zones. Dev. Neurobiol. 2010, 70, 693–713. [Google Scholar] [CrossRef] [PubMed]
- MacPhail, R.C.; Brooks, J.; Hunter, D.L.; Padnos, B.; Irons, T.D.; Padilla, S. Locomotion in larval zebrafish: Influence of time of day, lighting and ethanol. Neurotoxicology 2009, 30, 52–58. [Google Scholar] [CrossRef]
- Schnörr, S.J.; Steenbergen, P.J.; Richardson, M.K.; Champagne, D.L. Measuring thigmotaxis in larval zebrafish. Behav. Brain Res. 2012, 228, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Douek, A.M.; Amiri Khabooshan, M.; Henry, J.; Stamatis, S.A.; Kreuder, F.; Ramm, G.; Änkö, M.L.; Wlodkowic, D.; Kaslin, J. An Engineered sgsh Mutant Zebrafish Recapitulates Molecular and Behavioural Pathobiology of Sanfilippo Syndrome A/MPS IIIA. Int. J. Mol. Sci. 2021, 22, 5948. [Google Scholar] [CrossRef] [PubMed]
- Koppens, M.A.; Bounova, G.; Gargiulo, G.; Tanger, E.; Janssen, H.; Cornelissen-Steijger, P.; Blom, M.; Song, J.Y.; Wessels, L.F.; van Lohuizen, M. Deletion of Polycomb Repressive Complex 2 from Mouse Intestine Causes Loss of Stem Cells. Gastroenterology 2016, 151, 684–697. [Google Scholar] [CrossRef] [Green Version]
- Lucore, E.C.; Connaughton, V.P. Observational learning and irreversible starvation in first-feeding zebrafish larvae: Is it okay to copy from your friends? Zoology 2021, 145, 125896. [Google Scholar] [CrossRef]
- Shen, X.; Liu, Y.; Hsu, Y.J.; Fujiwara, Y.; Kim, J.; Mao, X.; Yuan, G.C.; Orkin, S.H. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell 2008, 32, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Holder, M.; Grau, D.; Saldaña-Meyer, R.; Yu, J.R.; Ganai, R.A.; Zhang, J.; Wang, M.; LeRoy, G.; Dobenecker, M.W.; et al. Distinct Stimulatory Mechanisms Regulate the Catalytic Activity of Polycomb Repressive Complex 2. Mol. Cell 2018, 70, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stützer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda, S.; Mas, G.; Di Croce, L. Regulation of gene transcription by Polycomb proteins. Sci. Adv. 2015, 1, e1500737. [Google Scholar] [CrossRef] [Green Version]
- Le Faou, P.; Völkel, P.; Angrand, P.O. The zebrafish genes encoding the Polycomb repressive complex (PRC) 1. Gene 2011, 475, 10–21. [Google Scholar] [CrossRef]
- Van der Velden, Y.U.; Wang, L.; van Lohuizen, M.; Haramis, A.P. The Polycomb group protein Ring1b is essential for pectoral fin development. Development 2012, 139, 2210–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Velden, Y.U.; Wang, L.; Querol Cano, L.; Haramis, A.P. The polycomb group protein ring1b/rnf2 is specifically required for craniofacial development. PLoS ONE 2013, 8, e73997. [Google Scholar] [CrossRef] [Green Version]
- Rougeot, J.; Chrispijn, N.D.; Aben, M.; Elurbe, D.M.; Andralojc, K.M.; Murphy, P.J.; Jansen, P.W.T.C.; Vermeulen, M.; Cairns, B.R.; Kamminga, L.M. Maintenance of spatial gene expression by Polycomb-mediated repression after formation of a vertebrate body plan. Development 2019, 146, dev178590. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wang, M.; Chang, L.; Yu, J.; Song, A.; Liu, C.; Huang, W.; Zhang, T.; Wu, X.; Shen, X.; et al. RYBP/YAF2-PRC1 complexes and histone H1-dependent chromatin compaction mediate propagation of H2AK119ub1 during cell division. Nat. Cell Biol. 2020, 22, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef] [Green Version]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herz, H.M.; Morgan, M.; Gao, X.; Jackson, J.; Rickels, R.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Eissenberg, J.C.; Shilatifard, A. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 2014, 345, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venneti, S.; Garimella, M.T.; Sullivan, L.M.; Martinez, D.; Huse, J.T.; Heguy, A.; Santi, M.; Thompson, C.B.; Judkins, A.R. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 2013, 23, 558–564. [Google Scholar] [CrossRef] [Green Version]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537. [Google Scholar] [CrossRef] [Green Version]
- Ballester, L.Y.; Wang, Z.; Shandilya, S.; Miettinen, M.; Burger, P.C.; Eberhart, C.G.; Rodriguez, F.J.; Raabe, E.; Nazarian, J.; Warren, K.; et al. Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am. J. Surg. Pathol. 2013, 37, 1357–1364. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.S.; Sengupta, S.; Lee, K.; Hura, N.; Fuller, C.; DeWire, M.; Stevenson, C.B.; Fouladi, M.; Drissi, R. BMI-1 is a potential therapeutic target in diffuse intrinsic pontine glioma. Oncotarget 2017, 8, 62962–62975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakrishnan, I.; Danis, E.; Pierce, A.; Madhavan, K.; Wang, D.; Dahl, N.; Sanford, B.; Birks, D.K.; Davidson, N.; Metselaar, D.S.; et al. Senescence Induced by BMI1 Inhibition Is a Therapeutic Vulnerability in H3K27M-Mutant DIPG. Cell Rep. 2020, 33, 108286. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.S.; Gibson, W.T. EED-associated overgrowth in a second male patient. J. Hum. Genet. 2016, 61, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Sequerra Amram Cohen, A.; Gibson, W.T. EED-Related Overgrowth. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2019; pp. 1993–2021. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raby, L.; Völkel, P.; Hasanpour, S.; Cicero, J.; Toillon, R.-A.; Adriaenssens, E.; Van Seuningen, I.; Le Bourhis, X.; Angrand, P.-O. Loss of Polycomb Repressive Complex 2 Function Alters Digestive Organ Homeostasis and Neuronal Differentiation in Zebrafish. Cells 2021, 10, 3142. https://doi.org/10.3390/cells10113142
Raby L, Völkel P, Hasanpour S, Cicero J, Toillon R-A, Adriaenssens E, Van Seuningen I, Le Bourhis X, Angrand P-O. Loss of Polycomb Repressive Complex 2 Function Alters Digestive Organ Homeostasis and Neuronal Differentiation in Zebrafish. Cells. 2021; 10(11):3142. https://doi.org/10.3390/cells10113142
Chicago/Turabian StyleRaby, Ludivine, Pamela Völkel, Shaghayegh Hasanpour, Julien Cicero, Robert-Alain Toillon, Eric Adriaenssens, Isabelle Van Seuningen, Xuefen Le Bourhis, and Pierre-Olivier Angrand. 2021. "Loss of Polycomb Repressive Complex 2 Function Alters Digestive Organ Homeostasis and Neuronal Differentiation in Zebrafish" Cells 10, no. 11: 3142. https://doi.org/10.3390/cells10113142
APA StyleRaby, L., Völkel, P., Hasanpour, S., Cicero, J., Toillon, R.-A., Adriaenssens, E., Van Seuningen, I., Le Bourhis, X., & Angrand, P.-O. (2021). Loss of Polycomb Repressive Complex 2 Function Alters Digestive Organ Homeostasis and Neuronal Differentiation in Zebrafish. Cells, 10(11), 3142. https://doi.org/10.3390/cells10113142