
Early Intervention and Lifelong Treatment with GLP1 Receptor Agonist Liraglutide in a Wolfram Syndrome Rat Model with an Emphasis on Visual Neurodegeneration, Sensorineural Hearing Loss and Diabetic Phenotype

 ,
,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Long-Term (Prolonged) Treatment with GLP1 Receptor Agonist Liraglutide
2.3. Glucose Tolerance Tests (IPGTT)
2.4. Two-Hour-Postprandial Blood Sugar Test
2.5. In Vivo Magnetic Resonance Imaging
2.6. Hearing Evaluation
2.7. Visual Acuity Analysis
2.8. Cataract Scoring
2.9. Intraocular Pressure Scoring
2.10. Tissue Collection and Preparation
2.11. Retinal Wholemount Immunohistochemistry
2.12. Electron Microscopy
2.13. Morphometric Evaluation of Optic Nerve Fibers
2.14. Data Analysis
3. Results
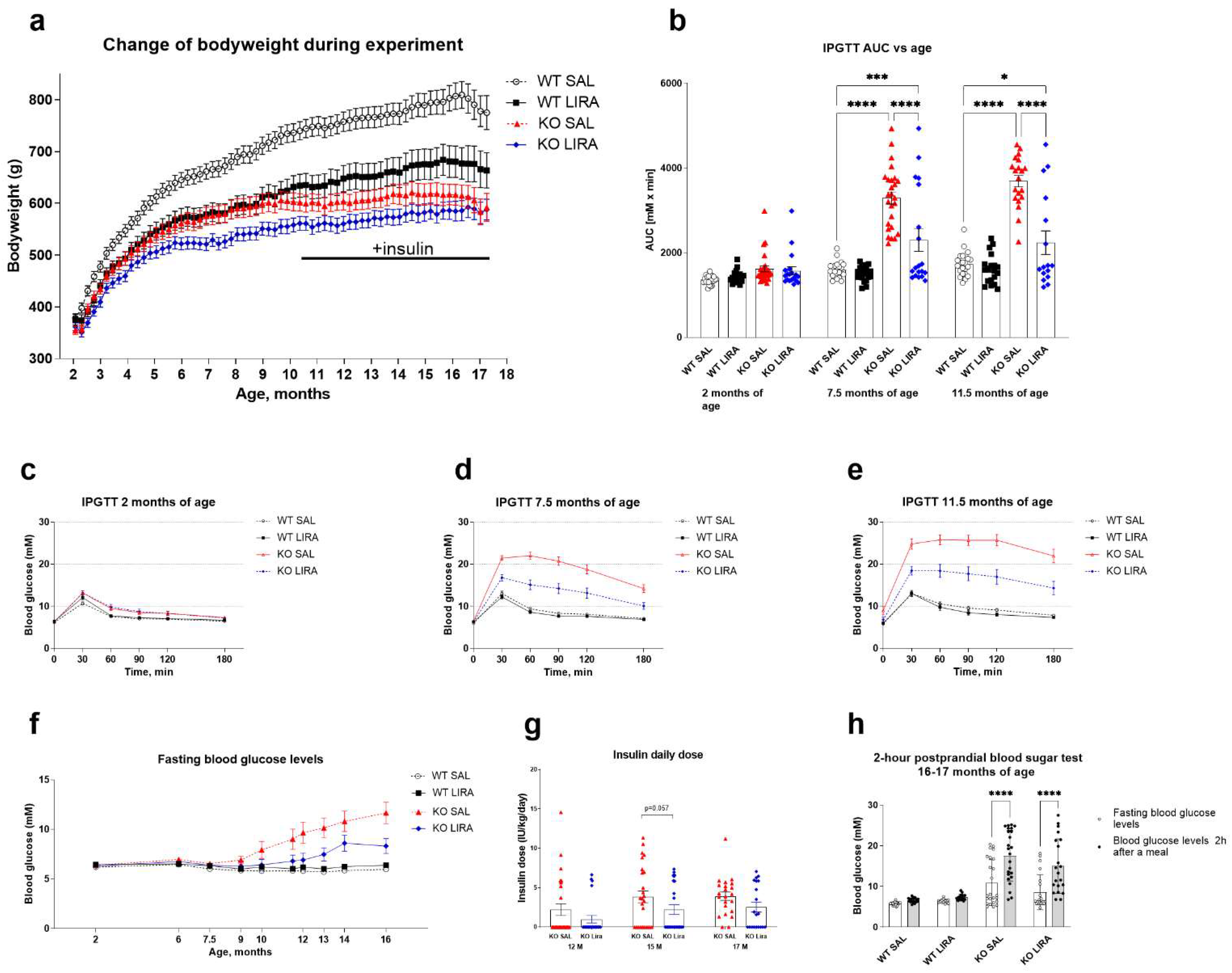
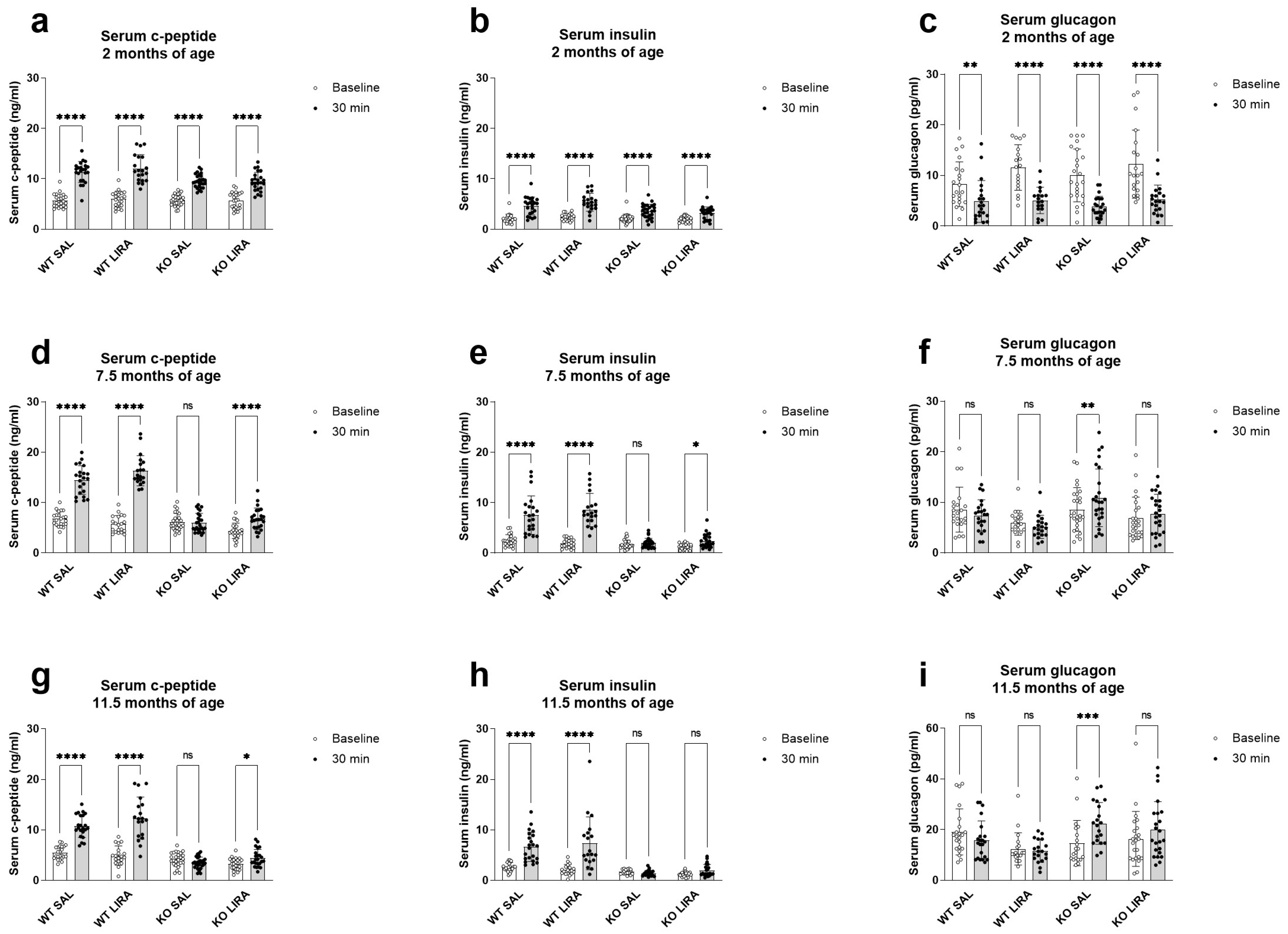
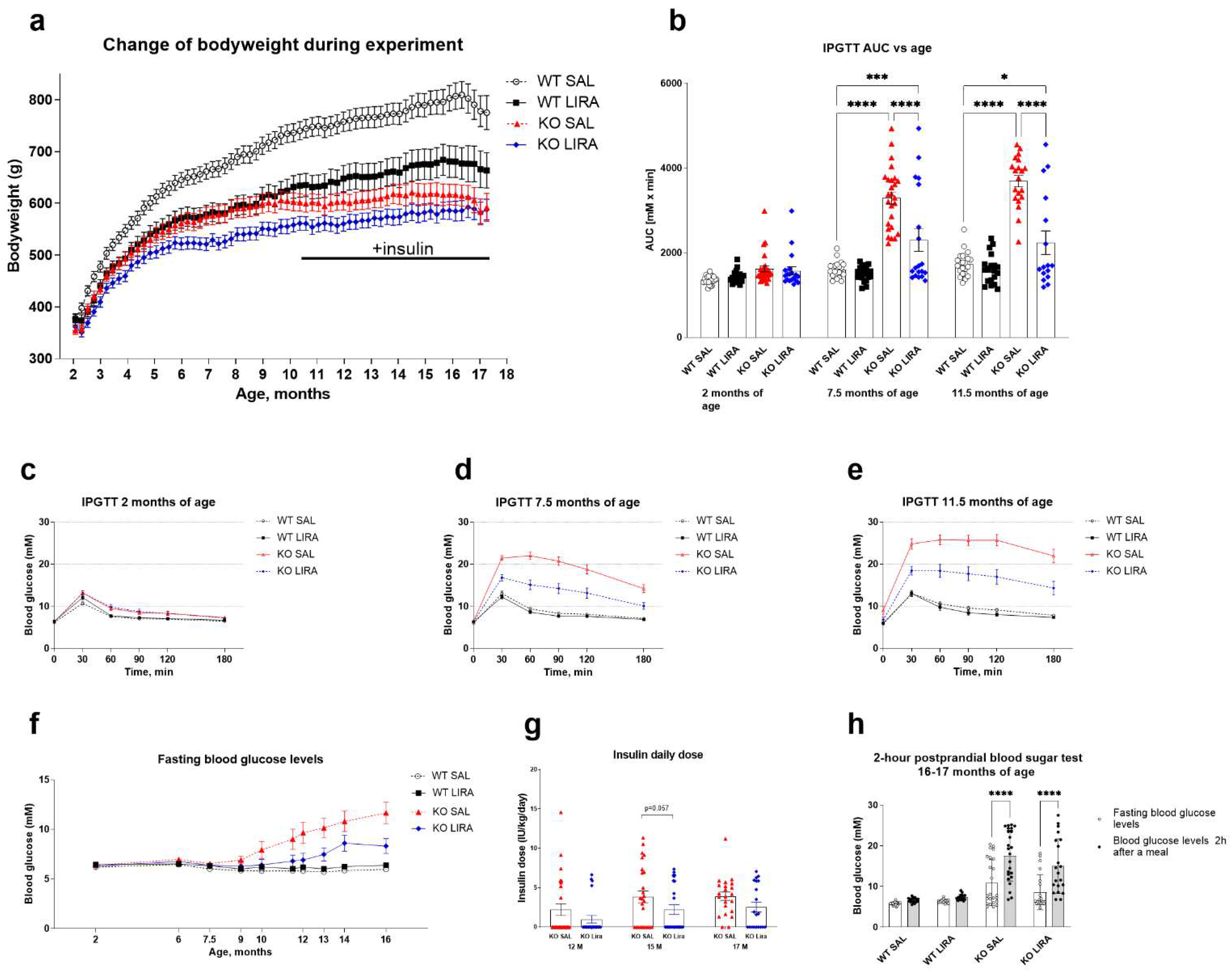
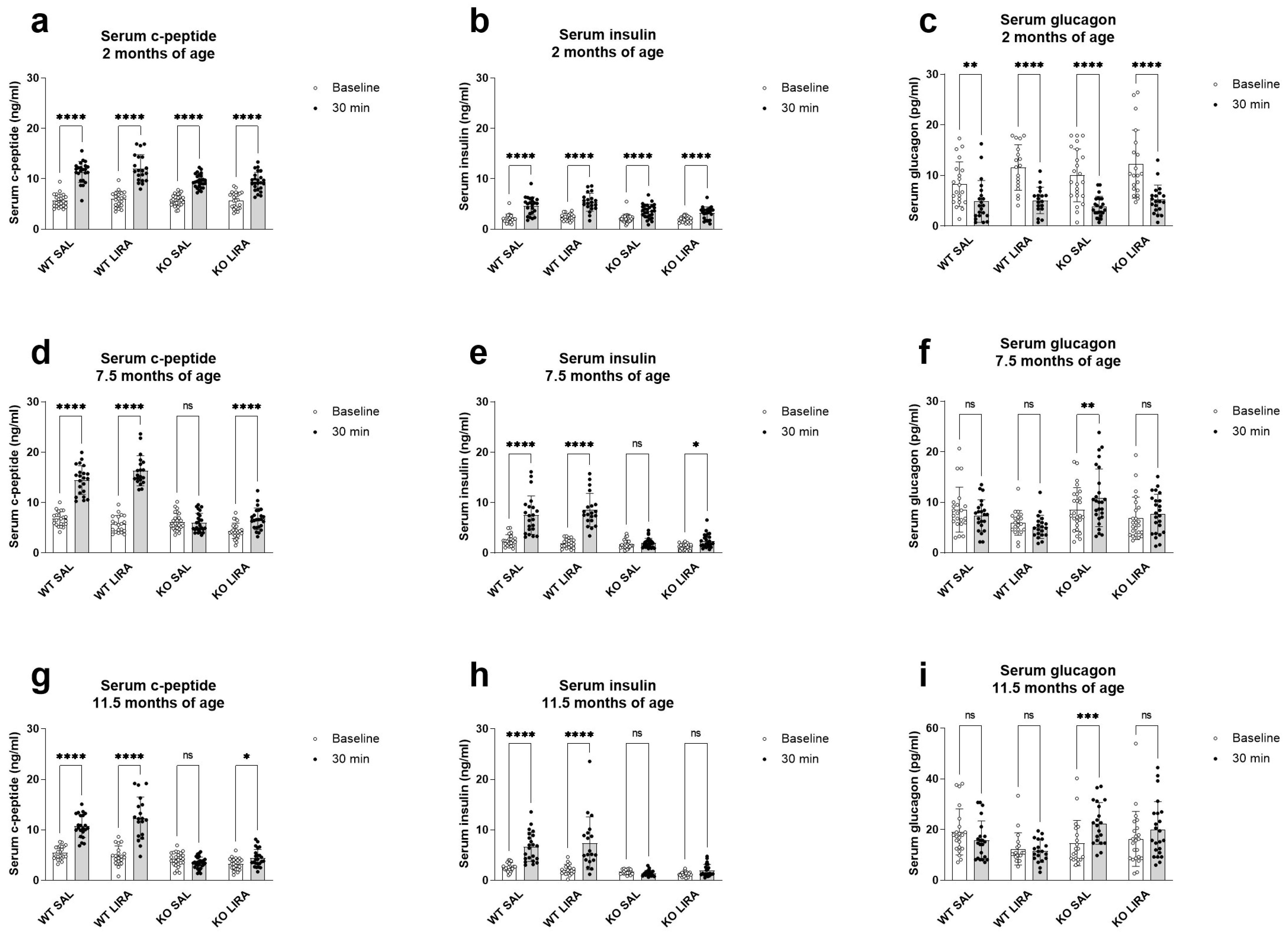
3.1. A 16-Month Treatment with Liraglutide Delays the Onset of Diabetes in Wfs1 KO Animals
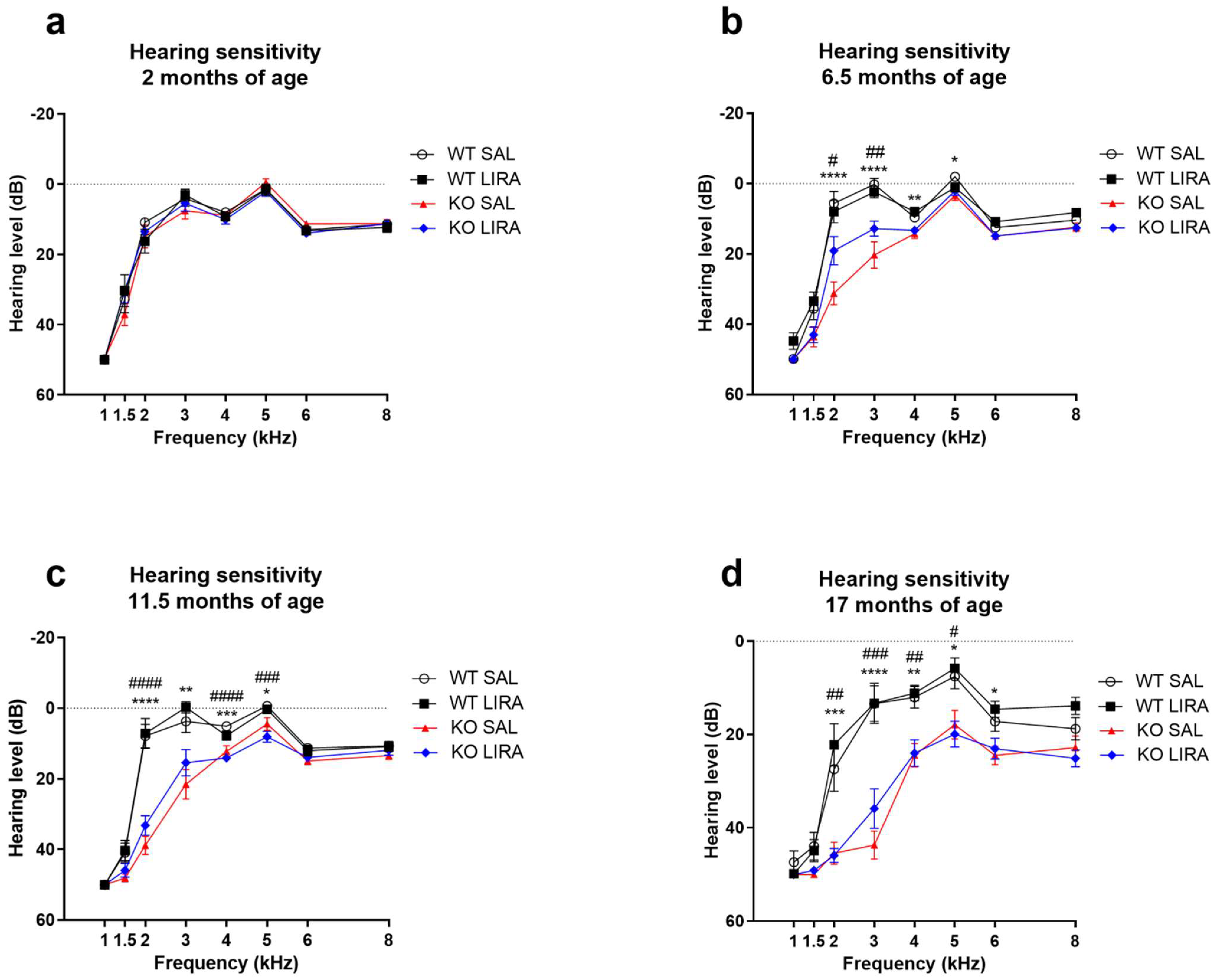
3.2. Liraglutide Treatment Did Not Prevent Progressive Sensorineural Hearing Loss of Wfs1 KO Rats
3.3. Sixteen-Month Treatment with Liraglutide Protects against Vision Loss in Wfs1 KO Animals
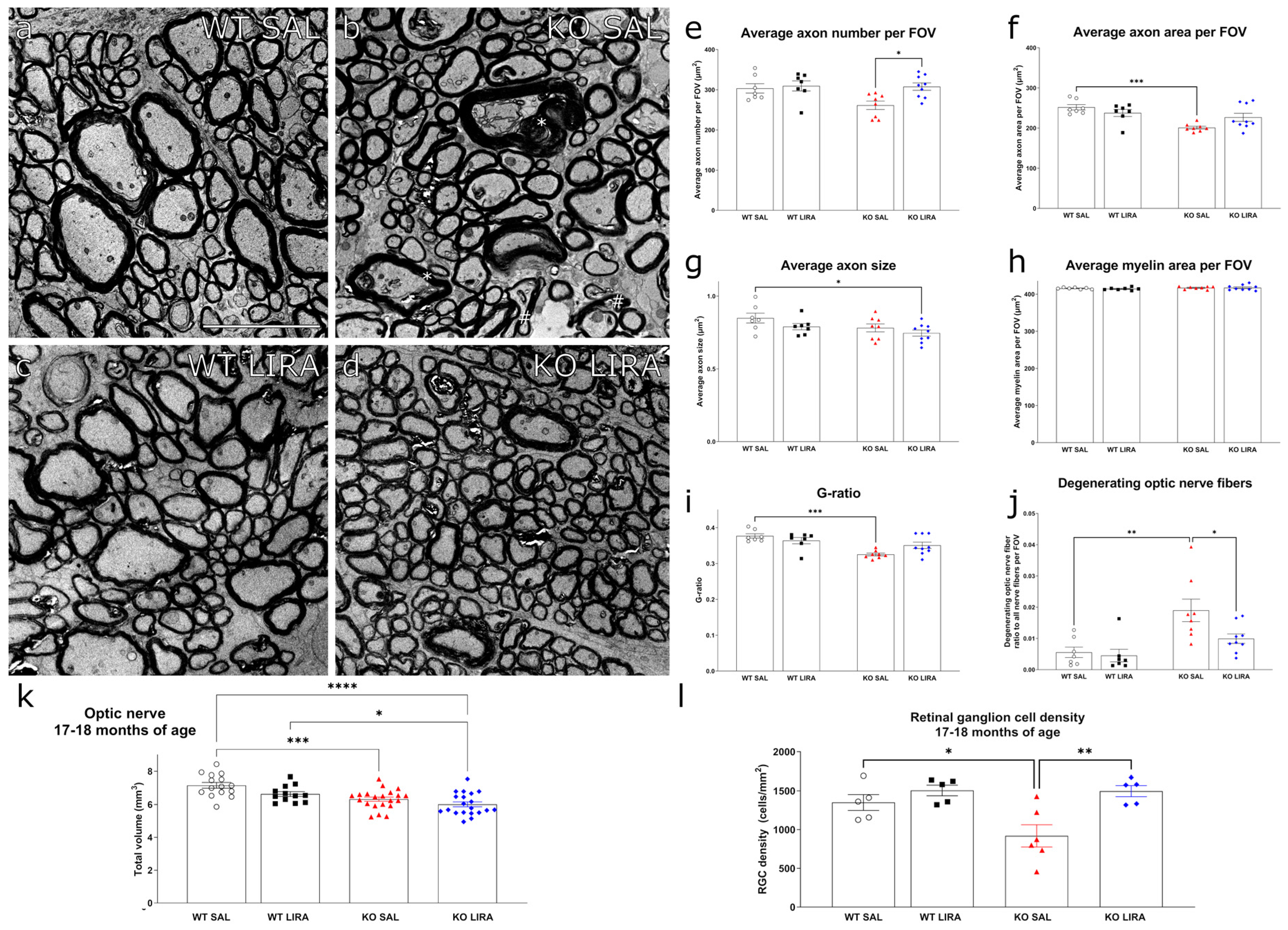
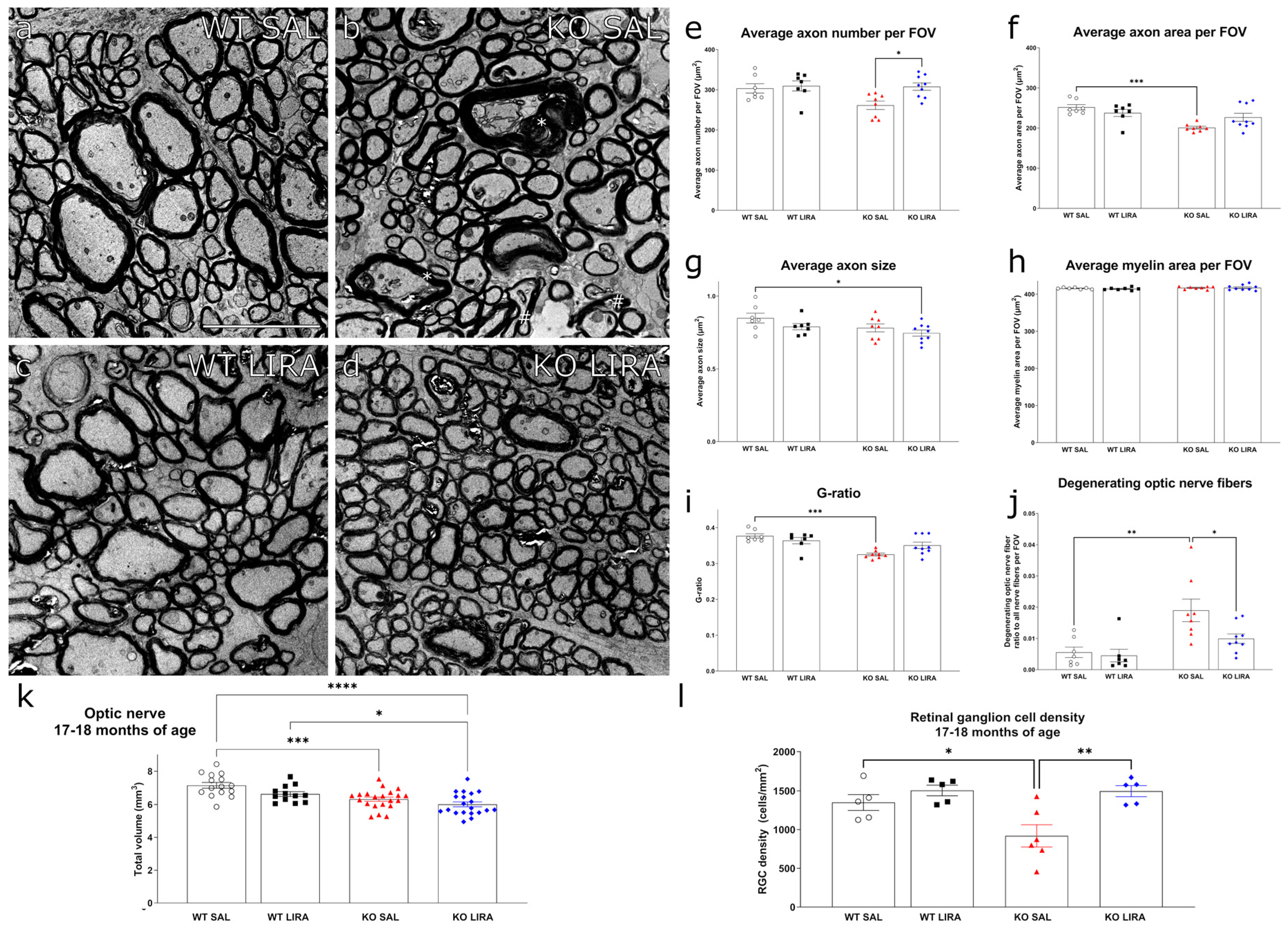
3.4. Sixteen-Month Treatment with Liraglutide Protected against the Development of Optic Nerve Atrophy in Wfs1 KO Animals
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barrett, T.G.; Bundey, S.E. Wolfram (DIDMOAD) syndrome. J. Med. Genet. 1997, 34, 838–841. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Bundey, S.E.; Macleod, A.F. (DIDMOAD) syndrome UK nationwide study of Wolfram. Lancet 1995, 346, 1458–1463. [Google Scholar] [CrossRef]
- Rigoli, L.; Lombardo, F.; Di Bella, C. Wolfram syndrome and WFS1 gene. Clin. Genet. 2011, 79, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Bueno, G.E.; Ruiz-Castañeda, D.; Martínez, J.R.; Muñoz, M.R.; Alascio, P.C. Natural history and clinical characteristics of 50 patients with Wolfram syndrome. Endocrine 2018, 61, 440–446. [Google Scholar] [CrossRef]
- Chaussenot, A.; Bannwarth, S.; Rouzier, C.; Vialettes, B.; Mkadem, S.A.E.I.; Chabrol, B.; Cano, A.; Labauge, P.; Paquis-Flucklinger, V. Neurologic features and genotype-phenotype correlation in Wolfram syndrome. Ann. Neurol. 2011, 69, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Li, Q.; Tong, A.L.; Mao, J.F.; Yu, M.; Yuan, T.; Chai, X.-F.; Gu, F. Clinical characteristics of wolfram syndrome in Chinese population and a novel frameshift mutation in WFS1. Front. Endocrinol. 2018, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Wilf-Yarkoni, A.; Shor, O.; Fellner, A.; Hellmann, M.A.; Pras, E.; Yonath, H.; Shkedi-Rafid, S.; Basel-Salmon, L.; Bazak, L.; Eliahou, R.; et al. Mild Phenotype of Wolfram Syndrome Associated With a Common Pathogenic Variant Is Predicted by a Structural Model of Wolframin. Neurol. Genet. 2021, 7, e578. [Google Scholar] [CrossRef] [PubMed]
- Urano, F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr. Diab. 2016, 16, 6. [Google Scholar] [CrossRef] [Green Version]
- Gharanei, S.; Zatyka, M.; Astuti, D.; Fenton, J.; Sik, A.; Nagy, Z.; Barrett, T.G. Vacuolar-type H+-ATPase V1A subunit is a molecular partner of Wolfram syndrome 1 (WFS1) protein, which regulates its expression and stability. Hum. Mol. Genet. 2013, 22, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Luuk, H.; Koks, S.; Plaas, M.; Hannibal, J.; Rehfeld, J.F.; Vasar, E. Distribution of Wfs1 protein in the central nervous system of the mouse and its relation to clinical symptoms of the Wolfram syndrome. J. Comp. Neurol. 2008, 509, 642–660. [Google Scholar] [CrossRef]
- Strom, T.M.; Hörtnagel, K.; Hofmann, S.; Gekeler, F.; Scharfe, C.; Rabl, W.; Gerbitz, K.D.; Meitinger, T. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum. Mol. Genet. 1998, 7, 2021–2028. [Google Scholar] [CrossRef]
- Suzuki, N.; Hosoya, M.; Oishi, N.; Okano, H.; Fujioka, M.; Ogawa, K. Expression pattern of wolframin, the WFS1 (Wolfram syndrome-1 gene) product, in common marmoset (Callithrix jacchus) cochlea. Neuroreport 2016, 27, 833–836. [Google Scholar] [CrossRef] [PubMed]
- Bonnet Wersinger, D.; Benkafadar, N.; Jagodzinska, J.; Hamel, C.; Tanizawa, Y.; Lenaers, G.; Delettre, C. Impairment of visual function and retinal ER stress activation in Wfs1-deficient mice. PLoS ONE 2014, 9, e97222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, S.G.; Fukuma, M.; Lipson, K.L.; Nguyen, L.X.; Allen, J.R.; Oka, Y.; Urano, F. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pacreatic β-cells. J. Biol. Chem. 2005, 280, 39609–39615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, D.; Ishihara, H.; Yamaguchi, S.; Yamada, T.; Tamura, A.; Katagiri, H.; Maruyama, Y.; Oka, Y. WFS1 protein modulates the free Ca2+ concentration in the endoplasmic reticulum. FEBS Lett. 2006, 580, 5635–5640. [Google Scholar] [CrossRef] [Green Version]
- Cagalinec, M.; Liiv, M.; Hodurova, Z.; Hickey, M.A.; Vaarmann, A.; Mandel, M.; Zeb, A.; Choubey, V.; Kuum, M.; Safiulina, D. Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome. PLoS Biol. 2016, 14, e1002511. [Google Scholar] [CrossRef]
- Delprat, B.; Maurice, T.; Delettre, C. Wolfram syndrome: MAMs’ connection? Review-article. Cell Death Dis. 2018, 9, 364. [Google Scholar] [CrossRef] [Green Version]
- Plaas, M.; Seppa, K.; Reimets, R.; Jagomäe, T.; Toots, M.; Koppel, T.; Vallisoo, T.; Nigul, M.; Heinla, I.; Meier, R.; et al. Wfs1-deficient rats develop primary symptoms of Wolfram syndrome: Insulin-dependent diabetes, optic nerve atrophy and medullary degeneration. Sci. Rep. 2017, 7, 10220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seppa, K.; Toots, M.; Reimets, R.; Jagomäe, T.; Koppel, T.; Pallase, M.; Hasselholt, S.; Mikkelsen, M.K.; Nyengaard, J.R.; Vasar, E.; et al. GLP-1 receptor agonist liraglutide has a neuroprotective effect on an aged rat model of Wolfram syndrome. Sci. Rep. 2019, 9, 15742. [Google Scholar] [CrossRef] [PubMed]
- Seppa, K.; Jagomäe, T.; Kukker, K.G.; Reimets, R.; Pastak, M.; Vasar, E.; Terasmaa, A.; Plaas, M. Liraglutide, 7,8-DHF and their co-treatment prevents loss of vision and cognitive decline in a Wolfram syndrome rat model. Sci. Rep. 2021, 11, 2275. [Google Scholar] [CrossRef]
- Toots, M.; Seppa, K.; Jagomäe, T.; Koppel, T.; Pallase, M.; Heinla, I.; Terasmaa, A.; Plaas, M.; Vasar, E. Preventive treatment with liraglutide protects against development of glucose intolerance in a rat model of Wolfram syndrome. Sci. Rep. 2018, 8, 10183. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, L.; Kaur, R.; Kumar, R.; Kumar, M.; Palta, K. GLP-1 Receptor Agonists in Type 2 Diabetes Mellitus. Curr. Diabetes Rev. 2020, 16, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Yusta, B.; Baggio, L.L.; Estall, J.L.; Koehler, J.A.; Holland, D.P.; Li, H.; Pipeleers, D.; Ling, Z.; Drucker, D.J. GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006, 4, 391–406. [Google Scholar] [CrossRef] [Green Version]
- Sedman, T.; Rünkorg, K.; Krass, M.; Luuk, H.; Vasar, E.; Volke, V. Exenatide Is an Effective Antihyperglycaemic Agent in a Mouse Model of Wolfram Syndrome. J. Diabetes. 2016, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, M.; Tanabe, K.; Amo-Shiinoki, K.; Hatanaka, M.; Morii, T.; Takahashi, H.; Seino, S.; Yamada, Y.; Tanizawa, Y. Activation of GLP-1 receptor signalling alleviates cellular stresses and improves beta cell function in a mouse model of Wolfram syndrome. Diabetologia 2018, 61, 2189–2201. [Google Scholar] [CrossRef] [Green Version]
- Scully, K.J.; Wolfsdorf, J.I. Efficacy of GLP-1 Agonist Therapy in Autosomal Dominant WFS1-Related Disorder: A Case Report. Horm. Res. Paediatr. 2021, 93, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Luippold, G.; Bedenik, J.; Voigt, A.; Grempler, R. Short-and Longterm Glycemic Control of Streptozotocin-Induced Diabetic Rats Using Different Insulin Preparations. PLoS ONE 2016, 11, e0156346. [Google Scholar] [CrossRef]
- Nordquist, L.; Sjöquist, M. Improvement of insulin response in the streptozotocin model of insulin-dependent diabetes mellitus. Insulin response with and without a long-acting insulin treatment. Animal 2009, 3, 685–689. [Google Scholar] [CrossRef]
- Chandra, M.; Riley, M.G.I.; Johnson, D.E. Spontaneous Neoplasms in Aged Sprague-Dawley Rats. Arch. Toxicol. 1992, 20, 327–340. [Google Scholar] [CrossRef]
- Kemp, D.T. Stimulated acoustic emissions from within the human auditory system. J. Acoust Soc. Am. 1978, 64, 1386–1391. [Google Scholar] [CrossRef]
- Kemp, D.T. Otoacoustic emissions, their origin in cochlear function, and use. Br. Med. Bull. 2002, 63, 223–241. [Google Scholar] [CrossRef]
- Pathme GmbH Whitepaper. Available online: https://www.pathme.de/download/whitepapers/Whitepaper_Short_RelationBetweenDPOAEs_and_behavioural_pure_tone_threshold.pdf (accessed on 6 October 2021).
- Sheppard, A.M.; Zhao, D.L.; Salvi, R. Isoflurane anesthesia suppresses distortion product otoacoustic emissions in rats. J. Otol. 2018, 13, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Livesey, J.C.; Wiens, L.W.; von Seggern, D.J.; Barlow, W.E.; Arnold, A. Inhibition of Radiation Cataractogenesis by WR-77913. Radiat. Res. 2014, 141, 99–104. [Google Scholar] [CrossRef]
- Nadal-Nicolás, F.M.; Jiménez-López, M.; Sobrado-Calvo, P.; Nieto-López, L.; Cánovas-Martinez, I.; Salinas-Navarro, M.; Vidal-Sanz, M.; Agudo, M. Brn3a as a marker of retinal ganglion cells: Qualitative and quantitative time course studies in naïve and optic nerve-injured retinas. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3860–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geeraerts, E.; Dekeyster, E.; Gaublomme, D.; Salinas-Navarro, M.; De Groef, L.; Moons, L. A freely available semi-automated method for quantifying retinal ganglion cells in entire retinal flatmounts. Exp. Eye Res. 2016, 147, 105–113. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carrera, I.; Frise, E.; Verena, K.; Mark, L.; Tobias, P.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji—An Open platform for biological image analysis. Nat. Methods 2009, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seynaeve, H.; Vermeiren, A.; Leys, A.; Dralands, L. Four cases of Wolfram syndrome: Ophthalmologic findings and complications. Bull. Soc. Belge Ophtalmol. 1994, 252, 75–80. [Google Scholar]
- De Heredia, M.L.; Clèries, R.; Nunes, V. Genotypic classification of patients with Wolfram syndrome: Insights into the natural history of the disease and correlation with phenotype. Genet. Med. 2013, 15, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Front. Endocrinol. 2019, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Falco, M.; Manente, L.; Lucariello, A.; Baldi, G.; Fiore, P.; Laforgia, P.; Baldi, A.; Iannaccone, A.; De Luca, A. Localization and distribution of wolframin in human tissues. Front. Biosci. 2012, 4, 1986–1998. [Google Scholar] [CrossRef]
- Sedman, T.; Krass, M.; Rünkorg, K.; Vasar, E.; Volke, V. Tolerance develops toward GLP-1 receptor agonists’ glucose-lowering effect in mice. Eur. J. Pharmacol. 2020, 885, 173443. [Google Scholar] [CrossRef]
- Katsuda, Y.; Sasase, T.; Tadaki, H.; Mera, Y. Contribution of hyperglycemia on diabetic complications in obese type 2 diabetic SDT fatty rats: Effects of SGLT inhibitor phlorizin. Exp. Anim. 2015, 64, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Degn, K.B.; Juhl, C.L.B.; Sturis, J.; Jakobsen, G.; Brock, B.; Chandramouli, V. One Week’s Treatment with the Long-Acting Glucose Release in Patients with Type 2 Diabetes. Diabetes 2004, 53, 1187–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harder, H.; Mscodont, N.L.; Thi, T.D.T.; Astrup, A. The Effect of Liraglutide, a Long-Acting 24-h Energy Expenditure in Patients Glycemic Control, Body Composition, and Glucagon-Like Peptide 1 Derivative, on with Type 2 Diabetes. Diabetes Care 2004, 27, 1915–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karzon, R.; Narayanan, A.; Chen, L.; Lieu, J.E.C.; Hershey, T. Longitudinal hearing loss in Wolfram syndrome. Orphanet J. Rare Dis. 2018, 13, 102. [Google Scholar] [CrossRef]
- Abdala, C.; Visser-Dumont, L. Distortion product otoacoustic emissions: A tool for hearing assessment and scientific study. Volta Rev. 2001, 103, 281–302. [Google Scholar] [PubMed]
- Eiberg, H.; Hansen, L.; Kjer, B.; Hansen, T.; Pedersen, O.; Bille, M.; Rosenberg, T.; Tranebjaerg, L. Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene. J. Med. Genet. 2006, 43, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Cheng, J.; Lu, Y.; Li, J.; Lu, Y.; Jin, Z.; Dai, P.; Wang, R.; Yuan, H. Identification of two novel missense WFS1 mutations, H696Y and R703H, in patients with non-syndromic low-frequency sensorineural hearing loss. J. Genet. Genom. 2011, 38, 71–76. [Google Scholar] [CrossRef]
- Lesperance, M.M.; Hall, J.W.; San Agustin, T.B.; Leal, S.M. Mutations in the Wolfram syndrome type 1 gene (WFS1) define a clinical entity of dominant low-frequency sensorineural hearing loss. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Escabi, C.D.; Frye, M.D.; Trevino, M.; Lobarinas, E. The rat animal model for noise-induced hearing loss. J. Acoust. Soc. Am. 2019, 146, 3692–3709. [Google Scholar] [CrossRef] [Green Version]
- Hilson, J.B.; Merchant, S.N.; Adams, J.C.; Joseph, J.T. Wolfram syndrome: A clinicopathologic correlation. Acta Neuropathol. 2009, 118, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Hoekel, J.; Chisholm, S.A.; Al-Lozi, A.; Hershey, T.; Earhart, G.; Hullar, T.; Tychsen, L.; Washington University Wolfram Study Group. Ophthalmologic correlates of disease severity in children and adolescents with Wolfram syndrome. J. Am. Assoc. Pediatric Ophthalmol. Strabismus 2014, 18, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Genís, D.; Dávalos, A.; Molins, A.; Ferrer, I. Wolfram syndrome: A neuropathological study. Acta Neuropathol. 1997, 93, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Zmyslowska, A.; Waszczykowska, A.; Baranska, D.; Stawiski, K.; Borowiec, M.; Jurowski, P.; Fendler, W.; Mlynarski, W. Optical coherence tomography and magnetic resonance imaging visual pathway evaluation in Wolfram syndrome. Dev. Med. Child Neurol. 2019, 61, 359–365. [Google Scholar] [CrossRef]
- Samara, A.; Lugar, H.M.; Hershey, T.; Shimony, J.S. Longitudinal assessment of neuroradiologic features in wolfram syndrome. Am. J. Neuroradiol. 2020, 41, 2364–2369. [Google Scholar] [CrossRef] [PubMed]
- Pierre, C.; Serge, P.; Raymond, R.; Johan, A.; Jose, S. The optomotor response: A robust first-line visual screening method for mice. Vis. Res. 2005, 45, 1439–1446. [Google Scholar]
- Coulter, J.B.; Eaton, D.K.; Marr, L.K. Effects of diabetes and insulin treatment on sorbitol and water of rat lenses. Ophthalmic Res. 1986, 18, 357–362. [Google Scholar] [CrossRef]
- Gault, V.A.; Hölscher, C. GLP-1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. Eur. J. Pharmacol. 2008, 587, 112–117. [Google Scholar] [CrossRef]
- Perry, T.A.; Holloway, H.W.; Weerasuriya, A.; Mouton, P.R.; Duffy, K.; Mattison, J.A.; Greig, N.H. Evidence of GLP-1-mediated neuroprotection in an animal model of pyridoxine-induced peripheral sensory neuropathy. Exp. Neurol. 2007, 203, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Maskery, M.P.; Holscher, C.; Jones, S.P.; Price, C.I.; Strain, W.D.; Watkins, C.L.; Werring, D.J.; Emsley, H.C.A. Glucagon-like peptide-1 receptor agonists as neuroprotective agents for ischemic stroke: A systematic scoping review. J. Cereb. Blood Flow Metab. 2021, 41, 14–30. [Google Scholar] [CrossRef]
- Hölscher, C. Potential Role of Glucagon-Like Peptide-1 (GLP-1) in Neuroprotection. CNS Drugs 2012, 26, 871–882. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jagomäe, T.; Seppa, K.; Reimets, R.; Pastak, M.; Plaas, M.; Hickey, M.A.; Kukker, K.G.; Moons, L.; De Groef, L.; Vasar, E.; et al. Early Intervention and Lifelong Treatment with GLP1 Receptor Agonist Liraglutide in a Wolfram Syndrome Rat Model with an Emphasis on Visual Neurodegeneration, Sensorineural Hearing Loss and Diabetic Phenotype. Cells 2021, 10, 3193. https://doi.org/10.3390/cells10113193
Jagomäe T, Seppa K, Reimets R, Pastak M, Plaas M, Hickey MA, Kukker KG, Moons L, De Groef L, Vasar E, et al. Early Intervention and Lifelong Treatment with GLP1 Receptor Agonist Liraglutide in a Wolfram Syndrome Rat Model with an Emphasis on Visual Neurodegeneration, Sensorineural Hearing Loss and Diabetic Phenotype. Cells. 2021; 10(11):3193. https://doi.org/10.3390/cells10113193
Chicago/Turabian StyleJagomäe, Toomas, Kadri Seppa, Riin Reimets, Marko Pastak, Mihkel Plaas, Miriam A. Hickey, Kaia Grete Kukker, Lieve Moons, Lies De Groef, Eero Vasar, and et al. 2021. "Early Intervention and Lifelong Treatment with GLP1 Receptor Agonist Liraglutide in a Wolfram Syndrome Rat Model with an Emphasis on Visual Neurodegeneration, Sensorineural Hearing Loss and Diabetic Phenotype" Cells 10, no. 11: 3193. https://doi.org/10.3390/cells10113193
APA StyleJagomäe, T., Seppa, K., Reimets, R., Pastak, M., Plaas, M., Hickey, M. A., Kukker, K. G., Moons, L., De Groef, L., Vasar, E., Kaasik, A., Terasmaa, A., & Plaas, M. (2021). Early Intervention and Lifelong Treatment with GLP1 Receptor Agonist Liraglutide in a Wolfram Syndrome Rat Model with an Emphasis on Visual Neurodegeneration, Sensorineural Hearing Loss and Diabetic Phenotype. Cells, 10(11), 3193. https://doi.org/10.3390/cells10113193