
The Interface between Cell Signaling Pathways and Pregnane X Receptor

 ,
,
Abstract
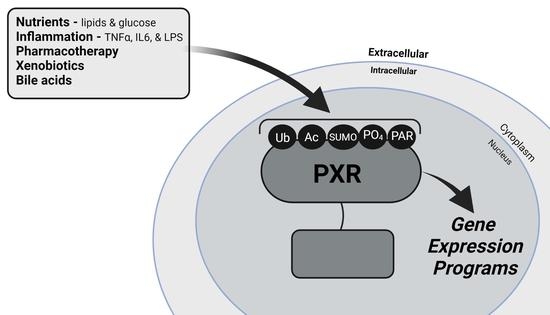
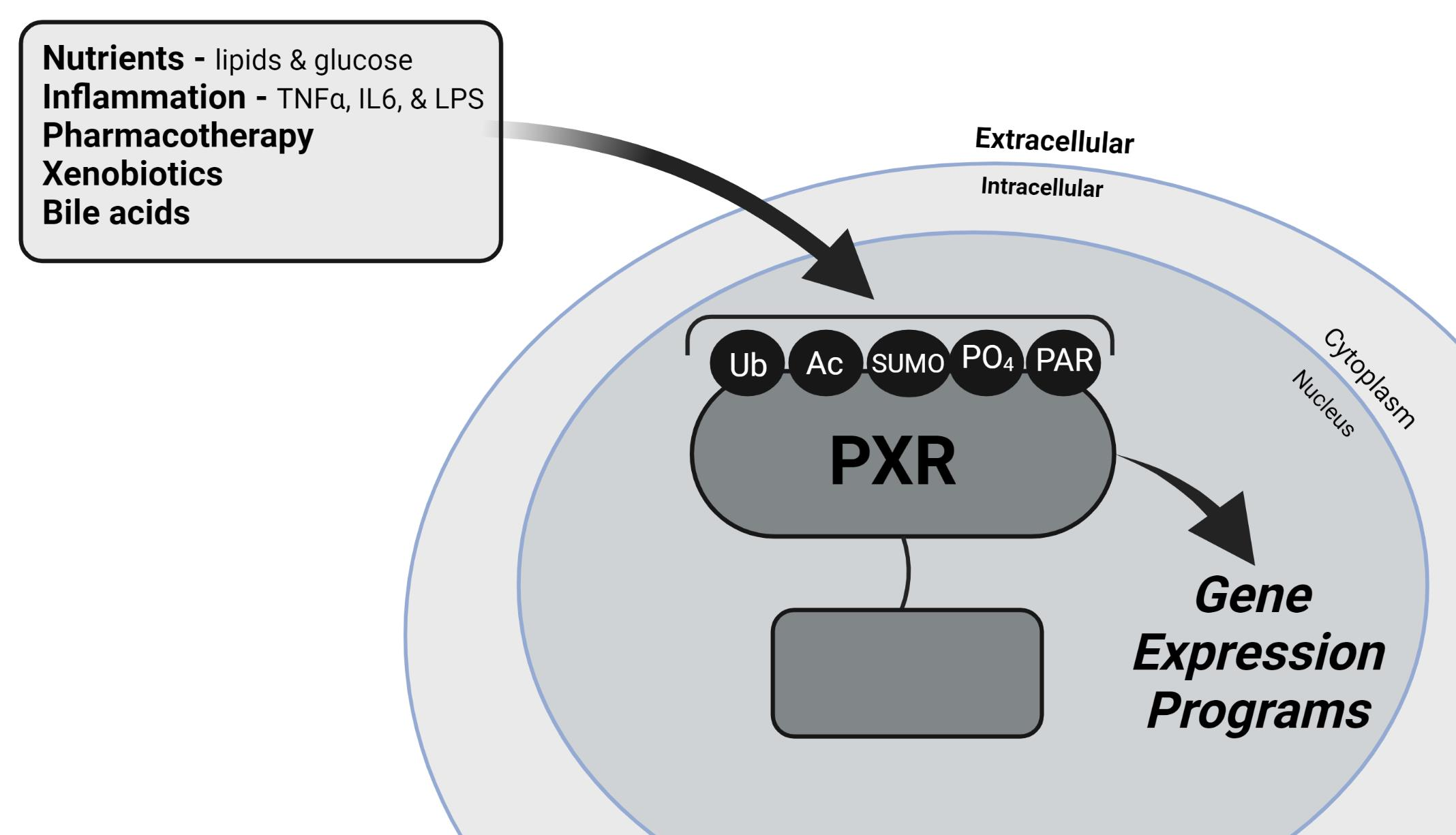{kind=link}
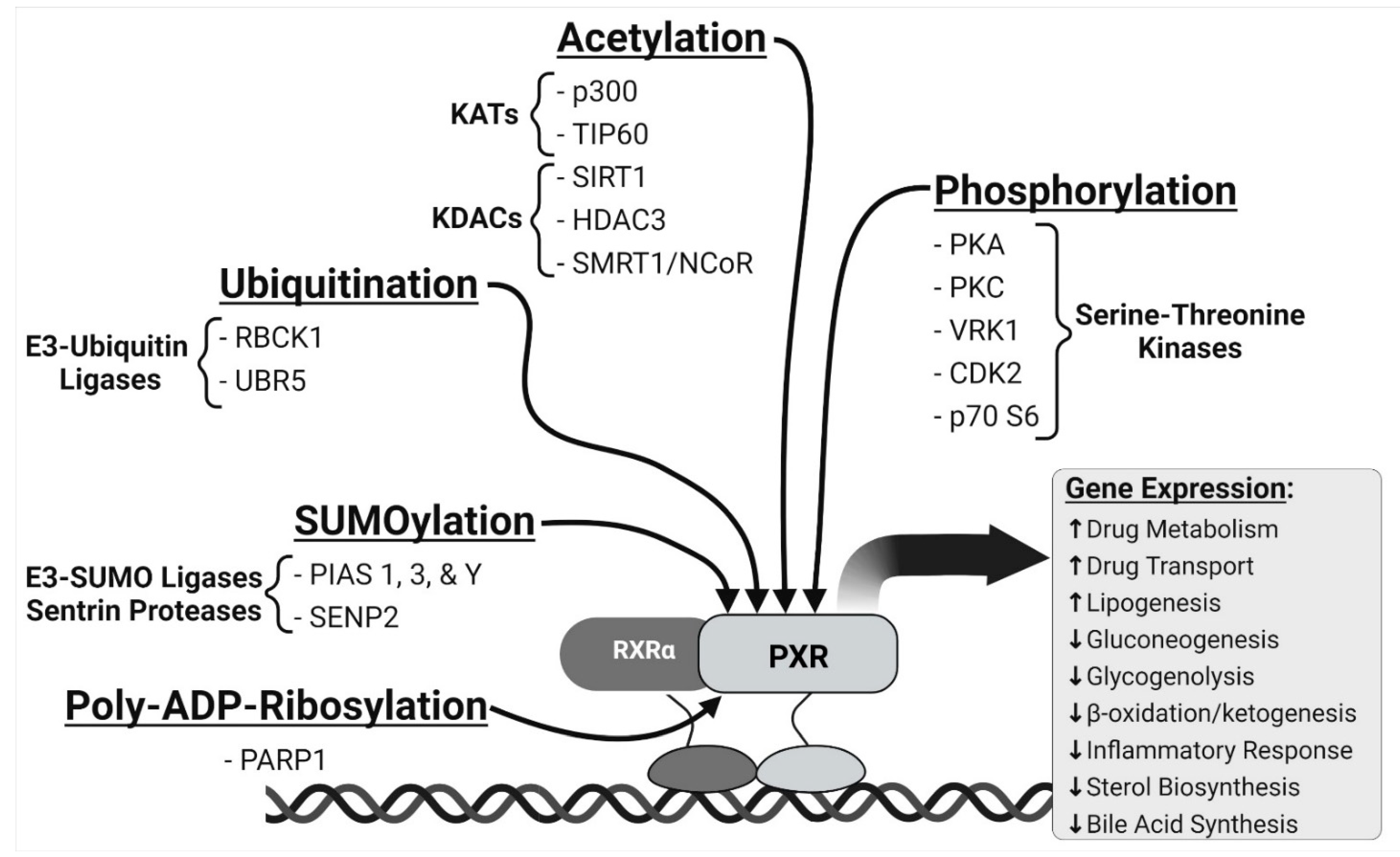{kind=link}
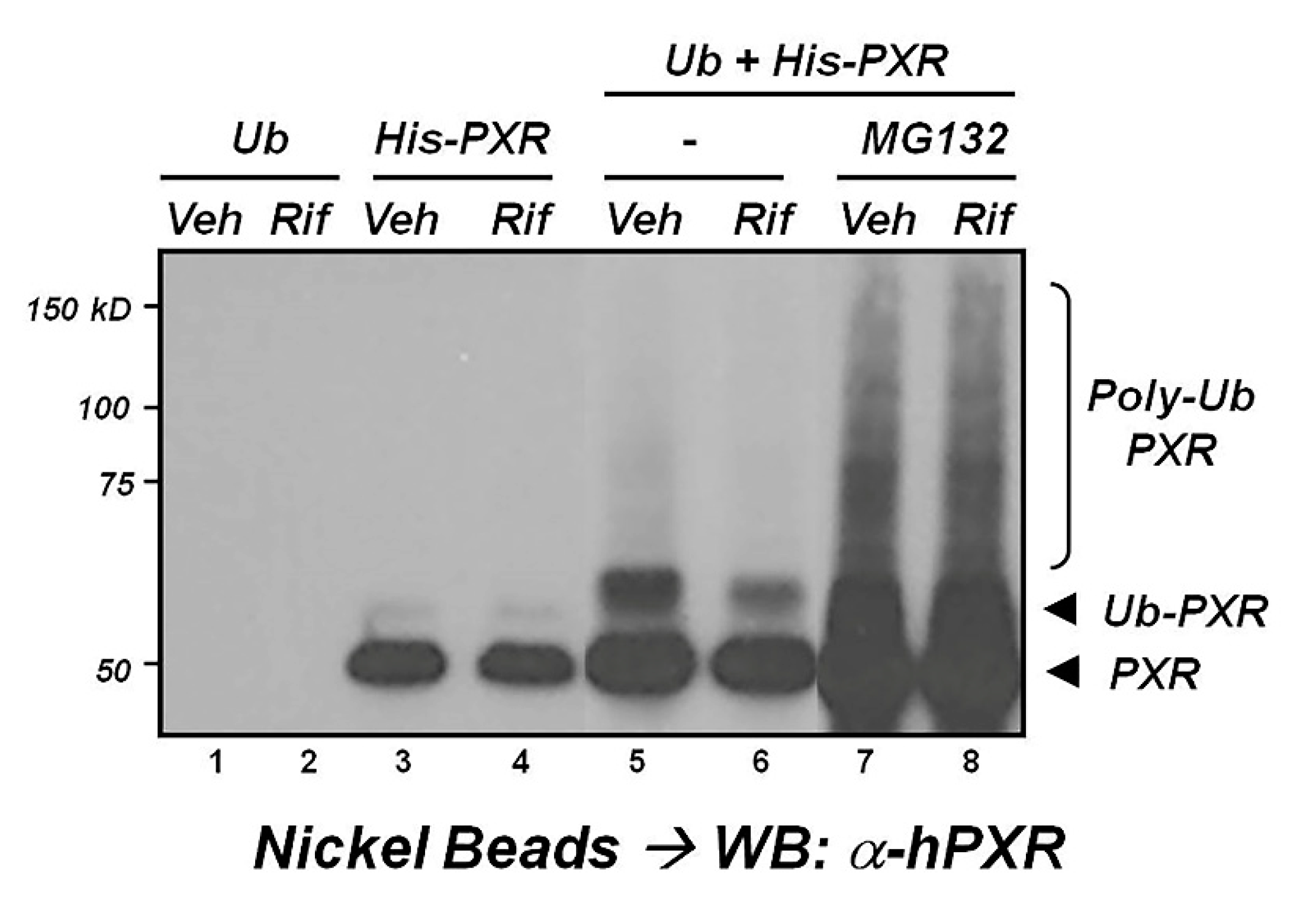{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. PXR and Cell Signaling Pathways
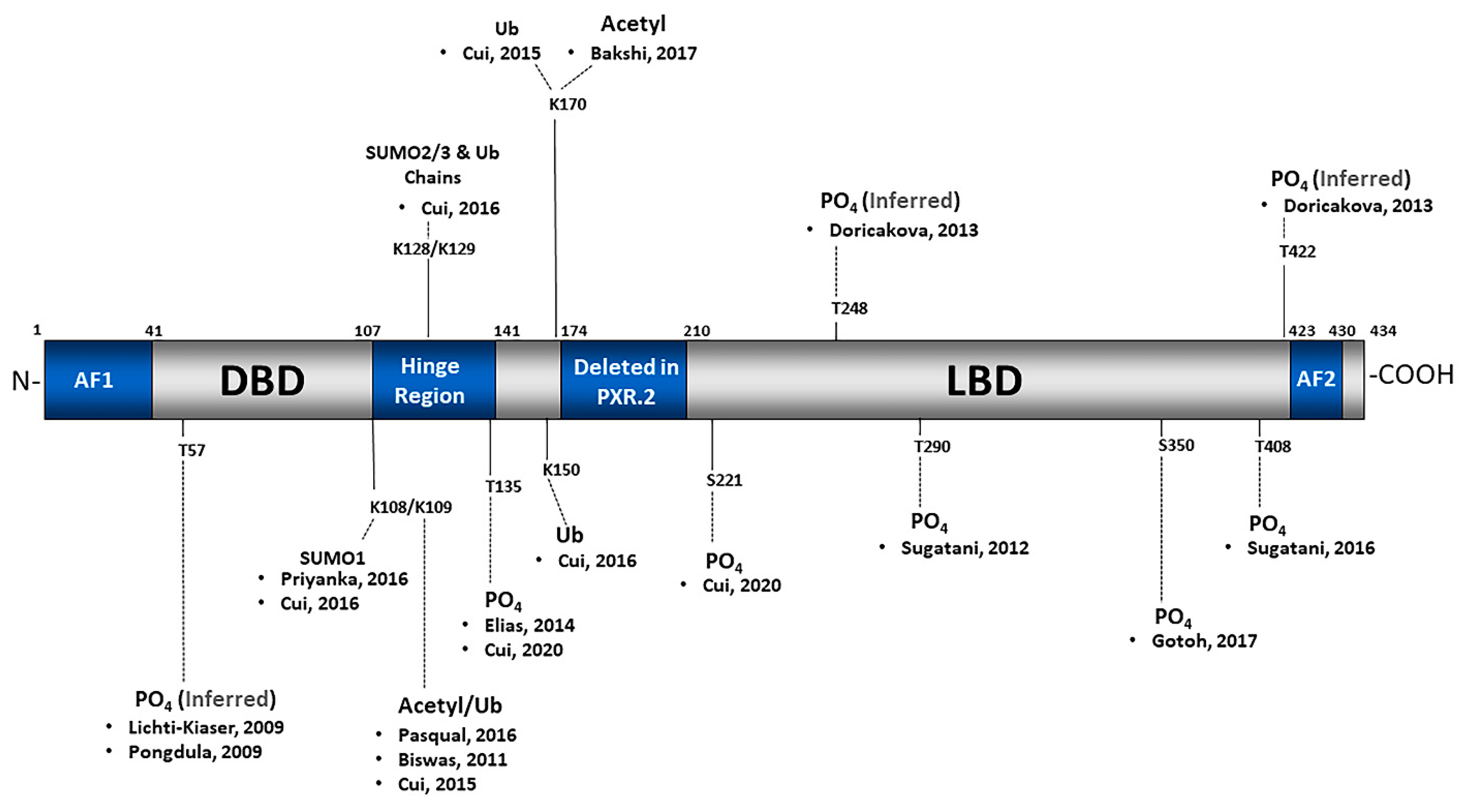
2.1. Phosphorylation
2.2. SUMOylation
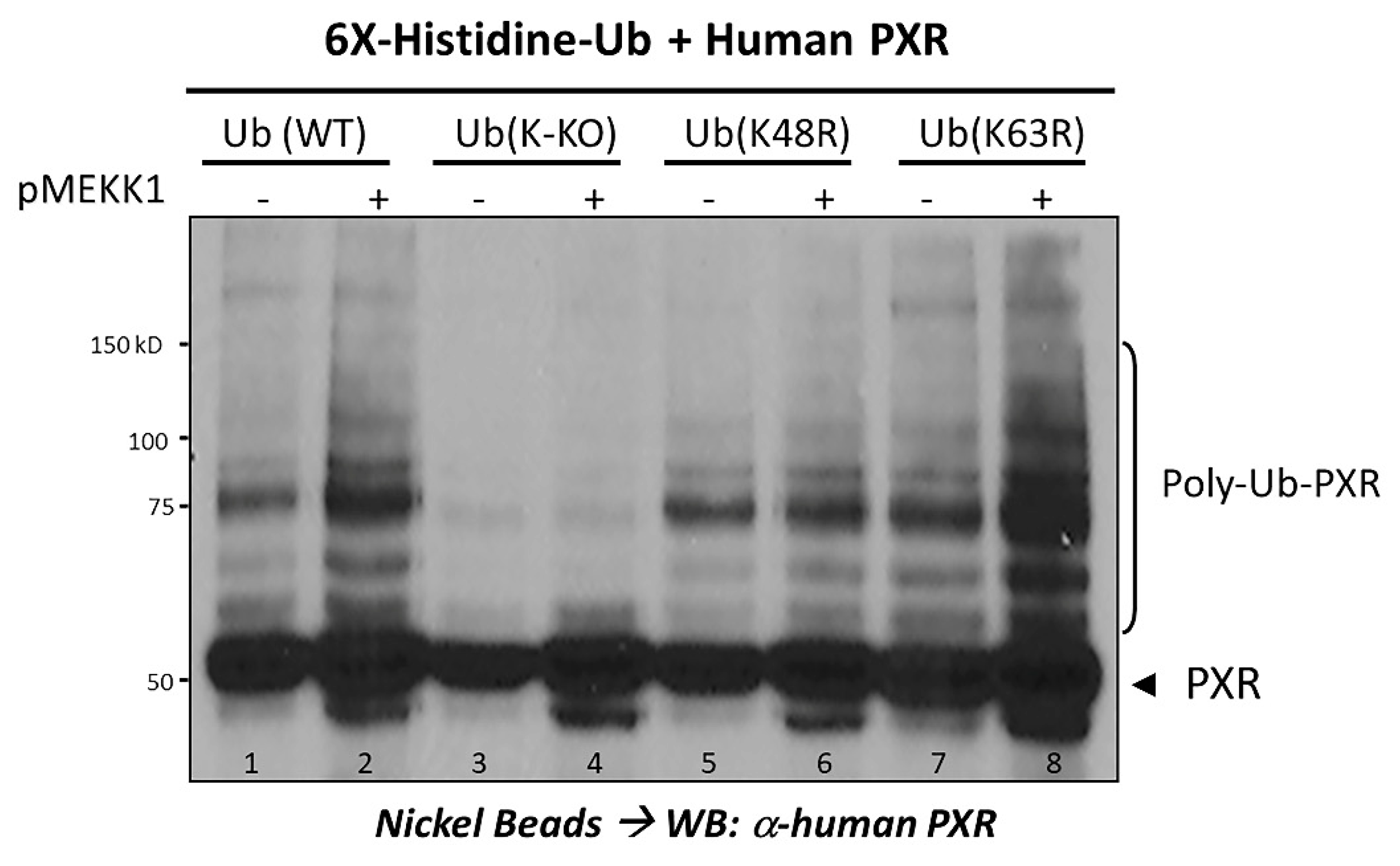
2.3. Ubiquitination
2.4. Acetylation
2.5. Poly(ADP-Ribosyl)ation
3. Conclusions
3.1. PXR and SUMOylation
3.2. A SUMO-Acetyl Switch
3.3. PXR, Phosphorylation, and Drug Metabolism
3.4. PXR and Glucose Metabolism
3.5. PXR and Lipid Metabolism
3.6. PXR and Inflammation
3.7. PXR and Cancer
3.8. PXR-Target Gene Activation and Poly(ADP-Ribosyl)ation (PARylation)
3.9. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bertilsson, G.; Heidrich, J.; Svensson, K.; Asman, M.; Jendeberg, L.; Sydow-Backman, M.; Ohlsson, R.; Postlind, H.; Blomquist, P.; Berkenstam, A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc. Natl. Acad. Sci. USA 1998, 95, 12208–12213. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, B.; Sabbagh, W., Jr.; Juguilon, H.; Bolado, J., Jr.; van Meter, C.M.; Ong, E.S.; Evans, R.M. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 1998, 12, 3195–3205. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Moore, J.T.; Wade, L.; Staudinger, J.L.; Watson, M.A.; Jones, S.A.; McKee, D.D.; Oliver, B.B.; Willson, T.M.; Zetterstrom, R.H.; et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 1998, 92, 73–82. [Google Scholar] [CrossRef]
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef]
- Romankiewicz, J.A.; Ehrman, M. Rifampin and warfarin: A drug interaction. Ann. Intern. Med. 1975, 82, 224–225. [Google Scholar] [CrossRef] [PubMed]
- Heimark, L.D.; Gibaldi, M.; Trager, W.F.; O’Reilly, R.A.; Goulart, D.A. The mechanism of the warfarin-rifampin drug interaction in humans. Clin. Pharm. 1987, 42, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Cytochromes P450, drugs, and diseases. Mol. Interv. 2003, 3, 194–204. [Google Scholar] [CrossRef]
- Staudinger, J.L. Clinical applications of small molecule inhibitors of Pregnane X receptor. Mol. Cell. Endocrinol. 2019, 485, 61–71. [Google Scholar] [CrossRef]
- Goodwin, B.; Gauthier, K.C.; Umetani, M.; Watson, M.A.; Lochansky, M.I.; Collins, J.L.; Leitersdorf, E.; Mangelsdorf, D.J.; Kliewer, S.A.; Repa, J.J. Identification of bile acid precursors as endogenous ligands for the nuclear xenobiotic pregnane X receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 223–228. [Google Scholar] [CrossRef]
- Goodwin, B.; Hodgson, E.; Liddle, C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol. Pharm. 1999, 56, 1329–1339. [Google Scholar] [CrossRef]
- Goodwin, B.; Moore, L.B.; Stoltz, C.M.; McKee, D.D.; Kliewer, S.A. Regulation of the human CYP2B6 gene by the nuclear pregnane X receptor. Mol. Pharm. 2001, 60, 427–431. [Google Scholar]
- Goodwin, B.; Redinbo, M.R.; Kliewer, S.A. Regulation of cyp3a gene transcription by the pregnane x receptor. Annu. Rev. Pharm. Toxicol. 2002, 42, 1–23. [Google Scholar] [CrossRef]
- Xie, W.; Barwick, J.L.; Simon, C.M.; Pierce, A.M.; Safe, S.; Blumberg, B.; Guzelian, P.S.; Evans, R.M. Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev. 2000, 14, 3014–3023. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef]
- Maglich, J.M.; Stoltz, C.M.; Goodwin, B.; Hawkins-Brown, D.; Moore, J.T.; Kliewer, S.A. Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharm. 2002, 62, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, J.; Liu, Y.; Madan, A.; Habeebu, S.; Klaassen, C.D. Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab. Dispos. 2001, 29, 1467–1472. [Google Scholar] [PubMed]
- Staudinger, J.L.; Ding, X.; Lichti, K. Pregnane X receptor and natural products: Beyond drug-drug interactions. Exp. Opin. Drug Metab. Toxicol. 2006, 2, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Zhang, J.; Dowhan, D.H.; Han, Y.; Moore, D.D. Specific and overlapping functions of the nuclear hormone receptors CAR and PXR in xenobiotic response. Pharm. J. 2002, 2, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, J.; Rosenfeld, J.M.; Xu, L.; Evans, R.M.; Xie, W. A nuclear receptor-mediated xenobiotic response and its implication in drug metabolism and host protection. Curr. Drug Metab. 2003, 4, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Fricker, G.; Bauer, B. Pregnane X receptor (PXR) regulates P-glycoprotein at the blood-brain barrier: Functional similarities between pig and human PXR. J. Pharm. Exp. 2009, 329, 141–149. [Google Scholar] [CrossRef]
- Geick, A.; Eichelbaum, M.; Burk, O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 2001, 276, 14581–14587. [Google Scholar] [CrossRef]
- von Richter, O.; Burk, O.; Fromm, M.F.; Thon, K.P.; Eichelbaum, M.; Kivisto, K.T. Cytochrome P450 3A4 and P-glycoprotein expression in human small intestinal enterocytes and hepatocytes: A comparative analysis in paired tissue specimens. Clin. Pharm. 2004, 75, 172–183. [Google Scholar] [CrossRef]
- Masuyama, H.; Nakamura, K.; Nobumoto, E.; Hiramatsu, Y. Inhibition of pregnane X receptor pathway contributes to the cell growth inhibition and apoptosis of anticancer agents in ovarian cancer cells. Int J. Oncol. 2016, 49, 1211–1220. [Google Scholar] [CrossRef]
- Masuyama, H.; Hiramatsu, Y.; Mizutani, Y.; Inoshita, H.; Kudo, T. The expression of pregnane X receptor and its target gene, cytochrome P450 3A1, in perinatal mouse. Mol. Cell. Endocrinol. 2001, 172, 47–56. [Google Scholar] [CrossRef]
- Schote, A.B.; Turner, J.D.; Schiltz, J.; Muller, C.P. Nuclear receptors in human immune cells: Expression and correlations. Mol. Immunol. 2007, 44, 1436–1445. [Google Scholar] [CrossRef] [PubMed]
- Meng, R.; Zhang, X.; Wang, H.; Zhang, D.; Zhao, X. Different Inductive Effects of Praziquantel Racemate and its Enantiomers on the Enzyme CYP3A4 Mediated by Pregnane X Receptor and its Variants. Curr. Drug Metab. 2021, 22, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Siest, G.; Jeannesson, E.; Marteau, J.B.; Samara, A.; Marie, B.; Pfister, M.; Visvikis-Siest, S. Transcription factor and drug-metabolizing enzyme gene expression in lymphocytes from healthy human subjects. Drug Metab. Dispos. 2008, 36, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Pavek, P. Pregnane X Receptor (PXR)-Mediated Gene Repression and Cross-Talk of PXR with Other Nuclear Receptors via Coactivator Interactions. Front. Pharm. 2016, 7, 456. [Google Scholar] [CrossRef]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef]
- Xie, W.; Radominska-Pandya, A.; Shi, Y.; Simon, C.M.; Nelson, M.C.; Ong, E.S.; Waxman, D.J.; Evans, R.M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci. USA 2001, 98, 3375–3380. [Google Scholar] [CrossRef]
- Kandel, B.A.; Thomas, M.; Winter, S.; Damm, G.; Seehofer, D.; Burk, O.; Schwab, M.; Zanger, U.M. Genomewide comparison of the inducible transcriptomes of nuclear receptors CAR, PXR and PPARalpha in primary human hepatocytes. Biochim. Biophys. Acta 2016, 1859, 1218–1227. [Google Scholar] [CrossRef]
- Kawana, K.; Ikuta, T.; Kobayashi, Y.; Gotoh, O.; Takeda, K.; Kawajiri, K. Molecular mechanism of nuclear translocation of an orphan nuclear receptor, SXR. Mol. Pharm. 2003, 63, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Squires, E.J.; Sueyoshi, T.; Negishi, M. Cytoplasmic localization of pregnane X receptor and ligand-dependent nuclear translocation in mouse liver. J. Biol. Chem. 2004, 279, 49307–49314. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.G.; Glass, C.K. Coregulator codes of transcriptional regulation by nuclear receptors. J. Biol. Chem. 2001, 276, 36865–36868. [Google Scholar] [CrossRef] [PubMed]
- Synold, T.W.; Dussault, I.; Forman, B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, A.; Koibuchi, N.; Oka, J.; Taguchi, M.; Shishiba, Y.; Ozawa, Y. Bisphenol-A, an environmental estrogen, activates the human orphan nuclear receptor, steroid and xenobiotic receptor-mediated transcription. Eur J. Endocrinol. 2001, 145, 513–517. [Google Scholar] [CrossRef]
- Takeshita, A.; Taguchi, M.; Koibuchi, N.; Ozawa, Y. Putative role of the orphan nuclear receptor SXR (steroid and xenobiotic receptor) in the mechanism of CYP3A4 inhibition by xenobiotics. J. Biol. Chem. 2002, 277, 32453–32458. [Google Scholar] [CrossRef]
- Wentworth, J.M.; Agostini, M.; Love, J.; Schwabe, J.W.; Chatterjee, V.K. St John’s wort, a herbal antidepressant, activates the steroid X receptor. J. Endocrinol. 2000, 166, R11–R16. [Google Scholar] [CrossRef]
- Bhalla, S.; Ozalp, C.; Fang, S.; Xiang, L.; Kemper, J.K. Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1alpha. Functional implications in hepatic cholesterol and glucose metabolism. J. Biol. Chem. 2004, 279, 45139–45147. [Google Scholar] [CrossRef]
- Johnson, D.R.; Li, C.W.; Chen, L.Y.; Ghosh, J.C.; Chen, J.D. Regulation and binding of pregnane X receptor by nuclear receptor corepressor silencing mediator of retinoid and thyroid hormone receptors (SMRT). Mol. Pharm. 2006, 69, 99–108. [Google Scholar] [CrossRef]
- Mottis, A.; Mouchiroud, L.; Auwerx, J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013, 27, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Tabb, M.M.; Sadatrafiei, A.; Grun, F.; Blumberg, B. Tocotrienols activate the steroid and xenobiotic receptor, SXR, and selectively regulate expression of its target genes. Drug Metab. Dispos. 2004, 32, 1075–1082. [Google Scholar] [CrossRef]
- Ding, X.; Staudinger, J.L. The ratio of constitutive androstane receptor to pregnane X receptor determines the activity of guggulsterone against the Cyp2b10 promoter. J. Pharm. Exp. 2005, 314, 120–127. [Google Scholar] [CrossRef]
- Ding, X.; Staudinger, J.L. Repression of PXR-mediated induction of hepatic CYP3A gene expression by protein kinase C. Biochem Pharm. 2005, 69, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Lin, W.; Wu, J.; Chen, T. Flavonoids activate pregnane x receptor-mediated CYP3A4 gene expression by inhibiting cyclin-dependent kinases in HepG2 liver carcinoma cells. Bmc Biochem 2010, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Lichti-Kaiser, K.; Brobst, D.; Xu, C.; Staudinger, J.L. A systematic analysis of predicted phosphorylation sites within the human pregnane X receptor protein. J. Pharm. Exp. 2009, 331, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Lichti-Kaiser, K.; Xu, C.; Staudinger, J.L. Cyclic AMP-dependent protein kinase signaling modulates pregnane x receptor activity in a species-specific manner. J. Biol. Chem. 2009, 284, 6639–6649. [Google Scholar] [CrossRef]
- Pondugula, S.R.; Brimer-Cline, C.; Wu, J.; Schuetz, E.G.; Tyagi, R.K.; Chen, T. A phosphomimetic mutation at threonine-57 abolishes transactivation activity and alters nuclear localization pattern of human pregnane x receptor. Drug Metab. Dispos. 2009, 37, 719–730. [Google Scholar] [CrossRef]
- Pondugula, S.R.; Dong, H.; Chen, T. Phosphorylation and protein-protein interactions in PXR-mediated CYP3A repression. Exp. Opin. Drug Metab. Toxicol. 2009, 5, 861–873. [Google Scholar] [CrossRef]
- Sivertsson, L.; Edebert, I.; Palmertz, M.P.; Ingelman-Sundberg, M.; Neve, E.P. Induced CYP3A4 expression in confluent Huh7 hepatoma cells as a result of decreased cell proliferation and subsequent pregnane X receptor activation. Mol. Pharm. 2013, 83, 659–670. [Google Scholar] [CrossRef]
- Sugatani, J.; Uchida, T.; Kurosawa, M.; Yamaguchi, M.; Yamazaki, Y.; Ikari, A.; Miwa, M. Regulation of pregnane X receptor (PXR) function and UGT1A1 gene expression by posttranslational modification of PXR protein. Drug Metab. Dispos. 2012, 40, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Hiramatsu, Y.; Kunitomi, M.; Kudo, T.; MacDonald, P.N. Endocrine disrupting chemicals, phthalic acid and nonylphenol, activate Pregnane X receptor-mediated transcription. Mol. Endocrinol. 2000, 14, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Inoshita, H.; Hiramatsu, Y.; Kudo, T. Ligands have various potential effects on the degradation of pregnane X receptor by proteasome. Endocrinology 2002, 143, 55–61. [Google Scholar] [CrossRef]
- Rana, R.; Coulter, S.; Kinyamu, H.; Goldstein, J.A. RBCK1, an E3 ubiquitin ligase, interacts with and ubiquinates the human pregnane X receptor. Drug Metab. Dispos. 2013, 41, 398–405. [Google Scholar] [CrossRef]
- Staudinger, J.L.; Xu, C.; Biswas, A.; Mani, S. Post-translational modification of pregnane x receptor. Pharm. Res. 2011, 64, 4–10. [Google Scholar] [CrossRef]
- Cui, W.; Sun, M.; Galeva, N.; Williams, T.D.; Azuma, Y.; Staudinger, J.L. SUMOylation and Ubiquitylation Circuitry Controls Pregnane X Receptor Biology in Hepatocytes. Drug Metab. Dispos. 2015, 43, 1316–1325. [Google Scholar] [CrossRef]
- Cui, W.; Sun, M.; Zhang, S.; Shen, X.; Galeva, N.; Williams, T.D.; Staudinger, J.L. A SUMO-acetyl switch in PXR biology. Biochim. Biophys. Acta 2016, 1859, 1170–1182. [Google Scholar] [CrossRef]
- Priyanka; Kotiya, D.; Rana, M.; Subbarao, N.; Puri, N.; Tyagi, R.K. Transcription regulation of nuclear receptor PXR: Role of SUMO-1 modification and NDSM in receptor function. Mol. Cell. Endocrinol. 2016, 420, 194–207. [Google Scholar] [CrossRef]
- Wang, C.; Xu, W.; Zhang, Y.; Huang, D.; Huang, K. Poly(ADP-ribosyl)ated PXR is a critical regulator of acetaminophen-induced hepatotoxicity. Cell Death Dis. 2018, 9, 819. [Google Scholar] [CrossRef]
- Ding, X.; Staudinger, J.L. Induction of drug metabolism by forskolin: The role of the pregnane X receptor and the protein kinase a signal transduction pathway. J. Pharm. Exp. 2005, 312, 849–856. [Google Scholar] [CrossRef]
- Doricakova, A.; Novotna, A.; Vrzal, R.; Pavek, P.; Dvorak, Z. The role of residues T248, Y249 and T422 in the function of human pregnane X receptor. Arch. Toxicol. 2013, 87, 291–301. [Google Scholar] [CrossRef]
- Gotoh, S.; Miyauchi, Y.; Moore, R.; Negishi, M. Glucose elicits serine/threonine kinase VRK1 to phosphorylate nuclear pregnane X receptor as a novel hepatic gluconeogenic signal. Cell Signal. 2017, 40, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, S.; Negishi, M. Serum- and glucocorticoid-regulated kinase 2 determines drug-activated pregnane X receptor to induce gluconeogenesis in human liver cells. J. Pharm. Exp. 2014, 348, 131–140. [Google Scholar] [CrossRef]
- Hakkola, J.; Rysa, J.; Hukkanen, J. Regulation of hepatic energy metabolism by the nuclear receptor PXR. Biochim. Biophys. Acta 2016, 1859, 1072–1082. [Google Scholar] [CrossRef]
- Lopez-Borges, S.; Lazo, P.A. The human vaccinia-related kinase 1 (VRK1) phosphorylates threonine-18 within the mdm-2 binding site of the p53 tumour suppressor protein. Oncogene 2000, 19, 3656–3664. [Google Scholar] [CrossRef]
- Cui, W.; Shen, X.; Agbas, E.; Tompkins, B.; Cameron-Carter, H.; Staudinger, J.L. Phosphorylation Modulates the Coregulatory Protein Exchange of the Nuclear Receptor Pregnane X Receptor. J. Pharm. Exp. 2020, 373, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Elias, A.; High, A.A.; Mishra, A.; Ong, S.S.; Wu, J.; Peng, J.; Chen, T. Identification and characterization of phosphorylation sites within the pregnane X receptor protein. Biochem. Pharm. 2014, 87, 360–370. [Google Scholar] [CrossRef]
- Sugatani, J.; Noguchi, Y.; Hattori, Y.; Yamaguchi, M.; Yamazaki, Y.; Ikari, A. Threonine-408 Regulates the Stability of Human Pregnane X Receptor through Its Phosphorylation and the CHIP/Chaperone-Autophagy Pathway. Drug Metab. Dispos. 2016, 44, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.J.; Feng, Y.H.; Gu, B.H.; Li, Y.M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef]
- Bossis, G.; Melchior, F. SUMO: Regulating the regulator. Cell Div. 2006, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Treuter, E.; Venteclef, N. Transcriptional control of metabolic and inflammatory pathways by nuclear receptor SUMOylation. Biochim. Biophys. Acta 2011, 1812, 909–918. [Google Scholar] [CrossRef]
- Rabellino, A.; Andreani, C.; Scaglioni, P.P. The Role of PIAS SUMO E3-Ligases in Cancer. Cancer Res. 2017, 77, 1542–1547. [Google Scholar] [CrossRef]
- Nayak, A.; Muller, S. SUMO-specific proteases/isopeptidases: SENPs and beyond. Genome Biol. 2014, 15, 422. [Google Scholar] [CrossRef]
- Hu, G.; Xu, C.; Staudinger, J.L. Pregnane X receptor is SUMOylated to repress the inflammatory response. J. Pharm. Exp. 2010, 335, 342–350. [Google Scholar] [CrossRef]
- Paunescu, E. In vivo and in vitro suppression of humoral and cellular immunological response by rifampicin. Nature 1970, 228, 1188–1190. [Google Scholar] [CrossRef]
- Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 1994, 79, 13–21. [Google Scholar] [CrossRef]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef]
- Ong, S.S.; Goktug, A.N.; Elias, A.; Wu, J.; Saunders, D.; Chen, T. Stability of the human pregnane X receptor is regulated by E3 ligase UBR5 and serine/threonine kinase DYRK2. Biochem. J. 2014, 459, 193–203. [Google Scholar] [CrossRef]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 2012, 125, 539–548. [Google Scholar] [CrossRef]
- Brown, J.L.; Roberts, W.K. Evidence that approximately eighty per cent of the soluble proteins from Ehrlich ascites cells are Nalpha-acetylated. J. Biol. Chem. 1976, 251, 1009–1014. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Mirsky, A.E. Structural Modifications of Histones and their Possible Role in the Regulation of RNA Synthesis. Science 1964, 144, 559. [Google Scholar] [CrossRef]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Marks, P.A.; Xu, W.S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef]
- Biswas, A.; Pasquel, D.; Tyagi, R.K.; Mani, S. Acetylation of pregnane X receptor protein determines selective function independent of ligand activation. Biochem. Biophys. Res. Commun. 2011, 406, 371–376. [Google Scholar] [CrossRef]
- Pasquel, D.; Doricakova, A.; Li, H.; Kortagere, S.; Krasowski, M.D.; Biswas, A.; Walton, W.G.; Redinbo, M.R.; Dvorak, Z.; Mani, S. Acetylation of lysine 109 modulates pregnane X receptor DNA binding and transcriptional activity. Biochim. Biophys. Acta 2016, 1859, 1155–1169. [Google Scholar] [CrossRef]
- McInerney, E.M.; Rose, D.W.; Flynn, S.E.; Westin, S.; Mullen, T.M.; Krones, A.; Inostroza, J.; Torchia, J.; Nolte, R.T.; Assa-Munt, N.; et al. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev. 1998, 12, 3357–3368. [Google Scholar] [CrossRef]
- Bakshi, K.; Ranjitha, B.; Dubey, S.; Jagannadham, J.; Jaiswal, B.; Gupta, A. Novel complex of HAT protein TIP60 and nuclear receptor PXR promotes cell migration and adhesion. Sci. Rep. 2017, 7, 3635. [Google Scholar] [CrossRef]
- Zaja, R.; Mikoc, A.; Barkauskaite, E.; Ahel, I. Molecular Insights into Poly(ADP-ribose) Recognition and Processing. Biomolecules 2012, 3, 1–17. [Google Scholar] [CrossRef]
- Oliver, F.J.; Menissier-de Murcia, J.; Nacci, C.; Decker, P.; Andriantsitohaina, R.; Muller, S.; de la Rubia, G.; Stoclet, J.C.; de Murcia, G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999, 18, 4446–4454. [Google Scholar] [CrossRef]
- Dou, W.; Mukherjee, S.; Li, H.; Venkatesh, M.; Wang, H.; Kortagere, S.; Peleg, A.; Chilimuri, S.S.; Wang, Z.T.; Feng, Y.; et al. Alleviation of gut inflammation by Cdx2/Pxr pathway in a mouse model of chemical colitis. PLoS ONE 2012, 7, e36075. [Google Scholar] [CrossRef]
- Ma, X.; Shah, Y.M.; Guo, G.L.; Wang, T.; Krausz, K.W.; Idle, J.R.; Gonzalez, F.J. Rifaximin is a gut-specific human pregnane X receptor activator. J. Pharm. Exp. 2007, 322, 391–398. [Google Scholar] [CrossRef]
- Shah, Y.M.; Ma, X.; Morimura, K.; Kim, I.; Gonzalez, F.J. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am. J. Physiol. Gastrointest Liver Physiol. 2007, 292, G1114–G1122. [Google Scholar] [CrossRef]
- Cheng, J.; Shah, Y.M.; Ma, X.; Pang, X.; Tanaka, T.; Kodama, T.; Krausz, K.W.; Gonzalez, F.J. Therapeutic role of rifaximin in inflammatory bowel disease: Clinical implication of human pregnane X receptor activation. J. Pharm. Exp. 2010, 335, 32–41. [Google Scholar] [CrossRef]
- Yan, T.; Luo, Y.; Xia, Y.; Hamada, K.; Wang, Q.; Yan, N.; Krausz, K.W.; Ward, J.M.; Hao, H.; Wang, P.; et al. John’s Wort alleviates dextran sodium sulfate-induced colitis through pregnane X receptor-dependent NFkappaB antagonism. FASEB J. 2021, 35, e21968. [Google Scholar] [CrossRef]
- Guo, G.L.; Moffit, J.S.; Nicol, C.J.; Ward, J.M.; Aleksunes, L.A.; Slitt, A.L.; Kliewer, S.A.; Manautou, J.E.; Gonzalez, F.J. Enhanced acetaminophen toxicity by activation of the pregnane X receptor. Toxicol. Sci. 2004, 82, 374–380. [Google Scholar] [CrossRef]
- Wolf, K.K.; Wood, S.G.; Hunt, J.A.; Walton-Strong, B.W.; Yasuda, K.; Lan, L.; Duan, S.X.; Hao, Q.; Wrighton, S.A.; Jeffery, E.H.; et al. Role of the nuclear receptor pregnane X receptor in acetaminophen hepatotoxicity. Drug Metab. Dispos. 2005, 33, 1827–1836. [Google Scholar] [CrossRef]
- Okamura, M.; Shizu, R.; Abe, T.; Kodama, S.; Hosaka, T.; Sasaki, T.; Yoshinari, K. PXR Functionally Interacts with NF-kappaB and AP-1 to Downregulate the Inflammation-Induced Expression of Chemokine CXCL2 in Mice. Cells 2020, 9, 2296. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, S.; Altmeyer, M. Interplay between Ubiquitin, SUMO, and Poly(ADP-Ribose) in the Cellular Response to Genotoxic Stress. Front. Genet. 2016, 7, 63. [Google Scholar] [CrossRef]
- Yang, S.H.; Sharrocks, A.D. SUMO promotes HDAC-mediated transcriptional repression. Mol. Cell 2004, 13, 611–617. [Google Scholar] [CrossRef]
- Tan, H.; Xu, C.; Zeng, H.; Wang, Y.; Li, Y.; Fan, X.; Chen, P.; Jiang, Y.; Chen, X.; Huang, M.; et al. SUMOylation of pregnane X receptor suppresses rifampicin-induced CYP3A4 and P-gp expression and activity in LS174T cells. J. Pharm. Sci. 2016, 130, 66–71. [Google Scholar] [CrossRef][Green Version]
- Gotoh, S.; Negishi, M. Statin-activated nuclear receptor PXR promotes SGK2 dephosphorylation by scaffolding PP2C to induce hepatic gluconeogenesis. Sci. Rep. 2015, 5, 14076. [Google Scholar] [CrossRef] [PubMed]
- Smutny, T.; Mani, S.; Pavek, P. Post-translational and post-transcriptional modifications of pregnane X receptor (PXR) in regulation of the cytochrome P450 superfamily. Curr. Drug Metab. 2013, 14, 1059–1069. [Google Scholar] [CrossRef]
- Kodama, S.; Koike, C.; Negishi, M.; Yamamoto, Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol. Cell Biol. 2004, 24, 7931–7940. [Google Scholar] [CrossRef]
- Li, L.; Li, H.; Garzel, B.; Yang, H.; Sueyoshi, T.; Li, Q.; Shu, Y.; Zhang, J.; Hu, B.; Heyward, S.; et al. SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol. Pharm. 2015, 87, 674–682. [Google Scholar] [CrossRef]
- Amer, A.O.; Probert, P.M.; Dunn, M.; Knight, M.; Vallance, A.E.; Flecknell, P.A.; Oakley, F.; Cameron, I.; White, S.A.; Blain, P.G.; et al. Sustained Isoprostane E2 Elevation, Inflammation and Fibrosis after Acute Ischaemia-Reperfusion Injury Are Reduced by Pregnane X Receptor Activation. PLoS ONE 2015, 10, e0136173. [Google Scholar] [CrossRef]
- Erickson, S.L.; Alston, L.; Nieves, K.; Chang, T.K.H.; Mani, S.; Flannigan, K.L.; Hirota, S.A. The xenobiotic sensing pregnane X receptor regulates tissue damage and inflammation triggered by C difficile toxins. FASEB J. 2020, 34, 2198–2212. [Google Scholar] [CrossRef]
- Garg, A.; Zhao, A.; Erickson, S.L.; Mukherjee, S.; Lau, A.J.; Alston, L.; Chang, T.K.; Mani, S.; Hirota, S.A. Pregnane X Receptor Activation Attenuates Inflammation-Associated Intestinal Epithelial Barrier Dysfunction by Inhibiting Cytokine-Induced Myosin Light-Chain Kinase Expression and c-Jun N-Terminal Kinase 1/2 Activation. J. Pharm. Exp. 2016, 359, 91–101. [Google Scholar] [CrossRef]
- Kim, S.; Choi, S.; Dutta, M.; Asubonteng, J.O.; Polunas, M.; Goedken, M.; Gonzalez, F.J.; Cui, J.Y.; Gyamfi, M.A. Pregnane X receptor exacerbates nonalcoholic fatty liver disease accompanied by obesity- and inflammation-prone gut microbiome signature. Biochem. Pharm. 2021, 114698. [Google Scholar] [CrossRef]
- Mencarelli, A.; D’Amore, C.; Renga, B.; Cipriani, S.; Carino, A.; Sepe, V.; Perissutti, E.; D’Auria, M.V.; Zampella, A.; Distrutti, E.; et al. Solomonsterol A, a marine pregnane-X-receptor agonist, attenuates inflammation and immune dysfunction in a mouse model of arthritis. Mar. Drugs 2013, 12, 36–53. [Google Scholar] [CrossRef]
- Qiu, Z.; Cervantes, J.L.; Cicek, B.B.; Mukherjee, S.; Venkatesh, M.; Maher, L.A.; Salazar, J.C.; Mani, S.; Khanna, K.M. Pregnane X Receptor Regulates Pathogen-Induced Inflammation and Host Defense against an Intracellular Bacterial Infection through Toll-like Receptor 4. Sci. Rep. 2016, 6, 31936. [Google Scholar] [CrossRef]
- Teng, S.; Piquette-Miller, M. The involvement of the pregnane X receptor in hepatic gene regulation during inflammation in mice. J. Pharm. Exp. 2005, 312, 841–848. [Google Scholar] [CrossRef]
- Sun, M.; Cui, W.; Woody, S.K.; Staudinger, J.L. Pregnane X receptor modulates the inflammatory response in primary cultures of hepatocytes. Drug Metab. Dispos. 2015, 43, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Austin, G.; Holcroft, A.; Rinne, N.; Wang, L.; Clark, R.E. Evidence that the pregnane X and retinoid receptors PXR, RAR and RXR may regulate transcription of the transporter hOCT1 in chronic myeloid leukaemia cells. Eur. J. Haematol. 2015, 94, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Hassen, W.; Kassambara, A.; Reme, T.; Sahota, S.; Seckinger, A.; Vincent, L.; Cartron, G.; Moreaux, J.; Hose, D.; Klein, B. Drug metabolism and clearance system in tumor cells of patients with multiple myeloma. Oncotarget 2015, 6, 6431–6447. [Google Scholar] [CrossRef] [PubMed]
- Pondugula, S.R.; Mani, S. Pregnane xenobiotic receptor in cancer pathogenesis and therapeutic response. Cancer Lett. 2013, 328, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Yogiashi, Y.; Mihara, M.; Takada, I.; Kitagawa, H.; Kato, S. Vitamin K induces osteoblast differentiation through pregnane X receptor-mediated transcriptional control of the Msx2 gene. Mol. Cell Biol. 2007, 27, 7947–7954. [Google Scholar] [CrossRef] [PubMed]
- Kameda, T.; Miyazawa, K.; Mori, Y.; Yuasa, T.; Shiokawa, M.; Nakamaru, Y.; Mano, H.; Hakeda, Y.; Kameda, A.; Kumegawa, M. Vitamin K2 inhibits osteoclastic bone resorption by inducing osteoclast apoptosis. Biochem. Biophys. Res. Commun. 1996, 220, 515–519. [Google Scholar] [CrossRef]
- Tabb, M.M.; Sun, A.; Zhou, C.; Grun, F.; Errandi, J.; Romero, K.; Pham, H.; Inoue, S.; Mallick, S.; Lin, M.; et al. Vitamin K2 regulation of bone homeostasis is mediated by the steroid and xenobiotic receptor SXR. J. Biol. Chem. 2003, 278, 43919–43927. [Google Scholar] [CrossRef]
- Gupta, A.; Mugundu, G.M.; Desai, P.B.; Thummel, K.E.; Unadkat, J.D. Intestinal human colon adenocarcinoma cell line LS180 is an excellent model to study pregnane X receptor, but not constitutive androstane receptor, mediated CYP3A4 and multidrug resistance transporter 1 induction: Studies with anti-human immunodeficiency virus protease inhibitors. Drug Metab. Dispos. 2008, 36, 1172–1180. [Google Scholar] [CrossRef]
- Masuyama, H.; Nakatsukasa, H.; Takamoto, N.; Hiramatsu, Y. Down-regulation of pregnane X receptor contributes to cell growth inhibition and apoptosis by anticancer agents in endometrial cancer cells. Mol. Pharm. 2007, 72, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Venkatesh, M.; Li, H.; Goetz, R.; Mukherjee, S.; Biswas, A.; Zhu, L.; Kaubisch, A.; Wang, L.; Pullman, J.; et al. Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice. J. Clin. Investig. 2011, 121, 3220–3232. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Singh, S.V.; Singh, S.P.; Mu, Y.; Lee, J.H.; Saini, S.P.; Toma, D.; Ren, S.; Kagan, V.E.; Day, B.W.; et al. Orphan nuclear receptor pregnane X receptor sensitizes oxidative stress responses in transgenic mice and cancerous cells. Mol. Endocrinol 2006, 20, 279–290. [Google Scholar] [CrossRef]
- Koutsounas, I.; Patsouris, E.; Theocharis, S. Pregnane X receptor and human malignancy. Histol. Histopathol. 2013, 28, 405–420. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, A.K.; McLaughlin, L.A.; Henderson, C.J.; Wolf, C.R. Activation status of the pregnane X receptor influences vemurafenib availability in humanized mouse models. Cancer Res. 2015, 75, 4573–4581. [Google Scholar] [CrossRef] [PubMed]
- Qiao, E.Q.; Yang, H.J. Effect of pregnane X receptor expression on drug resistance in breast cancer. Oncol. Lett. 2014, 7, 1191–1196. [Google Scholar] [CrossRef]
- Zhuo, W.; Hu, L.; Lv, J.; Wang, H.; Zhou, H.; Fan, L. Role of pregnane X receptor in chemotherapeutic treatment. Cancer Chemother. Pharm. 2014, 74, 217–227. [Google Scholar] [CrossRef]
- Zucchini-Pascal, N.; de Sousa, G.; Pizzol, J.; Rahmani, R. Pregnane X receptor activation protects rat hepatocytes against deoxycholic acid-induced apoptosis. Liver Int. 2010, 30, 284–297. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogers, R.S.; Parker, A.; Vainer, P.D.; Elliott, E.; Sudbeck, D.; Parimi, K.; Peddada, V.P.; Howe, P.G.; D’Ambrosio, N.; Ruddy, G.; et al. The Interface between Cell Signaling Pathways and Pregnane X Receptor. Cells 2021, 10, 3262. https://doi.org/10.3390/cells10113262
Rogers RS, Parker A, Vainer PD, Elliott E, Sudbeck D, Parimi K, Peddada VP, Howe PG, D’Ambrosio N, Ruddy G, et al. The Interface between Cell Signaling Pathways and Pregnane X Receptor. Cells. 2021; 10(11):3262. https://doi.org/10.3390/cells10113262
Chicago/Turabian StyleRogers, Robert S., Annemarie Parker, Phill D. Vainer, Elijah Elliott, Dakota Sudbeck, Kaushal Parimi, Venkata P. Peddada, Parker G. Howe, Nick D’Ambrosio, Gregory Ruddy, and et al. 2021. "The Interface between Cell Signaling Pathways and Pregnane X Receptor" Cells 10, no. 11: 3262. https://doi.org/10.3390/cells10113262
APA StyleRogers, R. S., Parker, A., Vainer, P. D., Elliott, E., Sudbeck, D., Parimi, K., Peddada, V. P., Howe, P. G., D’Ambrosio, N., Ruddy, G., Stackable, K., Carney, M., Martin, L., Osterholt, T., & Staudinger, J. L. (2021). The Interface between Cell Signaling Pathways and Pregnane X Receptor. Cells, 10(11), 3262. https://doi.org/10.3390/cells10113262