
Electrophysiological Consequences of Cardiac Fibrosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
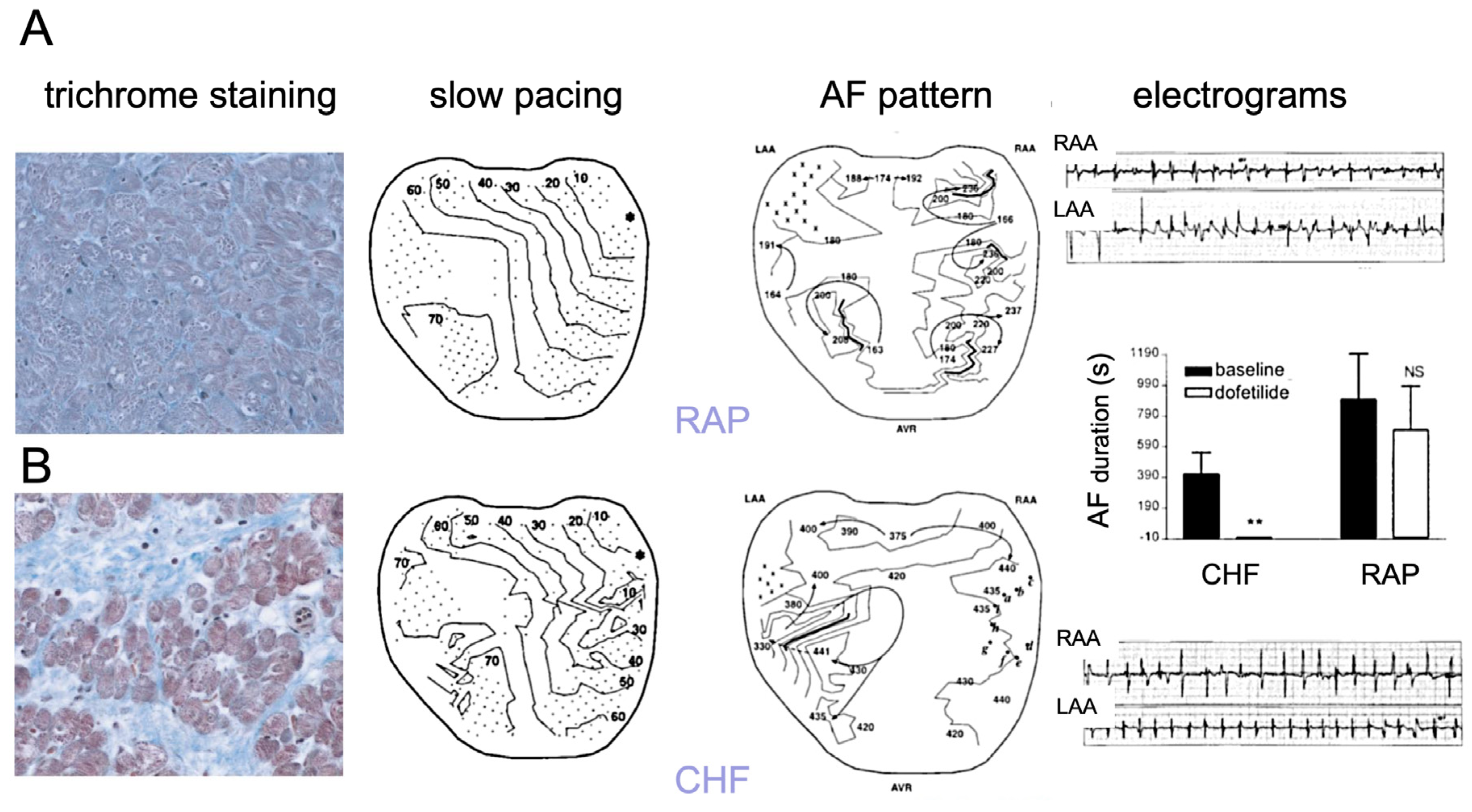{kind=link}
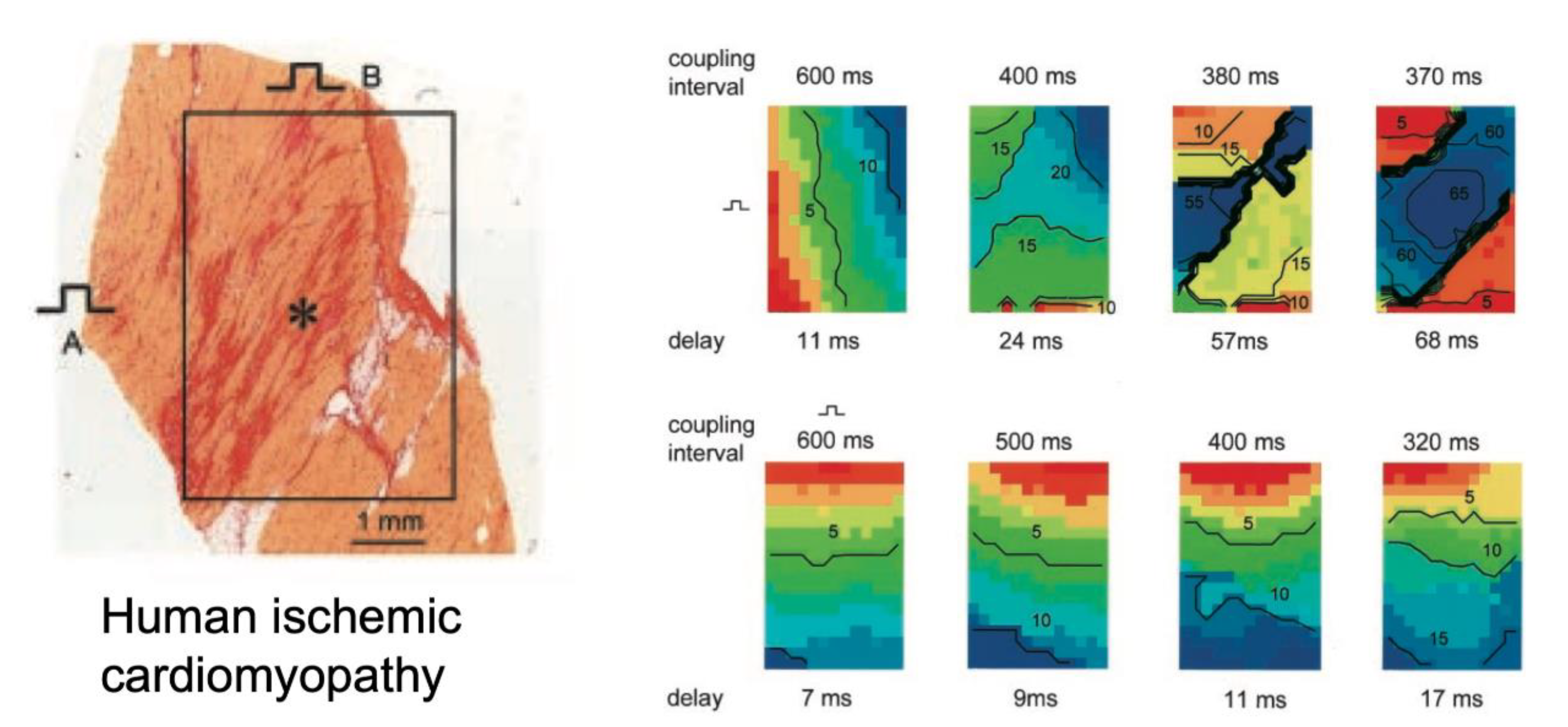{kind=link}
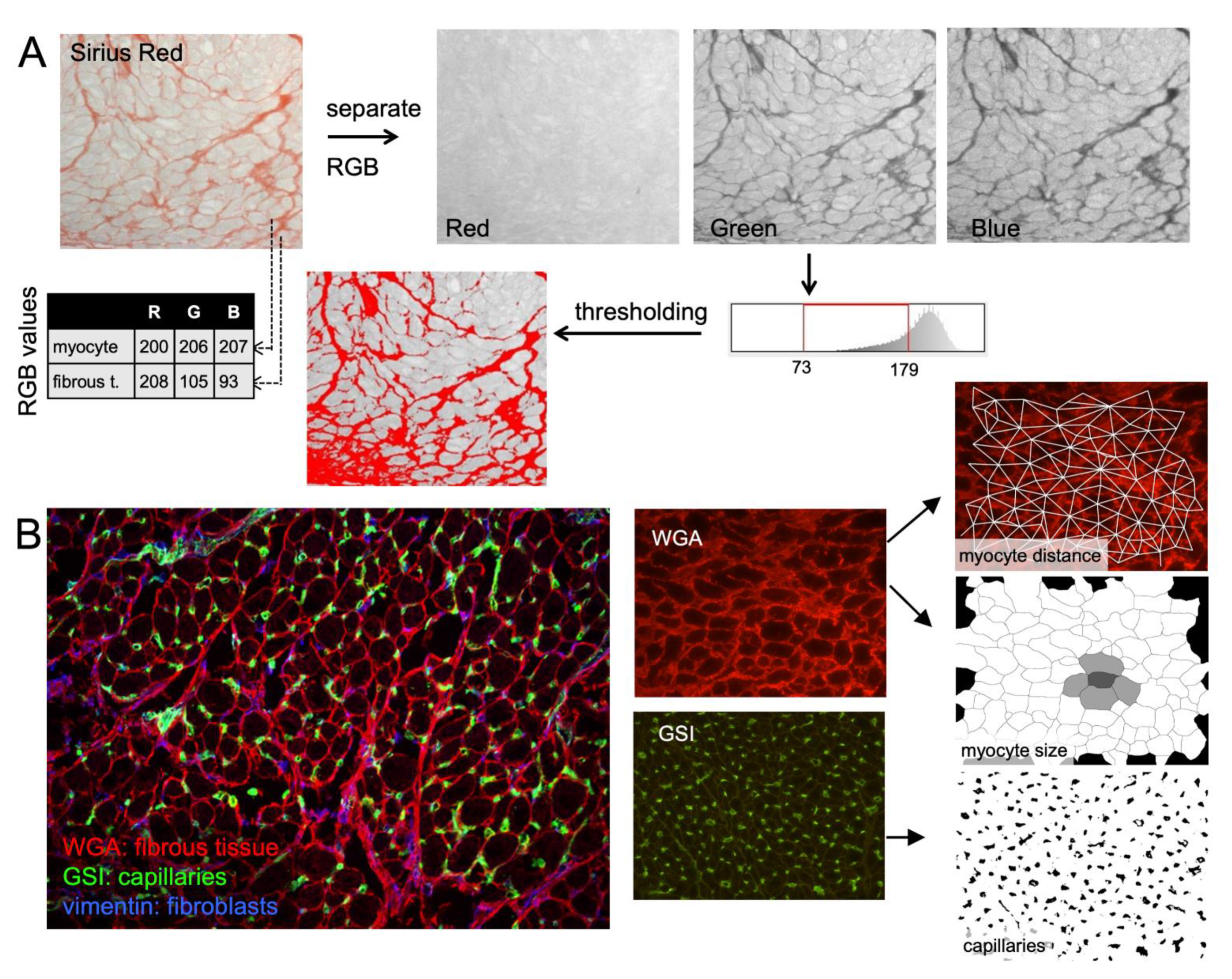{kind=link}
Abstract
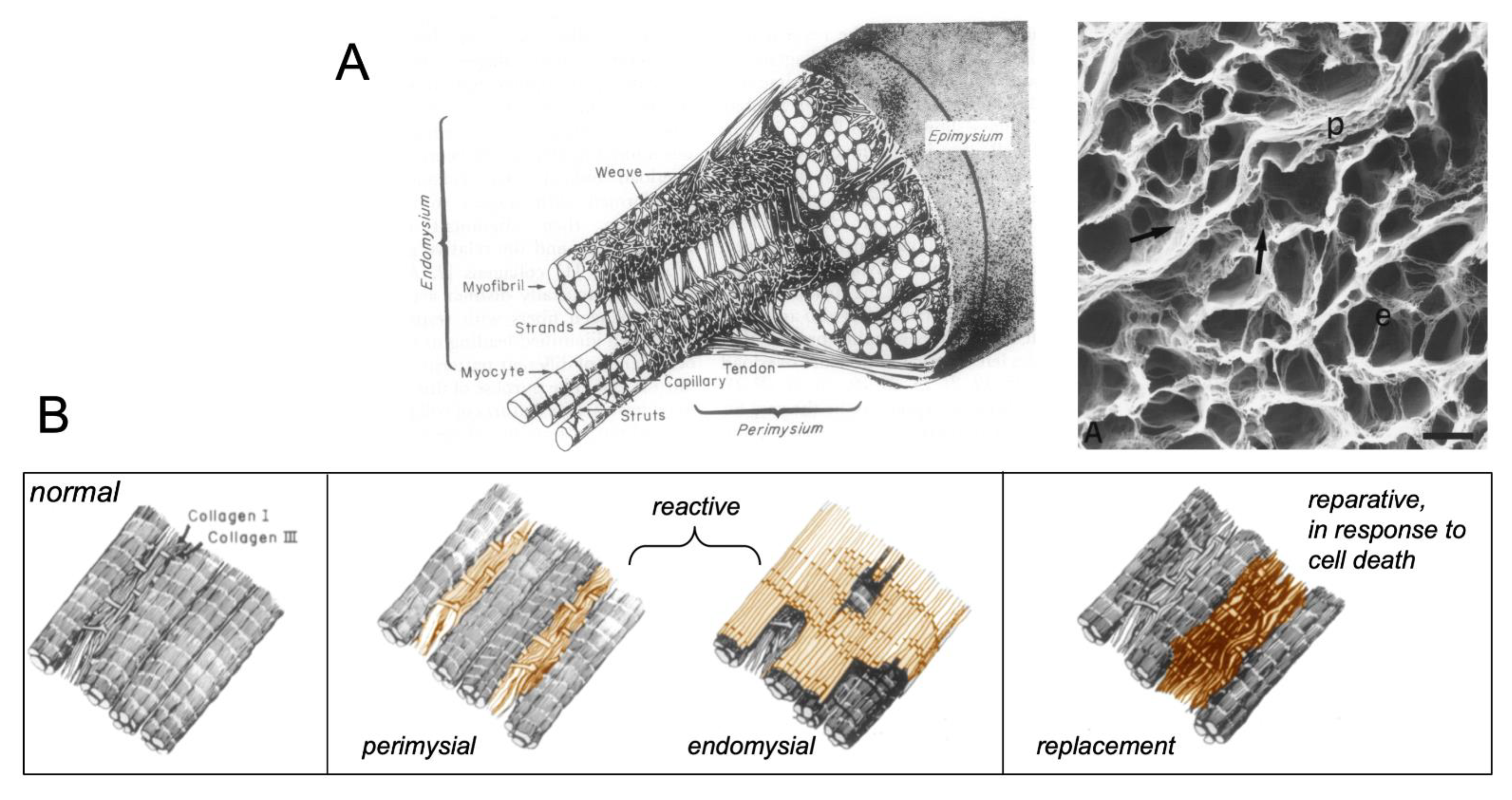
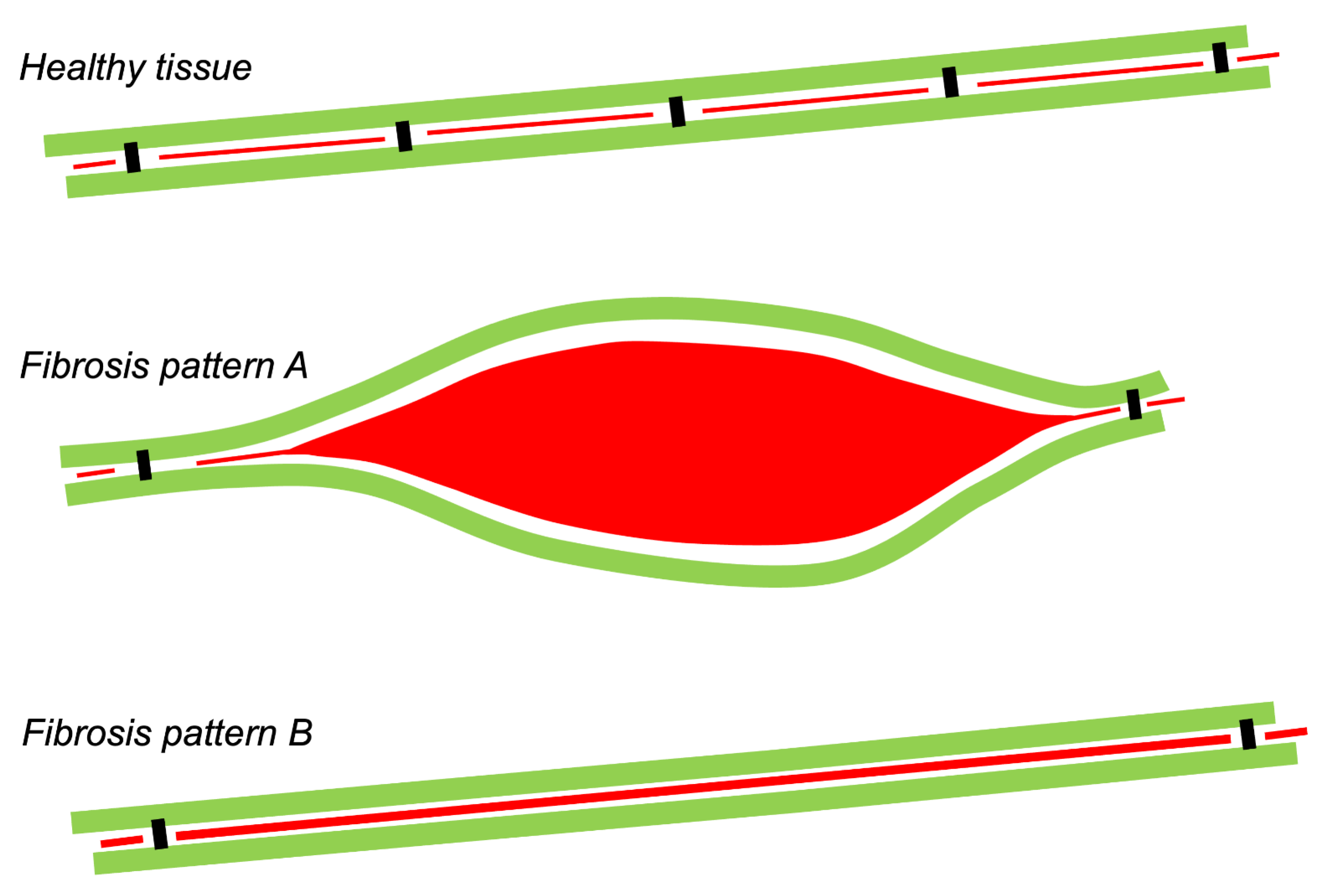
:1. Types of Fibrous Tissue and Fibrosis
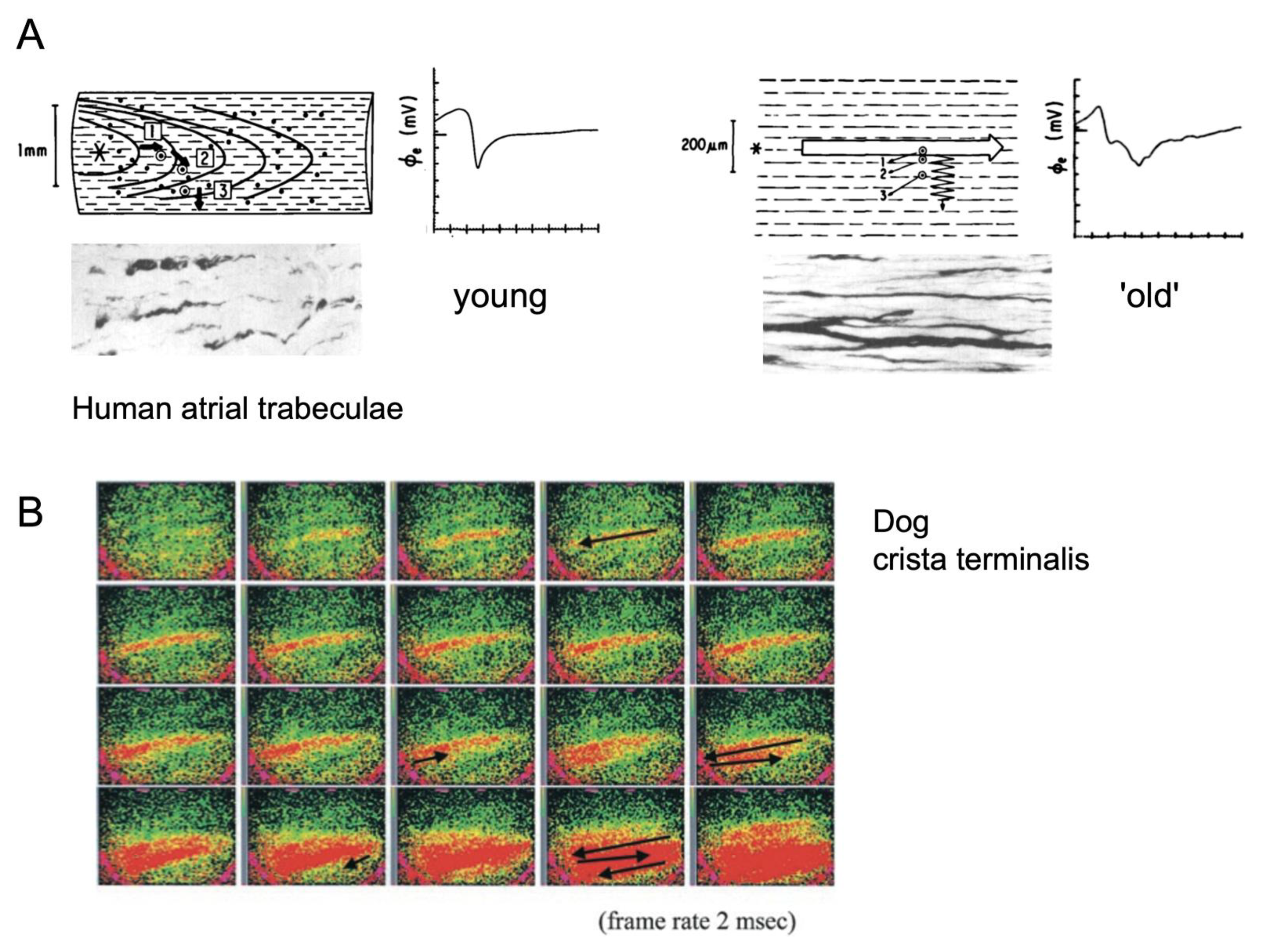
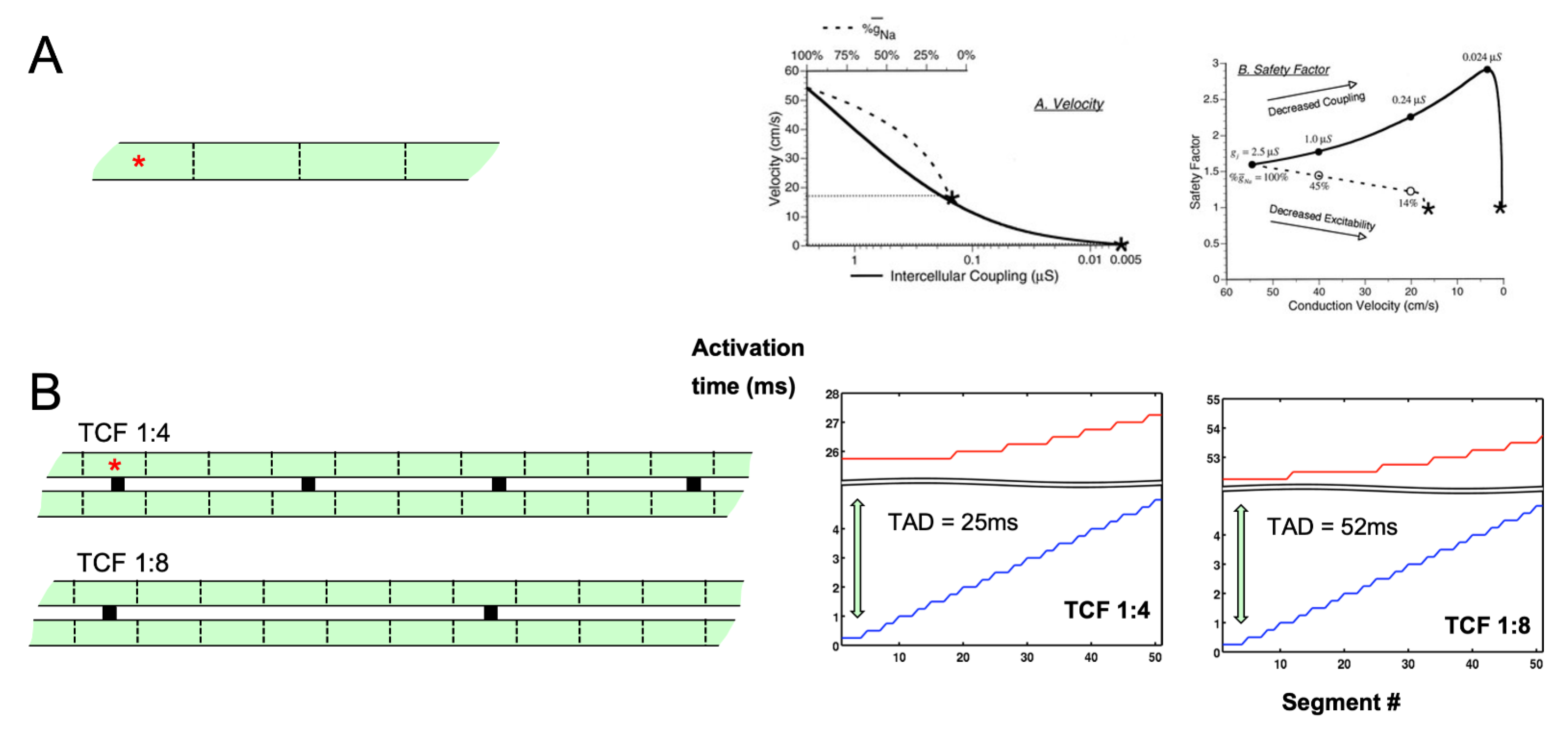
2. Electrical Connections and Propagation
3. Fibrosis as a Proarrhythmic Factor
4. Fibrosis and Ectopy
5. Fibrosis and Reentrant Activity
6. Association of Fibrosis with Arrhythmogenesis in Patients and Animal Models
7. Direct Correlation of Conduction Abnormalities to Fibrosis
8. Confounding Factors in Determining the Impact of Fibrosis on Propagation
8.1. Fatty Infiltration
8.2. Myocyte Hypertrophy
8.3. Connexin Expression
8.4. Fibroblast Density
8.5. Electrical Remodeling
9. Histological Quantification of Fibrosis
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weber, K.T.; Pick, R.; Jalil, J.E.; Janicki, J.S.; Carroll, E.P. Patterns of myocardial fibrosis. J. Mol. Cell. Cardiol. 1989, 21 (Suppl. S5), 121–131. [Google Scholar] [PubMed]
- Weber, K.T.; Sun, Y.; Tyagi, S.C.; Cleutjens, J.P. Collagen network of the myocardium: Function, structural remodeling and regulatory mechanisms. J. Mol. Cell. Cardiol. 1994, 26, 279–292. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Aoki, T.; Fukumoto, Y.; Shimokawa, H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J. Cardiol. 2012, 60, 416–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, F.Z.; Chu, P.Y.; Ziemann, M.; Kaspi, A.; Kiriazis, H.; Du, X.J.; El-Osta, A.; Kaye, D.M. Age-Related Differential Structural and Transcriptomic Responses in the Hypertensive Heart. Front. Physiol. 2018, 9, 817. [Google Scholar] [CrossRef]
- Dhein, S.; Salameh, A. Remodeling of Cardiac Gap Junctional Cell-Cell Coupling. Cells 2021, 10, 2422. [Google Scholar] [CrossRef]
- Koura, T.; Hara, M.; Takeuchi, S.; Ota, K.; Okada, Y.; Miyoshi, S.; Watanabe, A.; Shiraiwa, K.; Mitamura, H.; Kodama, I.; et al. Anisotropic conduction properties in canine atria analyzed by high-resolution optical mapping: Preferential direction of conduction block changes from longitudinal to transverse with increasing age. Circulation 2002, 105, 2092–2098. [Google Scholar] [CrossRef] [Green Version]
- Dolber, P.C.; Spach, M.S. Structure of canine Bachmann’s bundle related to propagation of excitation. Am. J. Physiol. 1989, 257, H1446–H1457. [Google Scholar]
- Clerc, L. Directional differences of impulse spread in trabecular muscle from mammalian heart. J. Physiol. 1976, 255, 335–346. [Google Scholar] [CrossRef]
- Hansson, A.; Holm, M.; Blomstrom, P.; Johansson, R.; Luhrs, C.; Brandt, J.; Olsson, S.B. Right atrial free wall conduction velocity and degree of anisotropy in patients with stable sinus rhythm studied during open heart surgery. Eur. Heart J. 1998, 19, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Spach, M.S.; Dolber, P.C.; Heidlage, J.F. Properties of discontinuous anisotropic propagation at a microscopic level. Ann. N. Y. Acad. Sci. 1990, 591, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Spach, M.S.; Dolber, P.C. Relating extracellular potentials and their derivatives to anisotropic propagation at a microscopic level in human cardiac muscle. Evidence for electrical uncoupling of side-to-side fiber connections with increasing age. Circ. Res. 1986, 58, 356–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spach, M.S.; Heidlage, J.F. The stochastic nature of cardiac propagation at a microscopic level. Electrical description of myocardial architecture and its application to conduction. Circ. Res. 1995, 76, 366–380. [Google Scholar] [CrossRef]
- Shaw, R.M.; Rudy, Y. Ionic Mechanisms of Propagation in Cardiac Tissue: Roles of the Sodium and L-type Calcium Currents During Reduced Excitability and Decreased Gap Junction Coupling. Circ. Res. 1997, 81, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Joyner, R.W.; Kumar, R.; Wilders, R.; Jongsma, H.J.; Verheijck, E.E.; Golod, D.A.; van Ginneken, A.C.; Wagner, M.B.; Goolsby, W.N. Modulating L-type calcium current affects discontinuous cardiac action potential conduction. Biophys. J. 1996, 71, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Rohr, S.; Kucera, J.P.; Fast, V.G.; Kléber, A.G. Paradoxical improvement of impulse conduction in cardiac tissue by partial cellular uncoupling. Science 1997, 275, 841–844. [Google Scholar] [CrossRef]
- Zhao, J.; Schotten, U.; Smaill, B.; Verheule, S. Loss of Side-to-Side Connections Affects the Relative Contributions of the Sodium and Calcium Current to Transverse Propagation Between Strands of Atrial Myocytes. Front. Physiol. 2018, 9, 1212. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.; Boyett, M.R.; Morris, G.M. Biology of the Sinus Node and its Disease. Arrhythm. Electrophysiol. Rev. 2015, 4, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Waller, B.F.; Gering, L.E.; Branyas, N.A.; Slack, J.D. Anatomy, histology, and pathology of the cardiac conduction system—Part V. Clin. Cardiol. 1993, 16, 565–569. [Google Scholar] [CrossRef]
- Xie, Y.; Sato, D.; Garfinkel, A.; Qu, Z.; Weiss, J.N. So little source, so much sink: Requirements for afterdepolarizations to propagate in tissue. Biophys. J. 2010, 99, 1408–1415. [Google Scholar] [CrossRef] [Green Version]
- Joyner, R.W.; van Capelle, F.J. Propagation through electrically coupled cells. How a small SA node drives a large atrium. Biophys. J. 1986, 50, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.N.; Qu, Z. The Sinus Node: Still Mysterious After All These Years. JACC Clin. Electrophysiol. 2020, 6, 1841–1843. [Google Scholar] [CrossRef] [PubMed]
- Jalife, J.; Michaels, D.C.; Delmar, M. Mechanisms of pacemaker synchronization in the sinus node. Prog Clin. Biol. Res. 1988, 275, 67–91. [Google Scholar]
- Wilders, R.; Wagner, M.B.; Golod, D.A.; Kumar, R.; Wang, Y.G.; Goolsby, W.N.; Joyner, R.W.; Jongsma, H.J. Effects of anisotropy on the development of cardiac arrhythmias associated with focal activity. Pflügers Arch. Eur. J. Physiol. 2000, 441, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.G.; Kumar, R.; Wagner, M.B.; Wilders, R.; Golod, D.A.; Goolsby, W.N.; Joyner, R.W. Electrical interactions between a real ventricular cell and an anisotropic two-dimensional sheet of model cells. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H452–H460. [Google Scholar] [CrossRef] [PubMed]
- Sato, D.; Xie, L.-H.; Sovari, A.A.; Tran, D.X.; Morita, N.; Xie, F.; Karagueuzian, H.; Garfinkel, A.; Weiss, J.N.; Qu, Z. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc. Natl. Acad. Sci. USA 2009, 106, 2983–2988. [Google Scholar] [CrossRef] [Green Version]
- Wiener, N.; Rosenblueth, A. The mathematical formulation of the problem of conduction of impulses in a network of connected excitable elements, specifically in cardiac muscle. Arch. Inst. Cardiol. Mex. 1946, 16, 205–265. [Google Scholar] [PubMed]
- de Bakker, J.; Coronel, R.; Tasseron, S.; Wilde, A.; Opthof, T.; Janse, M.; van Capelle, F.; Becker, A.; Jambroes, G. Ventricular tachycardia in the infarcted, Langendorff-perfused human heart: Role of the arrangement of surviving cardiac fibers. J. Am. Coll. Cardiol. 1990, 15, 1594–1607. [Google Scholar] [CrossRef] [Green Version]
- de Bakker, J.; van Capelle, F.; Janse, M.; Tasseron, S.; Vermeulen, J.; de Jonge, N.; Lahpor, J. Slow conduction in the infarcted human heart. ‘Zigzag’ course of activation. Circulation 1993, 88, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Pertsov, A.M.; Davidenko, J.M.; Salomonsz, R.; Baxter, W.T.; Jalife, J. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ. Res. 1993, 72, 631–650. [Google Scholar] [CrossRef] [Green Version]
- Kalifa, J.; Tanaka, K.; Zaitsev, A.V.; Warren, M.; Vaidyanathan, R.; Auerbach, D.; Pandit, S.; Vikstrom, K.L.; Ploutz-Snyder, R.; Talkachou, A.; et al. Mechanisms of wave fractionation at boundaries of high-frequency excitation in the posterior left atrium of the isolated sheep heart during atrial fibrillation. Circulation 2006, 113, 626–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlochiver, S.; Muñoz, V.; Vikstrom, K.L.; Taffet, S.M.; Berenfeld, O.; Jalife, J. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys. J. 2008, 95, 4469–4480. [Google Scholar] [CrossRef] [Green Version]
- Roney, C.H.; Bayer, J.D.; Zahid, S.; Meo, M.; Boyle, P.M.; Trayanova, N.A.; Haissaguerre, M.; Dubois, R.; Cochet, H.; Vigmond, E.J. Modelling methodology of atrial fibrosis affects rotor dynamics and electrograms. Europace 2016, 18, iv146–iv155. [Google Scholar] [CrossRef]
- Vandersickel, N.; Watanabe, M.; Tao, Q.; Fostier, J.; Zeppenfeld, K.; Panfilov, A.V. Dynamical anchoring of distant arrhythmia sources by fibrotic regions via restructuring of the activation pattern. PLoS Comput. Biol. 2018, 14, e1006637. [Google Scholar] [CrossRef] [Green Version]
- Moe, G.K.; Abildskov, J.A. Atrial fibrillation as a self-sustaining arrhythmia independent of focal discharge. Am. Heart J. 1959, 58, 59–70. [Google Scholar] [CrossRef]
- Spach, M.; Boineau, J. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: A major mechanism of structural heart disease arrhythmias. Pacing Clin. Electrophysiol. 1997, 20, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Vigmond, E.; Pashaei, A.; Amraoui, S.; Cochet, H.; Hassaguerre, M. Percolation as a mechanism to explain atrial fractionated electrograms and reentry in a fibrosis model based on imaging data. Heart Rhythm 2016, 13, 1536–1543. [Google Scholar] [CrossRef]
- Weiss, J.N.; Karma, A.; Shiferaw, Y.; Chen, P.-S.; Garfinkel, A.; Qu, Z. From pulsus to pulseless: The saga of cardiac alternans. Circ. Res. 2006, 98, 1244–1253. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Garfinkel, A.; Weiss, J.N.; Qu, Z. Cardiac alternans induced by fibroblast-myocyte coupling: Mechanistic insights from computational models. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H775–H784. [Google Scholar] [CrossRef] [Green Version]
- Majumder, R.; Engels, M.C.; de Vries, A.A.; Panfilov, A.V.; Pijnappels, D.A. Islands of spatially discordant APD alternans underlie arrhythmogenesis by promoting electrotonic dyssynchrony in models of fibrotic rat ventricular myocardium. Sci. Rep. 2016, 6, 24334. [Google Scholar] [CrossRef]
- Engelman, Z.J.; Trew, M.L.; Smaill, B.H. Structural heterogeneity alone is a sufficient substrate for dynamic instability and altered restitution. Circ. Arrhythm. Electrophysiol. 2010, 3, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Morita, N.; Lee, J.H.; Bapat, A.; Fishbein, M.C.; Mandel, W.J.; Chen, P.S.; Weiss, J.N.; Karagueuzian, H.S. Glycolytic inhibition causes spontaneous ventricular fibrillation in aged hearts. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H180–H191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glukhov, A.V.; Fedorov, V.V.; Kalish, P.W.; Ravikumar, V.K.; Lou, Q.; Janks, D.; Schuessler, R.B.; Moazami, N.; Efimov, I.R. Conduction remodeling in human end-stage nonischemic left ventricular cardiomyopathy. Circulation 2012, 125, 1835–1847. [Google Scholar] [CrossRef] [Green Version]
- Burstein, B.; Nattel, S. Atrial fibrosis: Mechanisms and clinical relevance in atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 802–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seydelmann, N.; Wanner, C.; Stork, S.; Ertl, G.; Weidemann, F. Fabry disease and the heart. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royer, A.; van Veen, T.A.; Le Bouter, S.; Marionneau, C.; Griol-Charhbili, V.; Leoni, A.L.; Steenman, M.; van Rijen, H.V.; Demolombe, S.; Goddard, C.A.; et al. Mouse model of SCN5A-linked hereditary Lenegre’s disease: Age-related conduction slowing and myocardial fibrosis. Circulation 2005, 111, 1738–1746. [Google Scholar] [CrossRef] [Green Version]
- Blok, M.; Boukens, B.J. Mechanisms of Arrhythmias in the Brugada Syndrome. Int. J. Mol. Sci. 2020, 21, 7051. [Google Scholar] [CrossRef]
- Mc, L.A.; Ellims, A.H.; Prabhu, S.; Voskoboinik, A.; Iles, L.M.; Hare, J.L.; Kaye, D.M.; Macciocca, I.; Mariani, J.A.; Kalman, J.M.; et al. Diffuse Ventricular Fibrosis on Cardiac Magnetic Resonance Imaging Associates With Ventricular Tachycardia in Patients With Hypertrophic Cardiomyopathy. J. Cardiovasc. Electrophysiol. 2016, 27, 571–580. [Google Scholar] [CrossRef]
- Disertori, M.; Mase, M.; Ravelli, F. Myocardial fibrosis predicts ventricular tachyarrhythmias. Trends Cardiovasc. Med. 2017, 27, 363–372. [Google Scholar] [CrossRef]
- Verheule, S.; Sato, T.; Everett, T.; Engle, S.K.; Otten, D.; Rubart-von der Lohe, M.; Nakajima, H.O.; Nakajima, H.; Field, L.J.; Olgin, J.E. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ. Res. 2004, 94, 1458–1465. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.K.; Chang, P.C.; Lee, Y.S.; Lin, S.F.; Zhu, W.; Maruyama, M.; Fishbein, M.C.; Chen, Z.; Rubart-von der Lohe, M.; Field, L.J.; et al. Triggered firing and atrial fibrillation in transgenic mice with selective atrial fibrosis induced by overexpression of TGF-beta1. Circ. J. 2012, 76, 1354–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Fareh, S.; Leung, T.; Nattel, S. Promotion of atrial fibrillation by heart failure in dogs: Atrial remodeling of a different sort. Circulation 1999, 100, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, N.; Cardin, S.; Leung, T.K.; Nattel, S. Differences in atrial versus ventricular remodeling in dogs with ventricular tachypacing-induced congestive heart failure. Cardiovasc. Res. 2004, 63, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Cardin, S.; Li, D.; Thorin-Trescases, N.; Leung, T.K.; Thorin, E.; Nattel, S. Evolution of the atrial fibrillation substrate in experimental congestive heart failure: Angiotensin-dependent and -independent pathways. Cardiovasc. Res. 2003, 60, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Benardeau, A.; Nattel, S. Contrasting efficacy of dofetilide in differing experimental models of atrial fibrillation. Circulation 2000, 102, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Shinagawa, K.; Pang, L.; Leung, T.K.; Cardin, S.; Wang, Z.; Nattel, S. Effects of angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation 2001, 104, 2608–2614. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.W.; Everett, T.H.; Rahmutula, D.; Guerra, J.M.; Wilson, E.; Ding, C.; Olgin, J.E. Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation 2006, 114, 1703–1712. [Google Scholar] [CrossRef] [Green Version]
- Shinagawa, K.; Shi, Y.-F.; Tardif, J.-C.; Leung, T.-K.; Nattel, S. Dynamic nature of atrial fibrillation substrate during development and reversal of heart failure in dogs. Circulation 2002, 105, 2672–2678. [Google Scholar] [CrossRef] [Green Version]
- Schoonderwoerd, B.A.; Ausma, J.; Crijns, H.J.; Van Veldhuisen, D.J.; Blaauw, E.H.; Van Gelder, I.C. Atrial ultrastructural changes during experimental atrial tachycardia depend on high ventricular rate. J. Cardiovasc. Electrophysiol. 2004, 15, 1167–1174. [Google Scholar] [CrossRef]
- Anne, W.; Willems, R.; Holemans, P.; Beckers, F.; Roskams, T.; Lenaerts, I.; Ector, H.; Heidbuchel, H. Self-terminating AF depends on electrical remodeling while persistent AF depends on additional structural changes in a rapid atrially paced sheep model. J. Mol. Cell Cardiol. 2007, 43, 148–158. [Google Scholar] [CrossRef]
- Verheule, S.; Tuyls, E.; van Hunnik, A.; Kuiper, M.; Schotten, U.; Allessie, M. Fibrillatory conduction in the atrial free walls of goats in persistent and permanent atrial fibrillation. Circ. Arrhythmia Electrophysiol. 2010, 3, 590–599. [Google Scholar] [CrossRef] [Green Version]
- Verheule, S.; Tuyls, E.; Gharaviri, A.; Hulsmans, S.; van Hunnik, A.; Kuiper, M.; Serroyen, J.; Zeemering, S.; Kuijpers, N.H.L.; Schotten, U. Loss of Continuity in the Thin Epicardial Layer Due to Endomysial Fibrosis Increases the Complexity of Atrial Fibrillatory Conduction. Circ. Arrhythmia Electrophysiol. 2013, 6, 202–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheule, S.; Eckstein, J.; Linz, D.; Maesen, B.; Bidar, E.; Gharaviri, A.; Schotten, U. Role of endo-epicardial dissociation of electrical activity and transmural conduction in the development of persistent atrial fibrillation. Prog. Biophys. Mol. Biol. 2014, 115, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Gharaviri, A.; Bidar, E.; Potse, M.; Zeemering, S.; Verheule, S.; Pezzuto, S.; Krause, R.; Maessen, J.G.; Auricchio, A.; Schotten, U. Epicardial Fibrosis Explains Increased Endo-Epicardial Dissociation and Epicardial Breakthroughs in Human Atrial Fibrillation. Front. Physiol. 2020, 11, 68. [Google Scholar] [CrossRef] [Green Version]
- Anné, W.; Willems, R.; Roskams, T.; Sergeant, P.; Herijgers, P.; Holemans, P.; Ector, H.; Heidbüchel, H. Matrix metalloproteinases and atrial remodeling in patients with mitral valve disease and atrial fibrillation. Cardiovasc. Res. 2005, 67, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Platonov, P.G.; Mitrofanova, L.B.; Orshanskaya, V.; Ho, S.Y. Structural abnormalities in atrial walls are associated with presence and persistency of atrial fibrillation but not with age. J. Am. Coll. Cardiol. 2011, 58, 2225–2232. [Google Scholar] [CrossRef] [Green Version]
- Verheule, S.; Wilson, E.; Everett, T.t.; Shanbhag, S.; Golden, C.; Olgin, J. Alterations in atrial electrophysiology and tissue structure in a canine model of chronic atrial dilatation due to mitral regurgitation. Circulation 2003, 107, 2615–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawara, T.; Derksen, R.; de Groot, J.R.; Coronel, R.; Tasseron, S.; Linnenbank, A.C.; Hauer, R.N.; Kirkels, H.; Janse, M.J.; de Bakker, J.M. Activation delay after premature stimulation in chronically diseased human myocardium relates to the architecture of interstitial fibrosis. Circulation 2001, 104, 3069–3075. [Google Scholar] [CrossRef] [Green Version]
- Krul, S.P.J.; Berger, W.R.; Smit, N.W.; van Amersfoorth, S.C.M.; Driessen, A.H.G.; van Boven, W.J.; Fiolet, J.W.T.; van Ginneken, A.C.G.; van der Wal, A.C.; de Bakker, J.M.T.; et al. Atrial fibrosis and conduction slowing in the left atrial appendage of patients undergoing thoracoscopic surgical pulmonary vein isolation for atrial fibrillation. Circ. Arrhythmia Electrophysiol. 2015, 8, 288–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, B.J.; Zhao, J.; Csepe, T.A.; Moore, B.T.; Li, N.; Jayne, L.A.; Kalyanasundaram, A.; Lim, P.; Bratasz, A.; Powell, K.A.; et al. Atrial fibrillation driven by micro-anatomic intramural re-entry revealed by simultaneous sub-epicardial and sub-endocardial optical mapping in explanted human hearts. Eur. Heart J. 2015, 36, 2390–2401. [Google Scholar] [CrossRef] [Green Version]
- McGann, C.; Akoum, N.; Patel, A.; Kholmovski, E.; Revelo, P.; Damal, K.; Wilson, B.; Cates, J.; Harrison, A.; Ranjan, R.; et al. Atrial fibrillation ablation outcome is predicted by left atrial remodeling on MRI. Circ. Arrhythmia Electrophysiol. 2014, 7, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Sohns, C.; Marrouche, N.F. Atrial fibrillation and cardiac fibrosis. Eur. Heart J. 2020, 41, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Zghaib, T.; Keramati, A.; Chrispin, J.; Huang, D.; Balouch, M.A.; Ciuffo, L.; Berger, R.D.; Marine, J.E.; Ashikaga, H.; Calkins, H.; et al. Multimodal Examination of Atrial Fibrillation Substrate: Correlation of Left Atrial Bipolar Voltage Using Multi-Electrode Fast Automated Mapping, Point-by-Point Mapping, and Magnetic Resonance Image Intensity Ratio. JACC Clin. Electrophysiol. 2018, 4, 59–68. [Google Scholar] [CrossRef]
- Chen, J.; Arentz, T.; Cochet, H.; Muller-Edenborn, B.; Kim, S.; Moreno-Weidmann, Z.; Minners, J.; Kohl, P.; Lehrmann, H.; Allgeier, J.; et al. Extent and spatial distribution of left atrial arrhythmogenic sites, late gadolinium enhancement at magnetic resonance imaging, and low-voltage areas in patients with persistent atrial fibrillation: Comparison of imaging vs. electrical parameters of fibrosis and arrhythmogenesis. Europace 2019, 21, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Cochet, H.; Dubois, R.; Yamashita, S.; Al Jefairi, N.; Berte, B.; Sellal, J.M.; Hooks, D.; Frontera, A.; Amraoui, S.; Zemoura, A.; et al. Relationship Between Fibrosis Detected on Late Gadolinium-Enhanced Cardiac Magnetic Resonance and Re-Entrant Activity Assessed With Electrocardiographic Imaging in Human Persistent Atrial Fibrillation. JACC Clin. Electrophysiol. 2018, 4, 17–29. [Google Scholar] [CrossRef]
- Boyle, P.M.; Zghaib, T.; Zahid, S.; Ali, R.L.; Deng, D.; Franceschi, W.H.; Hakim, J.B.; Murphy, M.J.; Prakosa, A.; Zimmerman, S.L.; et al. Computationally guided personalized targeted ablation of persistent atrial fibrillation. Nat. Biomed. Eng. 2019, 3, 870–879. [Google Scholar] [CrossRef]
- Prakosa, A.; Arevalo, H.J.; Deng, D.; Boyle, P.M.; Nikolov, P.P.; Ashikaga, H.; Blauer, J.J.E.; Ghafoori, E.; Park, C.J.; Blake, R.C., 3rd; et al. Personalized virtual-heart technology for guiding the ablation of infarct-related ventricular tachycardia. Nat. Biomed. Eng. 2018, 2, 732–740. [Google Scholar] [CrossRef]
- Saliani, A.; Irakoze, E.; Jacquemet, V. Simulation of diffuse and stringy fibrosis in a bilayer interconnected cable model of the left atrium. Europace 2021, 23, i169–i177. [Google Scholar] [CrossRef] [PubMed]
- Allessie, M.A.; de Groot, N.M.; Houben, R.P.; Schotten, U.; Boersma, E.; Smeets, J.L.; Crijns, H.J. Electropathological substrate of long-standing persistent atrial fibrillation in patients with structural heart disease: Longitudinal dissociation. Circ. Arrhythmia Electrophysiol. 2010, 3, 606–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Groot, N.M.; Houben, R.P.; Smeets, J.L.; Boersma, E.; Schotten, U.; Schalij, M.J.; Crijns, H.; Allessie, M.A. Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: Epicardial breakthrough. Circulation 2010, 122, 1674–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.Y.; Anderson, R.H.; Sanchez-Quintana, D. Atrial structure and fibres: Morphologic bases of atrial conduction. Cardiovasc. Res. 2002, 54, 325–336. [Google Scholar] [CrossRef]
- Zhao, J.; Butters, T.D.; Zhang, H.; Pullan, A.J.; LeGrice, I.J.; Sands, G.B.; Smaill, B.H. An image-based model of atrial muscular architecture: Effects of structural anisotropy on electrical activation. Circ. Arrhythmia Electrophysiol. 2012, 5, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Abed, H.S.; Samuel, C.S.; Lau, D.H.; Kelly, D.J.; Royce, S.G.; Alasady, M.; Mahajan, R.; Kuklik, P.; Zhang, Y.; Brooks, A.G.; et al. Obesity results in progressive atrial structural and electrical remodeling: Implications for atrial fibrillation. Heart Rhythm 2013, 10, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Azaouagh, A.; Churzidse, S.; Konorza, T.; Erbel, R. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: A review and update. Clin. Res. Cardiol. 2011, 100, 383–394. [Google Scholar] [CrossRef]
- Chaumont, C.; Suffee, N.; Gandjbakhch, E.; Balse, E.; Anselme, F.; Hatem, S.N. Epicardial origin of cardiac arrhythmias: Clinical evidences and pathophysiology. Cardiovasc. Res. 2021, cvab213. [Google Scholar] [CrossRef] [PubMed]
- Ausma, J.; Wijffels, M.; Thone, F.; Wouters, L.; Allessie, M.; Borgers, M. Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation 1997, 96, 3157–3163. [Google Scholar] [CrossRef]
- Boyden, P.A.; Hoffman, B.F. The effects on atrial electrophysiology and structure of surgically induced right atrial enlargement in dogs. Circ. Res. 1981, 49, 1319–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuberger, H.R.; Schotten, U.; Verheule, S.; Eijsbouts, S.; Blaauw, Y.; van Hunnik, A.; Allessie, M. Development of a substrate of atrial fibrillation during chronic atrioventricular block in the goat. Circulation 2005, 111, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Wiegerinck, R.F.; Verkerk, A.O.; Belterman, C.N.; van Veen, T.A.B.; Baartscheer, A.; Opthof, T.; Wilders, R.; de Bakker, J.M.T.; Coronel, R. Larger cell size in rabbits with heart failure increases myocardial conduction velocity and QRS duration. Circulation 2006, 113, 806–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spach, M.S.; Heidlage, J.F.; Barr, R.C.; Dolber, P.C. Cell size and communication: Role in structural and electrical development and remodeling of the heart. Heart Rhythm 2004, 1, 500–515. [Google Scholar] [CrossRef]
- Spach, M.S.; Heidlage, J.F.; Dolber, P.C.; Barr, R.C. Changes in anisotropic conduction caused by remodeling cell size and the cellular distribution of gap junctions and Na(+) channels. J. Electrocardiol. 2001, 34, 69–76. [Google Scholar] [CrossRef]
- Dhein, S.; Hammerath, S.B. Aspects of the intercellular communication in aged hearts: Effects of the gap junction uncoupler palmitoleic acid. Naunyn Schmiedebergs Arch. Pharm. 2001, 364, 397–408. [Google Scholar] [CrossRef]
- Dhein, S.; Seidel, T.; Salameh, A.; Jozwiak, J.; Hagen, A.; Kostelka, M.; Hindricks, G.; Mohr, F.W. Remodeling of cardiac passive electrical properties and susceptibility to ventricular and atrial arrhythmias. Front. Physiol. 2014, 5, 424. [Google Scholar] [CrossRef]
- De Smet, M.A.; Lissoni, A.; Nezlobinsky, T.; Wang, N.; Dries, E.; Perez-Hernandez, M.; Lin, X.; Amoni, M.; Vervliet, T.; Witschas, K.; et al. Cx43 hemichannel microdomain signaling at the intercalated disc enhances cardiac excitability. J. Clin. Investig. 2021, 131, e137752. [Google Scholar] [CrossRef]
- Polontchouk, L.; Haefliger, J.A.; Ebelt, B.; Schaefer, T.; Stuhlmann, D.; Mehlhorn, U.; Kuhn-Regnier, F.; De Vivie, E.R.; Dhein, S. Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J. Am. Coll. Cardiol. 2001, 38, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Verheule, S.; van Batenburg, C.A.; Coenjaerts, F.E.; Kirchhoff, S.; Willecke, K.; Jongsma, H.J. Cardiac conduction abnormalities in mice lacking the gap junction protein connexin40. J. Cardiovasc. Electrophysiol. 1999, 10, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Kanagaratnam, P.; Rothery, S.; Patel, P.; Severs, N.J.; Peters, N.S. Relative expression of immunolocalized connexins 40 and 43 correlates with human atrial conduction properties. J. Am. Coll. Cardiol. 2002, 39, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, P.S.; Chowdhury, R.A.; Patel, P.M.; Jabr, R.; Momin, A.U.; Vecht, J.; Gray, R.; Shipolini, A.; Fry, C.H.; Peters, N.S. Relationship between connexin expression and gap-junction resistivity in human atrial myocardium. Circ. Arrhythmia Electrophysiol. 2014, 7, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rook, M.B.; van Ginneken, A.C.; de Jonge, B.; el Aoumari, A.; Gros, D.; Jongsma, H.J. Differences in gap junction channels between cardiac myocytes, fibroblasts, and heterologous pairs. Am. J. Physiol. 1992, 263, C959–C977. [Google Scholar] [CrossRef]
- Gaudesius, G.; Miragoli, M.; Thomas, S.P.; Rohr, S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ. Res. 2003, 93, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Camelliti, P.; Green, C.R.; LeGrice, I.; Kohl, P. Fibroblast network in rabbit sinoatrial node: Structural and functional identification of homogeneous and heterogeneous cell coupling. Circ. Res. 2004, 94, 828–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, T.A.; Camelliti, P.; Rog-Zielinska, E.A.; Siedlecka, U.; Poggioli, T.; O’Toole, E.T.; Knöpfel, T.; Kohl, P. Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc. Natl. Acad. Sci. USA 2016, 113, 14852–14857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubart, M.; Tao, W.; Lu, X.L.; Conway, S.J.; Reuter, S.P.; Lin, S.F.; Soonpaa, M.H. Electrical coupling between ventricular myocytes and myofibroblasts in the infarcted mouse heart. Cardiovasc. Res. 2018, 114, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Greisas, A.; Zlochiver, S. The Multi-Domain Fibroblast/Myocyte Coupling in the Cardiac Tissue: A Theoretical Study. Cardiovasc. Eng. Technol. 2016, 7, 290–304. [Google Scholar] [CrossRef]
- Schuessler, R.B.; Grayson, T.M.; Bromberg, B.I.; Cox, J.L.; Boineau, J.P. Cholinergically mediated tachyarrhythmias induced by a single extrastimulus in the isolated canine right atrium. Circ. Res. 1992, 71, 1254–1267. [Google Scholar] [CrossRef] [Green Version]
- Kneller, J.; Zou, R.; Vigmond, E.J.; Wang, Z.; Leon, L.J.; Nattel, S. Cholinergic atrial fibrillation in a computer model of a two-dimensional sheet of canine atrial cells with realistic ionic properties. Circ. Res. 2002, 90, E73–E87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spach, M.S.; Dolber, P.C.; Heidlage, J.F. Interaction of inhomogeneities of repolarization with anisotropic propagation in dog atria. A mechanism for both preventing and initiating reentry. Circ. Res. 1989, 65, 1612–1631. [Google Scholar] [CrossRef] [Green Version]
- Winters, J.; von Braunmuhl, M.E.; Zeemering, S.; Gilbers, M.; Brink, T.T.; Scaf, B.; Guasch, E.; Mont, L.; Batlle, M.; Sinner, M.; et al. JavaCyte, a novel open-source tool for automated quantification of key hallmarks of cardiac structural remodeling. Sci. Rep. 2020, 10, 20074. [Google Scholar] [CrossRef]
- Avitall, B.; Bi, J.; Mykytsey, A.; Chicos, A. Atrial and ventricular fibrosis induced by atrial fibrillation: Evidence to support early rhythm control. Heart Rhythm 2008, 5, 839–845. [Google Scholar] [CrossRef]
- Guerra, J.M.; Everett, T.H.t.; Lee, K.W.; Wilson, E.; Olgin, J.E. Effects of the gap junction modifier rotigaptide (ZP123) on atrial conduction and vulnerability to atrial fibrillation. Circulation 2006, 114, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Zlochiver, S.; Vikstrom, K.L.; Yamazaki, M.; Moreno, J.; Klos, M.; Zaitsev, A.V.; Vaidyanathan, R.; Auerbach, D.S.; Landas, S.; et al. Spatial distribution of fibrosis governs fibrillation wave dynamics in the posterior left atrium during heart failure. Circ. Res. 2007, 101, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verheule, S.; Schotten, U. Electrophysiological Consequences of Cardiac Fibrosis. Cells 2021, 10, 3220. https://doi.org/10.3390/cells10113220
Verheule S, Schotten U. Electrophysiological Consequences of Cardiac Fibrosis. Cells. 2021; 10(11):3220. https://doi.org/10.3390/cells10113220
Chicago/Turabian StyleVerheule, Sander, and Ulrich Schotten. 2021. "Electrophysiological Consequences of Cardiac Fibrosis" Cells 10, no. 11: 3220. https://doi.org/10.3390/cells10113220
APA StyleVerheule, S., & Schotten, U. (2021). Electrophysiological Consequences of Cardiac Fibrosis. Cells, 10(11), 3220. https://doi.org/10.3390/cells10113220