
A Boron Delivery Antibody (BDA) with Boronated Specific Residues: New Perspectives in Boron Neutron Capture Therapy from an In Silico Investigation

 ,
,  , ,
, ,  , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
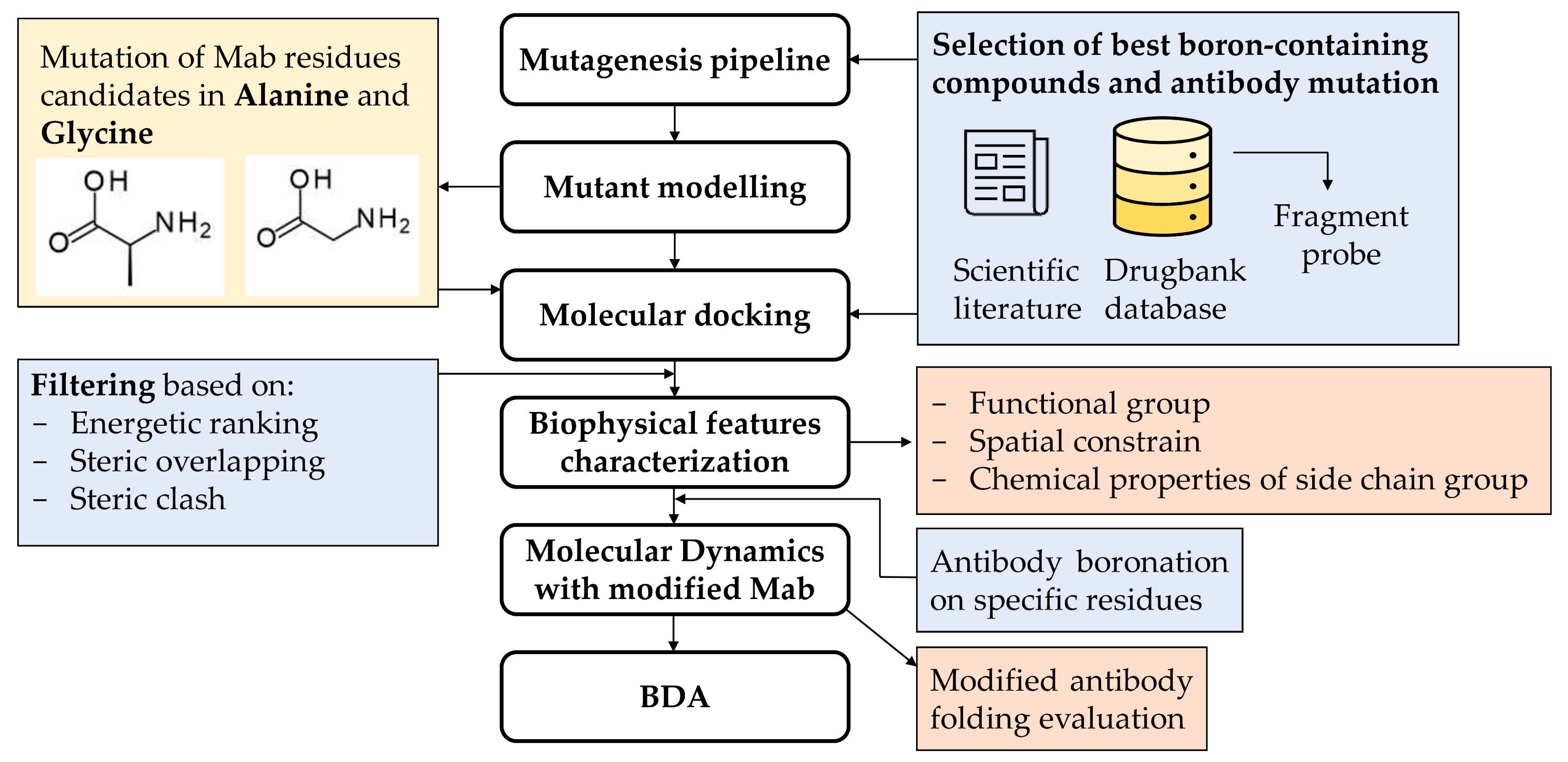
2.1. Pipeline Description
2.2. Fragment Probe for Docking
2.3. Case Study of Cetuximab Fab
2.4. Fab Mutagenesis
2.5. Docking Analysis
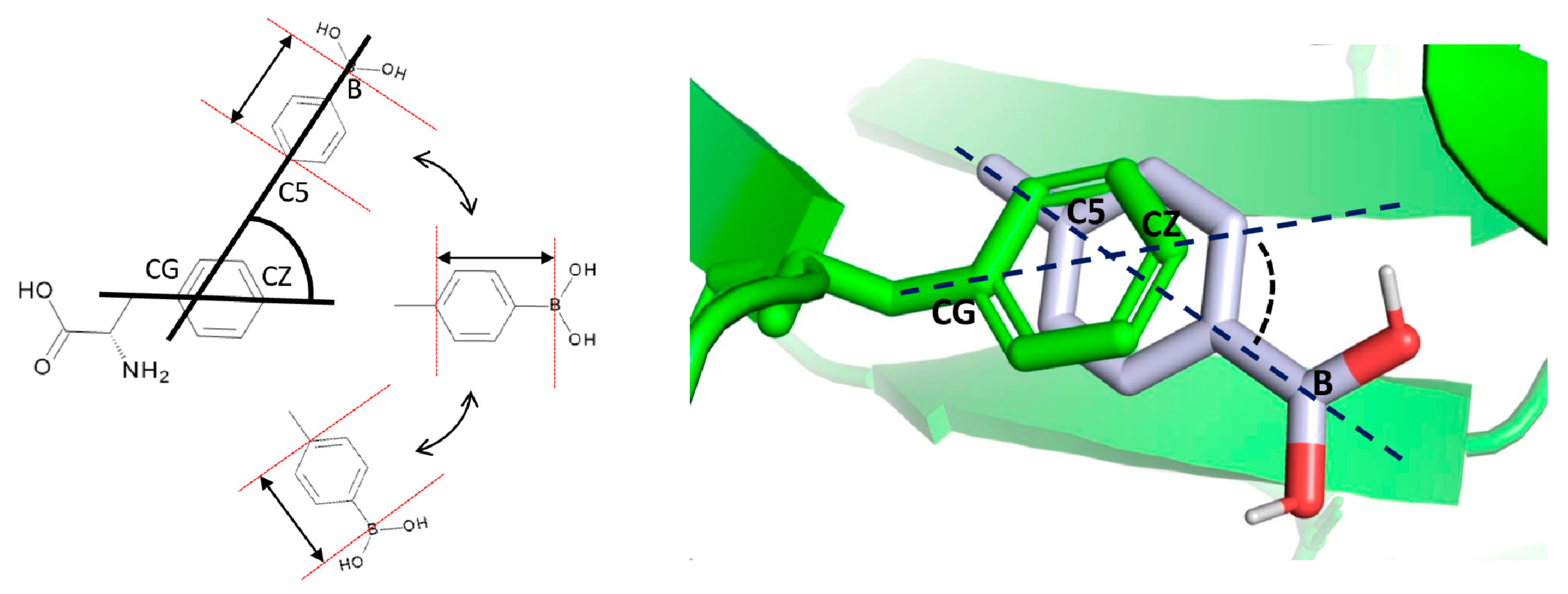
2.6. Boronated Amino Acid Residue Parametrization
2.7. MD Simulations
3. Results
3.1. Pipeline Description
3.1.1. Selection of Best Boronated Compounds and Antibody Mutation
3.1.2. Molecular Docking
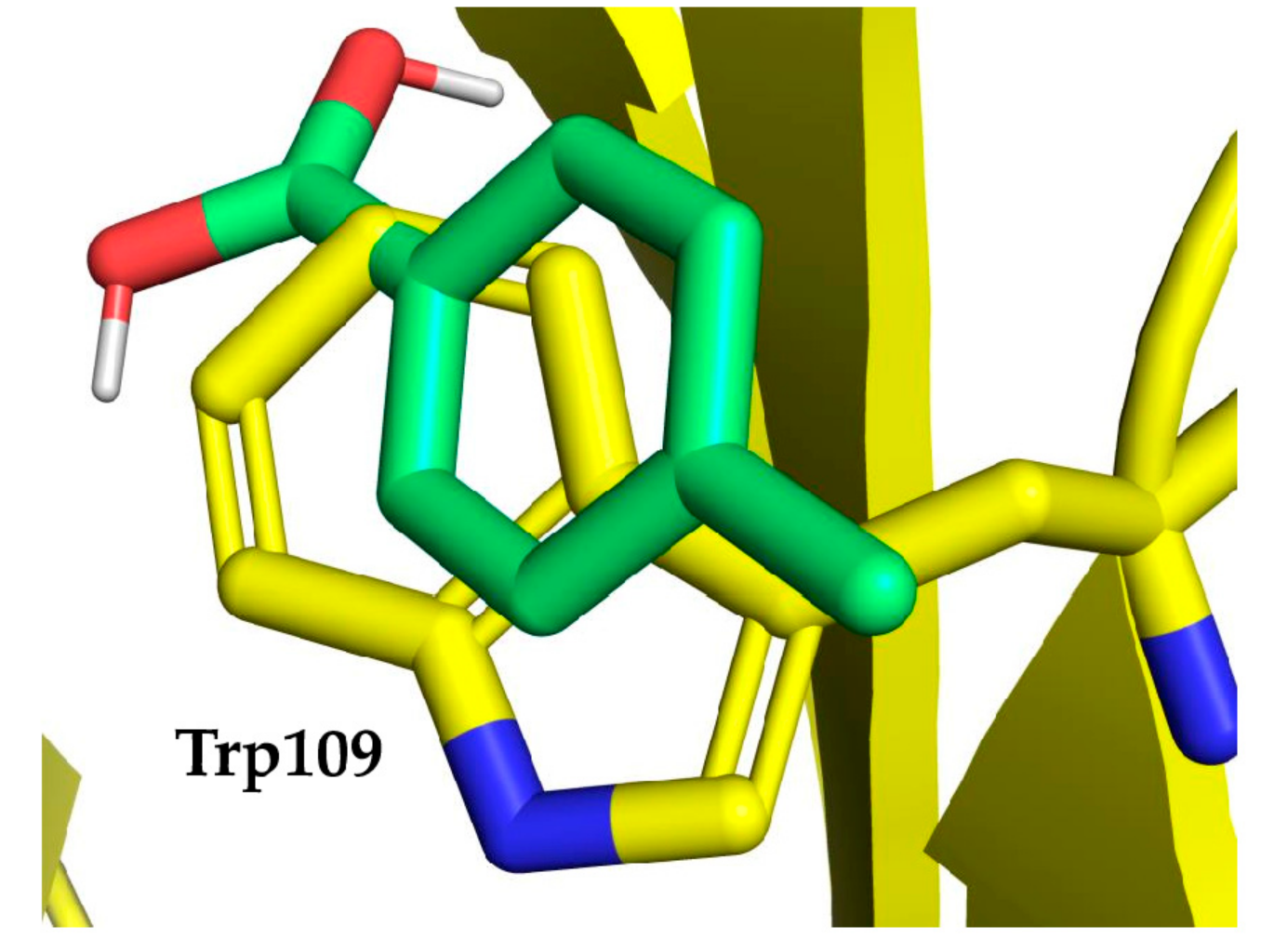
3.1.3. Antibody Boronation on Specific Residues
3.1.4. Modified Antibody Folding Evaluation
3.2. Case Study
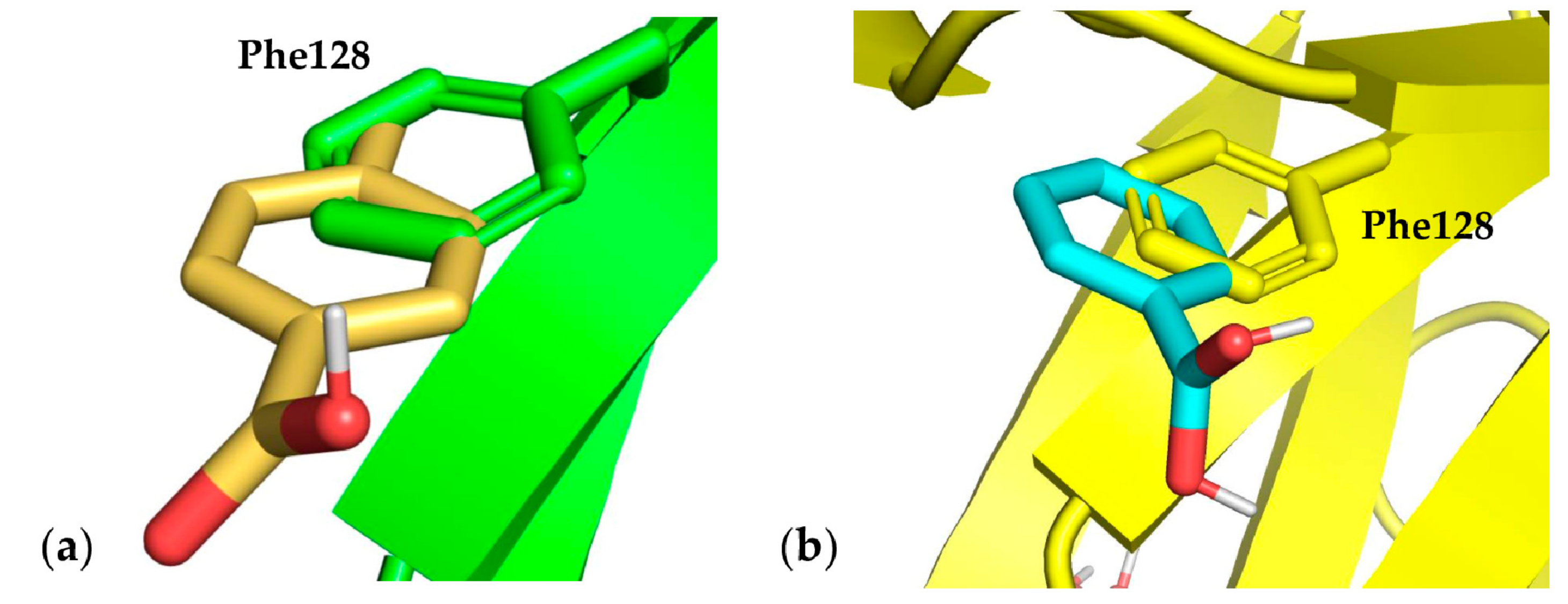
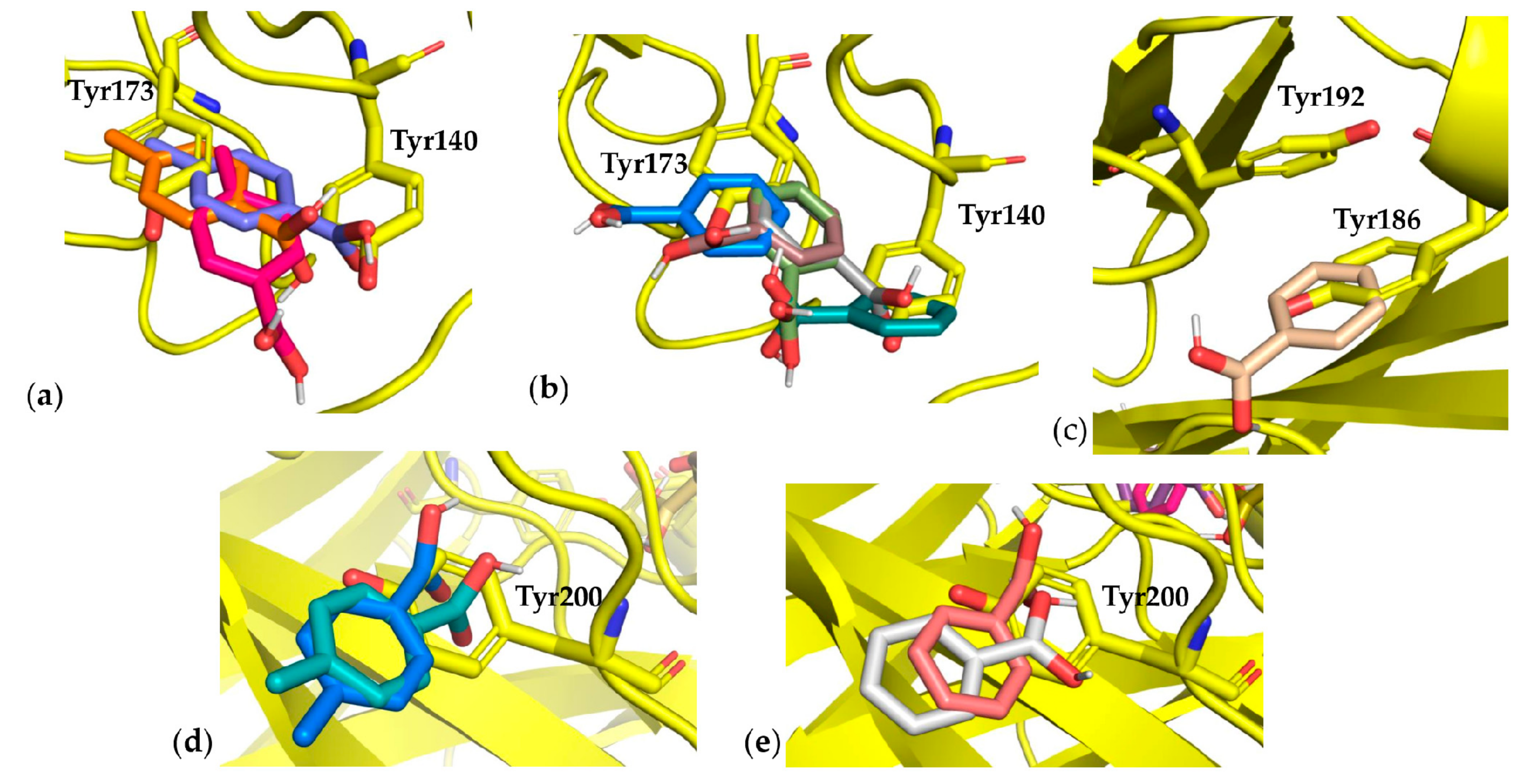
3.3. Docking Results
3.4. Monoclonal Antibody Folding Evaluation Using MD Simulations
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Worm, D.J.; Hoppenz, P.; Els-Heindl, S.; Kellert, M.; Kuhnert, R.; Saretz, S.; Köbberling, J.; Riedl, B.; Hey-Hawkins, E.; Beck-Sickinger, A.G. Selective Neuropeptide Y Conjugates with Maximized Carborane Loading as Promising Boron Delivery Agents for Boron Neutron Capture Therapy. J. Med. Chem. 2020, 63, 2358–2371. [Google Scholar] [CrossRef] [PubMed]
- Sauerwein, W.A.G.; Bet, P.M.; Wittig, A. Neutron Capture Therapy; Wittig, A., Moss, R., Nakagawa, Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar] [CrossRef]
- Miyatake, S.I.; Kawabata, S.; Hiramatsu, R.; Kuroiwa, T.; Suzuki, M.; Kondo, N.; Ono, K. Boron neutron capture therapy for malignant brain tumors. Neurol. Med. Chir. (Tokyo) 2016, 56, 361–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, K.; Konno, A.; Hiratsuka, J.; Yoshimoto, S.; Kato, T.; Ono, K.; Otsuki, N.; Hatazawa, J.; Tanaka, H.; Takayama, K.; et al. Boron neutron capture therapy using cyclotron-based epithermal neutron source and borofalan (10B) for recurrent or locally advanced head and neck cancer (JHN002): An open-label phase II trial. Radiother Oncol. 2021, 155, 182–187. [Google Scholar] [CrossRef] [PubMed]
- STELLA PHARMA. Available online: https://stella-pharma.co.jp/cp-bin/wordpress5/wp-content/uploads/2020/05/Steboronine-launched_ENG.pdf (accessed on 19 October 2021).
- Yang, W.; Barth, R.F.; Wu, G.; Kawabata, S.; Sferra, T.J.; Bandyopadhyaya, A.K.; Tjarks, W.; Ferketich, A.K.; Moeschberger, M.L.; Binns, P.J.; et al. Molecular targeting and treatment of EGFRvIII-positive gliomas using boronated monoclonal antibody L8A4. Clin. Cancer Res. 2006, 12, 3792–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Wu, G.; Barth, R.F.; Swindall, M.R.; Bandyopadhyaya, A.K.; Tjarks, W.; Tordoff, K.; Moeschberger, M.; Sferra, T.J.; Binns, P.J.; et al. Molecular targeting and treatment of composite EGFR and EGFRvIII-positive gliomas using boronated monoclonal antibodies. Clin. Cancer Res. 2008, 14, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Barth, R.F.; Wu, G.; Tjarks, W.; Binns, P.; Riley, K. Boron neutron capture therapy of EGFR or EGFRvIII positive gliomas using either boronated monoclonal antibodies or epidermal growth factor as molecular targeting agents. Appl. Radiat Isot. 2009, 67, S328–S331. [Google Scholar] [CrossRef]
- Torres-Sánchez, P.; Porras, I.; Ramos-Chernenko, N.; Arias de Saavedra, F.; Praena, J. Optimized beam shaping assembly for a 2.1-MeV proton-accelerator-based neutron source for boron neutron capture therapy. Sci. Rep. 2021, 11, 7576. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Barth, R.F.; Yang, W.; Chatterjee, M.; Tjarks, W.; Ciesielski, M.J.; Fenstermaker, R.A. Site-specific conjugation of boron-containing dendrimers to anti-EGF receptor monoclonal antibody cetuximab (IMC-C225) and its evaluation as a potential delivery agent for neutron capture therapy. Bioconjug. Chem. 2004, 15, 185–194. [Google Scholar] [CrossRef]
- Wu, G.; Yang, W.; Barth, R.F.; Kawabata, S.; Swindall, M.; Bandyopadhyaya, A.K.; Tjarks, W.; Khorsandi, B.; Blue, T.E.; Ferketich, A.K.; et al. Molecular targeting and treatment of an epidermal growth factor receptor-positive glioma using boronated cetuximab. Clin. Cancer Res. 2007, 13, 1260–1268. [Google Scholar] [CrossRef] [Green Version]
- Sauerwein, W.A.G.; Sancey, L.; Hey-Hawkins, E.; Kellert, M.; Panza, L.; Imperio, D.; Balcerzyk, M.; Rizzo, G.; Scalco, E.; Herrmann, K.; et al. Theranostics in Boron Neutron Capture Therapy. Life (Basel) 2021, 11, 330. [Google Scholar] [CrossRef]
- Capala, J.; Barth, R.F.; Bendayan, M.; Lauzon, M.; Adams, D.; Soloway, A.H.; Carlsson, J. Boronated epidermal growth factor as a potential targeting agent for boron neutron capture therapy of brain tumors. Bioconjug. Chem. 1996, 7, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Achmad, A.; Hanaoka, H.; Heryanto, Y.D.; Bhattarai, A.; Khongorzul, E.; Shintawati, R.; Kartamihardja, A.A.P.; Kanai, A.; Sugo, Y.; et al. Immuno-PET imaging for non-invasive assessment of cetuximab accumulation in non-small cell lung cancer. BMC Cancer 2019, 19, 1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, R.F.; Yang, W.; Adams, D.M.; Rotaru, J.H.; Shukla, S.; Sekido, M.; Tjarks, W.; Fenstermaker, R.A.; Ciesielski, M.; Nawrocky, M.M.; et al. Molecular targeting of the epidermal growth factor receptor for neutron capture therapy of gliomas. Cancer Res. 2002, 62, 3159–3166. [Google Scholar]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Rettig, S.J.; Trotter, J. Crystal and molecular structure of phenylboronic acid, C6H5B(OH)2. Can. J. Chem. 2011, 55, 3071–3075. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, S.R.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Kurt, B.; Temel, H. Parameterization of Boronates Using VFFDT and Paramfit for Molecular Dynamics Simulation. Molecules 2020, 25, 2196. [Google Scholar] [CrossRef]
- Kurt, B.; Temel, H. Development of AMBER parameters for molecular dynamics simulations of boron compounds containing aromatic structure. Chem. Phys. Lett. 2021, 775, 138656. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Machado, M.R.; Pantano, S. Split the Charge Difference in Two! A Rule of Thumb for Adding Proper Amounts of Ions in MD Simulations. J Chem Theory Comput. 2020, 16, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, G.; Cohen, S. Epidermal growth factor. Annu. Rev. Biochem. 1979, 48, 193–216. [Google Scholar] [CrossRef] [PubMed]
- Gullick, W.J.; Marsden, J.J.; Whittle, N.; Ward, B.; Bobrow, L.; Waterfield, M.D. Expression of epidermal growth factor receptors on human cervical, ovarian, and vulval carcinomas. Cancer Res. 1986, 46, 285–292. [Google Scholar] [PubMed]
- Soor, H.S.; Hansen, J.; Diaz, D.B.; Appavoo, S.; Yudin, A.K. Solid-phase synthesis of peptide β-aminoboronic acids. Pept. Sci. 2019, 111, e24072. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rondina, A.; Fossa, P.; Orro, A.; Milanesi, L.; De Palma, A.; Perico, D.; Mauri, P.L.; D’Ursi, P. A Boron Delivery Antibody (BDA) with Boronated Specific Residues: New Perspectives in Boron Neutron Capture Therapy from an In Silico Investigation. Cells 2021, 10, 3225. https://doi.org/10.3390/cells10113225
Rondina A, Fossa P, Orro A, Milanesi L, De Palma A, Perico D, Mauri PL, D’Ursi P. A Boron Delivery Antibody (BDA) with Boronated Specific Residues: New Perspectives in Boron Neutron Capture Therapy from an In Silico Investigation. Cells. 2021; 10(11):3225. https://doi.org/10.3390/cells10113225
Chicago/Turabian StyleRondina, Alessandro, Paola Fossa, Alessandro Orro, Luciano Milanesi, Antonella De Palma, Davide Perico, Pier Luigi Mauri, and Pasqualina D’Ursi. 2021. "A Boron Delivery Antibody (BDA) with Boronated Specific Residues: New Perspectives in Boron Neutron Capture Therapy from an In Silico Investigation" Cells 10, no. 11: 3225. https://doi.org/10.3390/cells10113225