Nogo-A Is Critical for Pro-Inflammatory Gene Regulation in Myocytes and Macrophages

 ,
, 
 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. C2C12 Cell Culture
2.2. Recombinant Adenovirus and Si-RNA Transfection of C2C12 Cells
2.3. Isolation, Culture, and Activation of Bone Marrow-Derived Macrophages
2.4. Animal Models Used in This Study
2.5. Muscle Injury
2.6. Induction of Endoplasmic Reticulum (ER) Stress Using Tunicamycin
2.7. Human Myopathy
2.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis
2.9. Western Blot Analysis
2.10. Immunofluorescence (IF) Assay
2.11. Histological Analysis
2.12. Measurement of Cytokine Levels
2.13. Flow Cytometry Analysis
2.14. Migration Assay
2.15. Phagocytosis Assay
2.16. Statistical Analysis
3. Results
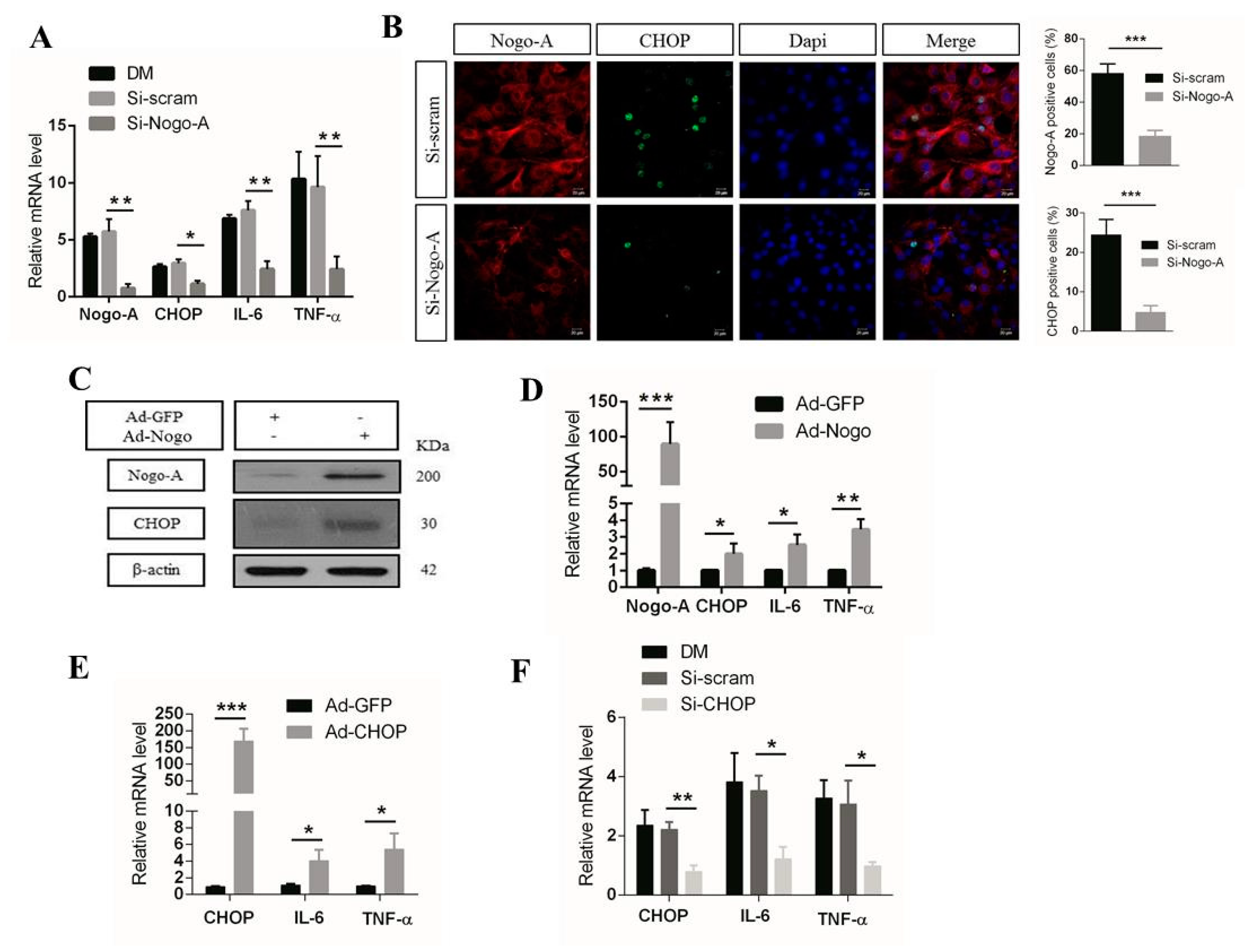
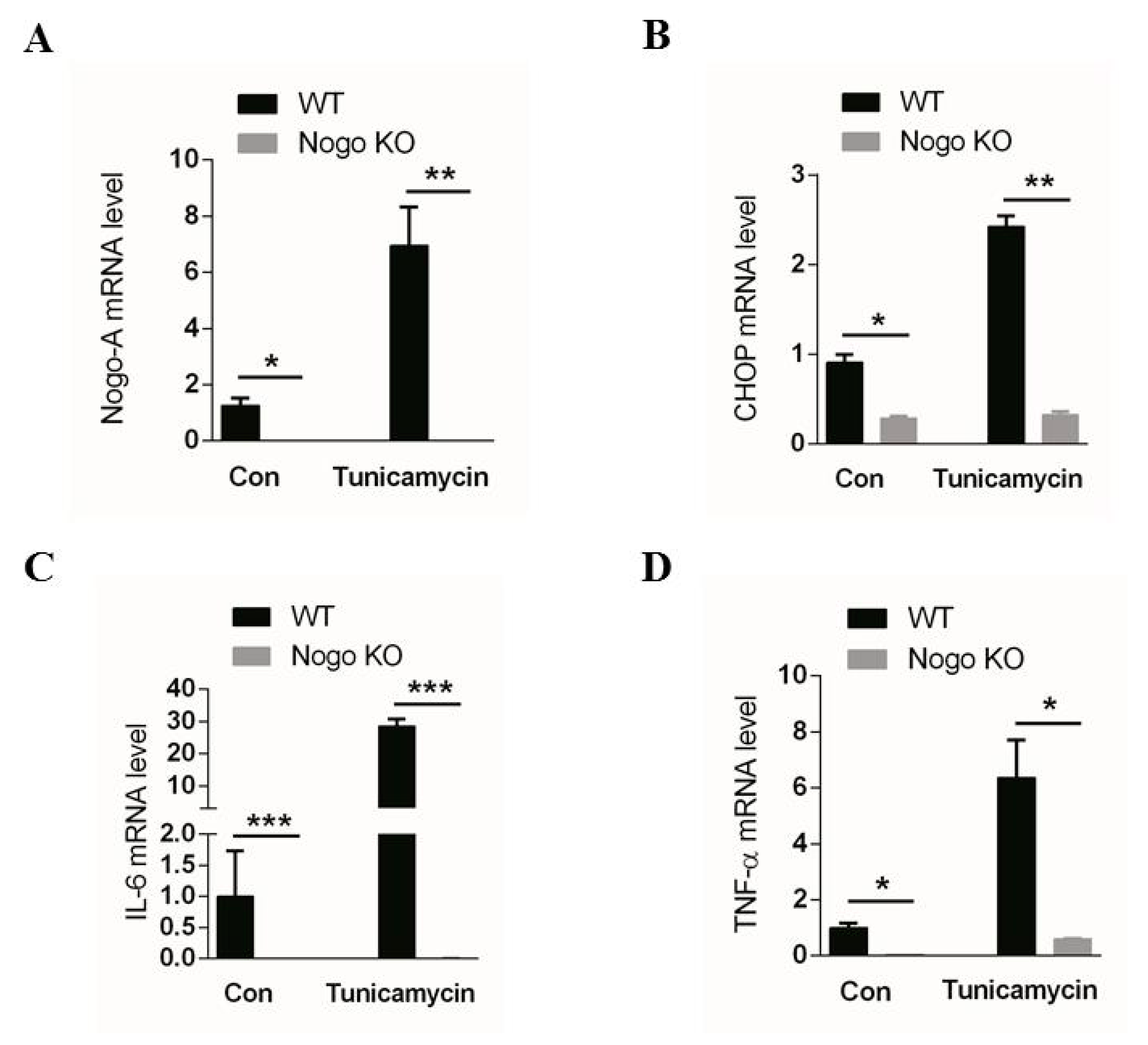
3.1. Nogo-A Regulates CHOP-Mediated Pro-Inflammatory Factor Expression in C2C12 Cells
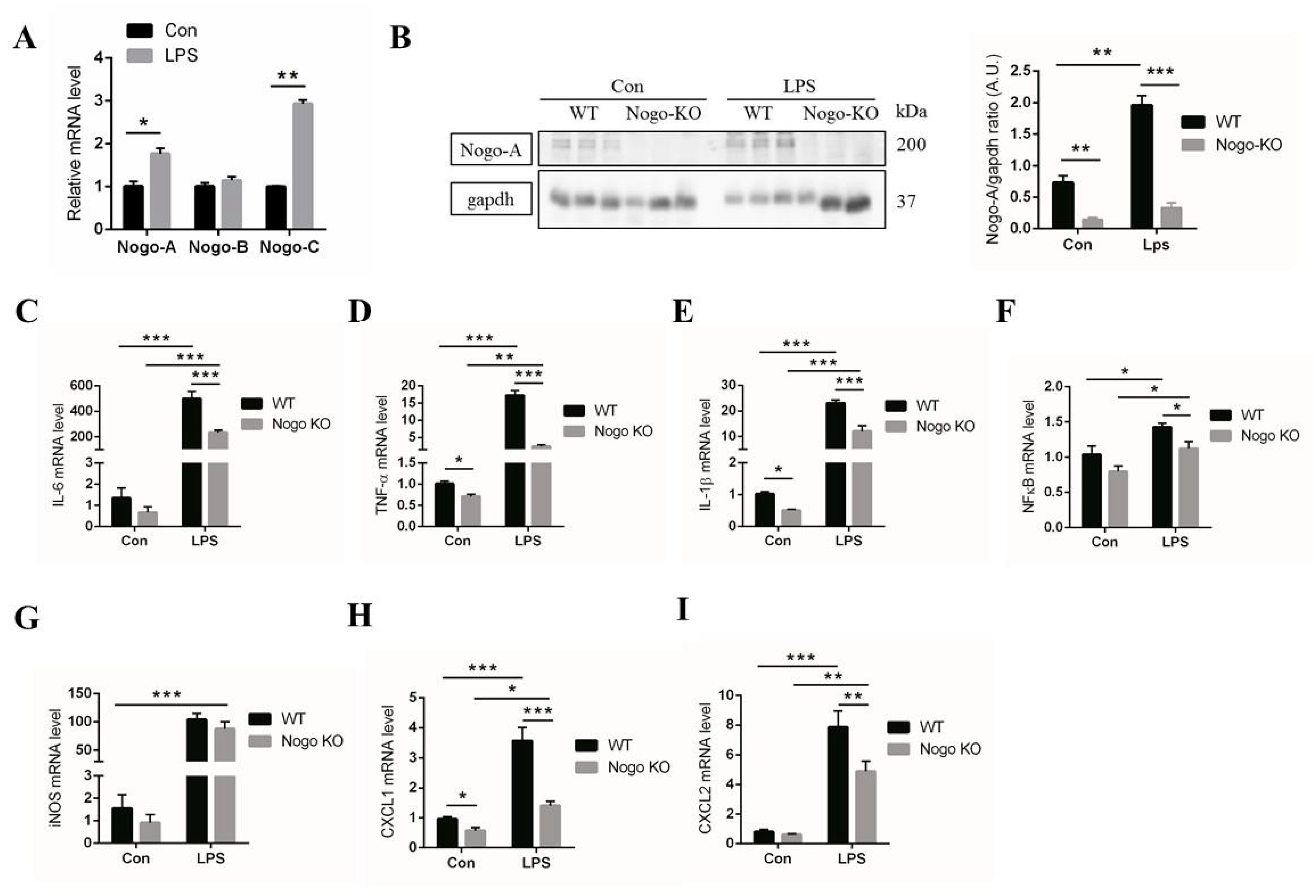
3.2. Nogo Deficiency Suppresses Expression of Pro-Inflammatory Factors in BMDM
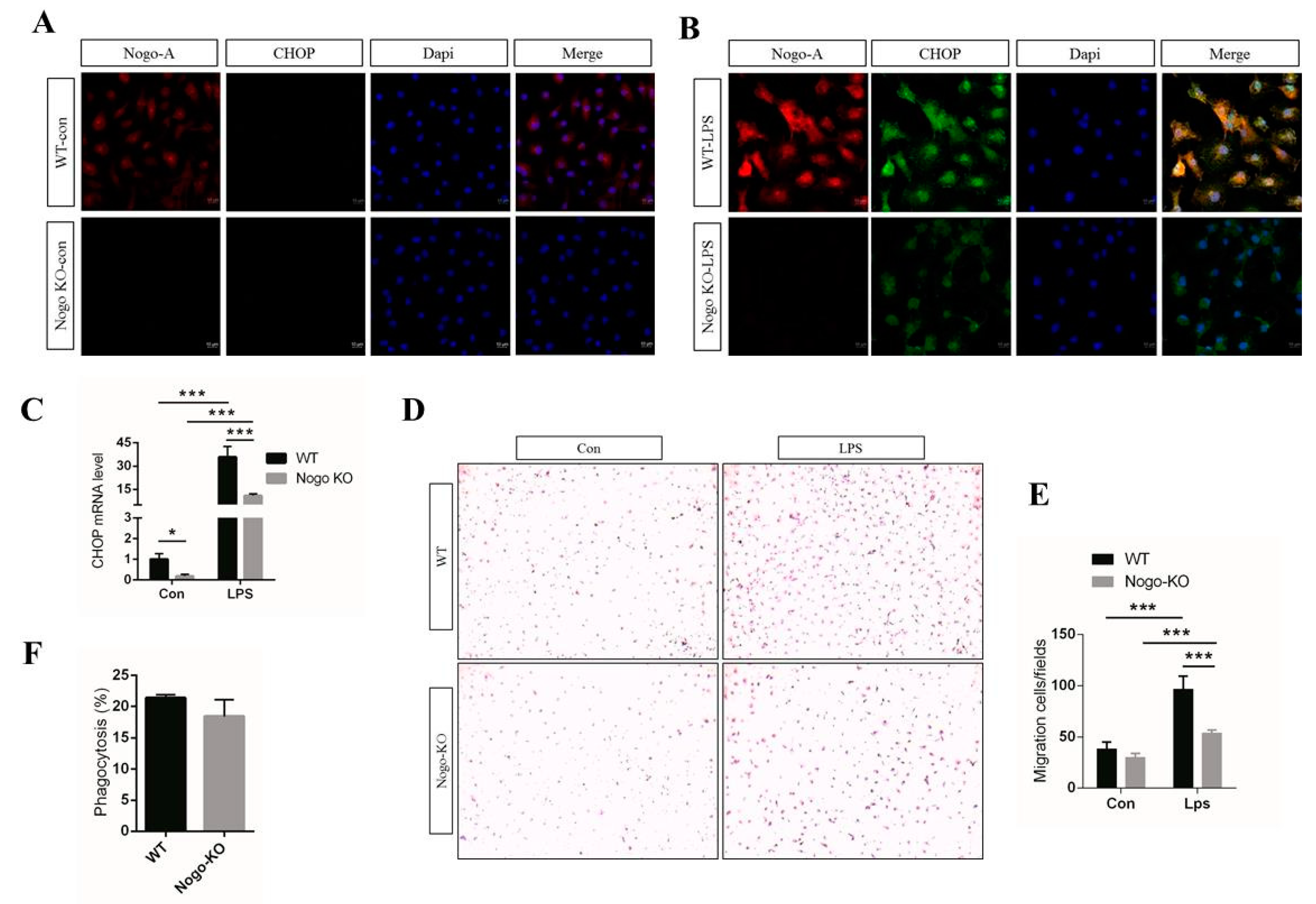
3.3. Nogo Deficiency Suppresses CHOP Signaling and Migration of BMDM
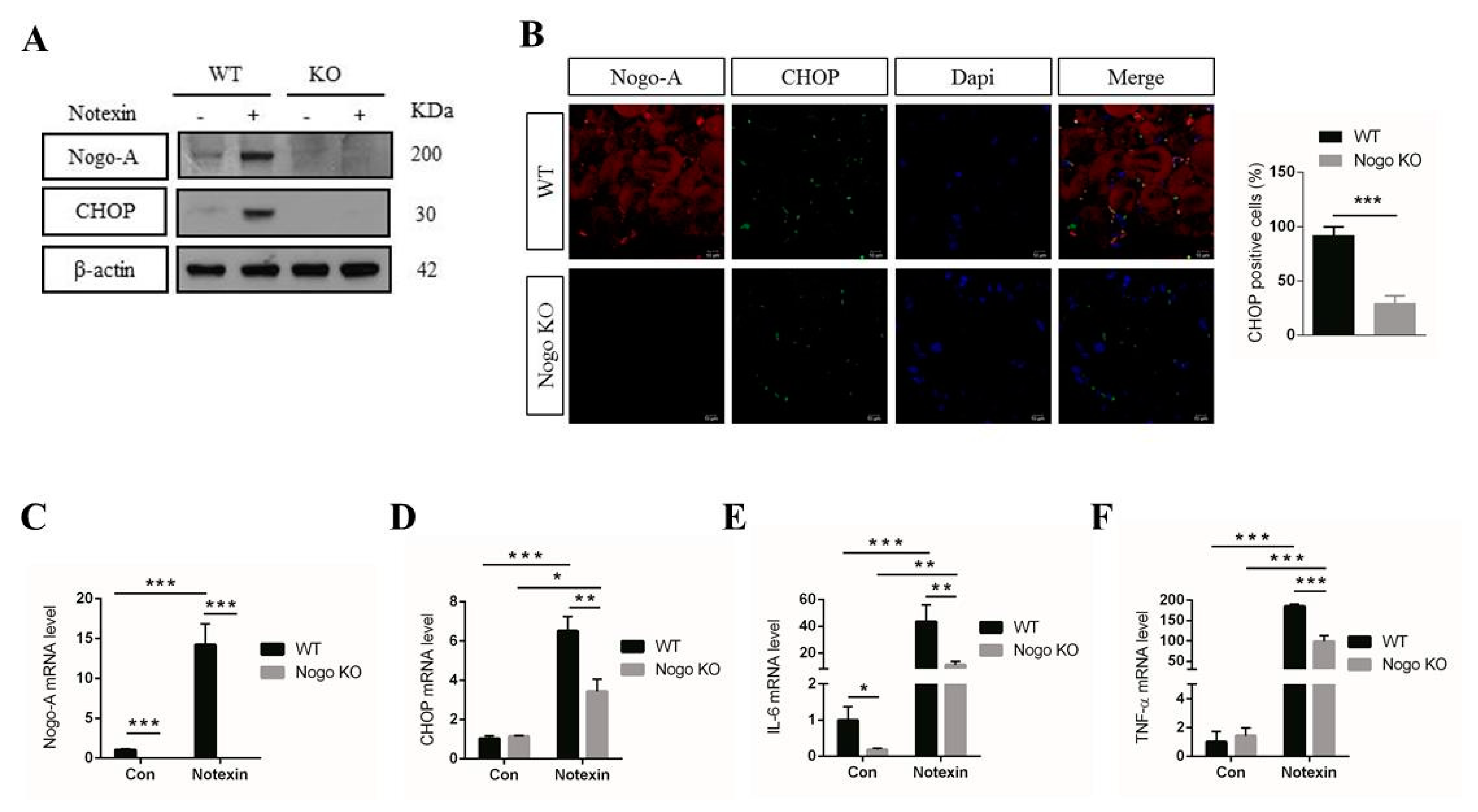
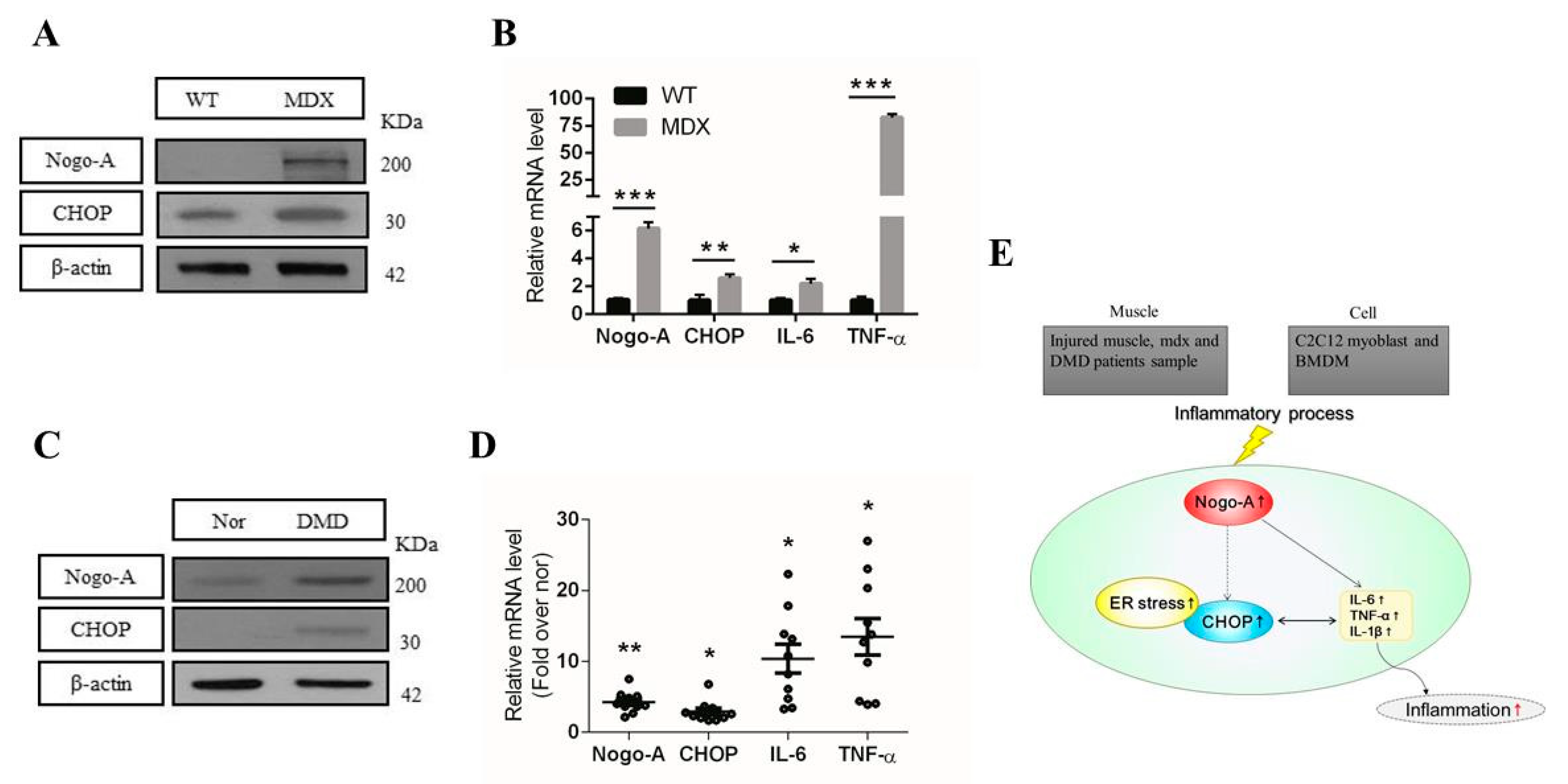
3.4. Nogo-A, CHOP, and Pro-Inflammatory Factors Are Upregulated in Injured Muscle
3.5. Pro-Inflammatory Factor Expression Mediated by CHOP Signaling IS Nogo-A-Dependent
3.6. Expression of Nogo-A, CHOP, and Pro-Inflammatory Factors Is Increased in Mdx Mice and Human DMD Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. 2017, 2, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Ullah, H.A.; Zaman, S.; Juhara, F.; Akter, L.; Tareq, S.M.; Masum, E.H.; Bhattacharjee, R. Evaluation of antinociceptive, in-vivo & in-vitro anti-inflammatory activity of ethanolic extract of Curcuma zedoaria rhizome. BMC Complement. Altern. Med. 2014, 14, 346. [Google Scholar]
- Porter, J.D.; Khanna, S.; Kaminski, H.J.; Rao, J.S.; Merriam, A.P.; Richmonds, C.R.; Leahy, P.; Li, J.; Guo, W.; Andrade, F.H. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2002, 11, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wu, G.; Shi, D.; Zhu, R.; Zeng, H.; Cao, B.; Huang, M.; Liao, H. Effects of nitric oxide on notexin-induced muscle inflammatory responses. Int. J. Biol. Sci. 2015, 11, 156. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.Y.; Sung, B.; Jung, K.J.; Zou, Y.; Yu, B.P. The molecular inflammatory process in aging. Antioxid. Redox Signal. 2006, 8, 572–581. [Google Scholar] [CrossRef]
- Arnold, L.; Perrin, H.; De Chanville, C.B.; Saclier, M.; Hermand, P.; Poupel, L.; Guyon, E.; Licata, F.; Carpentier, W.; Vilar, J. CX3CR1 deficiency promotes muscle repair and regeneration by enhancing macrophage ApoE production. Nat. Commun. 2015, 6, 8972. [Google Scholar] [CrossRef] [Green Version]
- Park, J.K.; Shao, M.; Kim, M.Y.; Baik, S.K.; Cho, M.Y.; Utsumi, T.; Satoh, A.; Ouyang, X.; Chung, C.; Iwakiri, Y. An endoplasmic reticulum protein, Nogo-B, facilitates alcoholic liver disease through regulation of kupffer cell polarization. Hepatology 2017, 65, 1720–1734. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Schultze, J.L.; Schmieder, A.; Goerdt, S. Macrophage activation in human diseases. Semin. Immunol. 2015, 27, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Lee, B.-S.; Jeon, S.-H.; Nam, H.-J.; Lee, G.; Kim, C.-H.; Cho, H.; Lee, J.-H. A novel isoform of met receptor tyrosine kinase blocks hepatocyte growth factor/Met signaling and stimulates skeletal muscle cell differentiation. J. Biol. Chem. 2015, 290, 1804–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schertzer, J.; Lynch, G. Comparative evaluation of IGF-I gene transfer and IGF-I protein administration for enhancing skeletal muscle regeneration after injury. Gene Ther. 2006, 13, 1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Jeong, J.S.; Kim, S.R.; Park, S.Y.; Chae, H.J.; Lee, Y.C. Inhibition of endoplasmic reticulum stress alleviates lipopolysaccharide-induced lung inflammation through modulation of NF-κB/HIF-1α signaling pathway. Sci. Rep. 2013, 3, 1–10. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Filipe, A.; Chernorudskiy, A.; Arbogast, S.; Varone, E.; Villar-Quiles, R.-N.; Pozzer, D.; Moulin, M.; Fumagalli, S.; Cabet, E.; Dudhal, S. Defective endoplasmic reticulum-mitochondria contacts and bioenergetics in SEPN1-related myopathy. Cell Death Differ. 2020, 28, 1–16. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Šereš, M.; Pavlíková, L.; Boháčová, V.; Kyca, T.; Borovská, I.; Lakatoš, B.; Breier, A.; Sulová, Z. Overexpression of GRP78/BiP in P-Glycoprotein-Positive L1210 Cells is Responsible for Altered Response of Cells to Tunicamycin as a Stressor of the Endoplasmic Reticulum. Cells 2020, 9, 890. [Google Scholar] [CrossRef] [Green Version]
- Gorman, A.M.; Healy, S.J.; Jäger, R.; Samali, A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol. Ther. 2012, 134, 306–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Shi, Q.; Song, X.; Wang, Y.; Wang, Y.; Song, E.; Song, Y. Activating transcription factor 4 (ATF4)-ATF3-C/EBP homologous protein (CHOP) cascade shows an essential role in the ER stress-induced sensitization of tetrachlorobenzoquinone-challenged PC12 cells to ROS-mediated apoptosis via death receptor 5 (DR5) signaling. Chem. Res. Toxicol. 2016, 29, 1510–1518. [Google Scholar] [PubMed]
- Endo, M.; Mori, M.; Akira, S.; Gotoh, T. C/EBP Homologous Protein (CHOP) Is Crucial for the Induction of Caspase-11 and the Pathogenesis of Lipopolysaccharide-Induced Inflammatio. J. Immunol. 2006, 176, 6245–6253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Sutendra, G.; Dromparis, P.; Wright, P.; Bonnet, S.; Haromy, A.; Hao, Z.; McMurtry, M.S.; Michalak, M.; Vance, J.E.; Sessa, W.C. The role of Nogo and the mitochondria–endoplasmic reticulum unit in pulmonary hypertension. Sci. Transl. Med. 2011, 3, ra55–ra88. [Google Scholar] [CrossRef] [Green Version]
- Voeltz, G.K.; Prinz, W.A.; Shibata, Y.; Rist, J.M.; Rapoport, T.A. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 2006, 124, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Weng, L.; Jia, S.; Xu, C.; Ye, J.; Cao, Y.; Liu, Y.; Zheng, M. Nogo-C regulates post myocardial infarction fibrosis through the interaction with ER Ca2+ leakage channel Sec61α in mouse hearts. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Dodd, D.A.; Niederoest, B.; Bloechlinger, S.; Dupuis, L.; Loeffler, J.-P.; Schwab, M.E. Nogo-A,-B, and-C are found on the cell surface and interact together in many different cell types. J. Biol. Chem. 2005, 280, 12494–12502. [Google Scholar] [CrossRef] [Green Version]
- Oertle, T.; Klinger, M.; Stuermer, C.A.; Schwab, M.E. A reticular rhapsody: Phylogenic evolution and nomenclature of the RTN/Nogo gene family. FASEB J. 2003, 17, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Wälchli, T.; Pernet, V.; Weinmann, O.; Shiu, J.-Y.; Guzik-Kornacka, A.; Decrey, G.; Yüksel, D.; Schneider, H.; Vogel, J.; Ingber, D.E. Nogo-A is a negative regulator of CNS angiogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, E1943–E1952. [Google Scholar] [CrossRef] [Green Version]
- Schwab, M.E. Nogo and axon regeneration. Curr. Opin. Neurobiol. 2004, 14, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Huber, A.B.; van der Haar, M.E.; Frank, M.; Schnell, L.; Spillmann, A.A.; Christ, F.; Schwab, M.E. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 2000, 403, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.E. Functions of Nogo proteins and their receptors in the nervous system. Nat. Rev. Neurosci. 2010, 11, 799–811. [Google Scholar] [CrossRef]
- Huber, A.B.; Weinmann, O.; Brösamle, C.; Oertle, T.; Schwab, M.E. Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J. Neurosci. 2002, 22, 3553–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acevedo, L.; Yu, J.; Erdjument-Bromage, H.; Miao, R.Q.; Kim, J.-E.; Fulton, D.; Tempst, P.; Strittmatter, S.M.; Sessa, W.C. A new role for Nogo as a regulator of vascular remodeling. Nat. Med. 2004, 10, 382–388. [Google Scholar] [CrossRef]
- Kritz, A.B.; Yu, J.; Wright, P.L.; Wan, S.; George, S.J.; Halliday, C.; Kang, N.; Sessa, W.C.; Baker, A.H. In vivo modulation of Nogo-B attenuates neointima formation. Mol. Ther. 2008, 16, 1798–1804. [Google Scholar] [CrossRef]
- Jia, S.; Qiao, X.; Ye, J.; Fang, X.; Xu, C.; Cao, Y.; Zheng, M. Nogo-C regulates cardiomyocyte apoptosis during mouse myocardial infarction. Cell Death Dis. 2016, 7, e2432. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Tang, X.; Zhang, X.; Zhuang, L. New mutations of Nogo-C in hepatocellular carcinoma. Mol. Biol. Rep. 2009, 36, 377–380. [Google Scholar] [CrossRef]
- Dupuis, L.; de Aguilar, J.-L.G.; di Scala, F.; Rene, F.; de Tapia, M.; Pradat, P.-F.; Lacomblez, L.; Seihlan, D.; Prinjha, R.; Walsh, F.S. Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis. Neurobiol. Dis. 2002, 10, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Jokic, N.; Gonzalez de Aguilar, J.L.; Pradat, P.F.; Dupuis, L.; Echaniz-Laguna, A.; Muller, A.; Dubourg, O.; Seilhean, D.; Hauw, J.J.; Loeffler, J.P. Nogo expression in muscle correlates with amyotrophic lateral sclerosis severity. Ann. Neurol. 2005, 57, 553–556. [Google Scholar] [CrossRef]
- Chen, X.; El Gazzar, M.; Yoza, B.K.; McCall, C.E. The NF-κB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 2009, 284, 27857–27865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horibata, Y.; Mitsuhashi, S.; Shimizu, H.; Maejima, S.; Sakamoto, H.; Aoyama, C.; Ando, H.; Sugimoto, H. The phosphatidylcholine transfer protein StarD7 is important for myogenic differentiation in mouse myoblast C2C12 cells and human primary skeletal myoblasts. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, E.-G.; Ko, H.J.; Cho, Y.-R.; Kim, H.-J.; Ma, Z.; Tim, Y.Y.; Friedline, R.H.; Kurt-Jones, E.; Finberg, R.; Fischer, M.A. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 2009, 58, 2525–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, S.-E.; Hwang, M.; Kim, A.-Y.; Lee, E.-M.; Lee, E.-J.; Hwang, S.-K.; Kim, S.-Y.; Kim, H.-K.; Jeong, K.-S. Myod overexpressed equine adipose-derived stem cells enhanced myogenic differentiation potential. Cell Transplant. 2016, 25, 2017–2026. [Google Scholar] [CrossRef]
- Bertan, F.; Wischhof, L.; Sosulina, L.; Mittag, M.; Dalügge, D.; Fornarelli, A.; Gardoni, F.; Marcello, E.; Di Luca, M.; Fuhrmann, M. Loss of Ryanodine Receptor 2 impairs neuronal activity-dependent remodeling of dendritic spines and triggers compensatory neuronal hyperexcitability. Cell Death Differ. 2020, 27, 3354–3373. [Google Scholar] [CrossRef]
- Chen, W.; Wang, W.; Sun, X.; Xie, S.; Xu, X.; Liu, M.; Yang, C.; Li, M.; Zhang, W.; Liu, W. NudCL2 regulates cell migration by stabilizing both myosin-9 and LIS1 with Hsp90. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Mdzomba, J.B.; Joly, S.; Rodriguez, L.; Dirani, A.; Lassiaz, P.; Behar-Cohen, F.; Pernet, V. Nogo-A-targeting antibody promotes visual recovery and inhibits neuroinflammation after retinal injury. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.X.; Zhang, W.M.; Zhang, H.J.; Li, T.T.; Wang, Y.L.; Qin, Y.W.; Gu, H.; Du, J. Mechanical stretch-induced endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. J. Pathol. 2015, 236, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Fan, S.; Yan, Z.; Xiao, S.; Wan, L.; Chen, C.; Zhong, S.; Liu, L.; Liu, J. Effects of Nogo-A silencing on TNF-α and IL-6 secretion and TH downregulation in lipopolysaccharide-stimulated PC12 cells. Biomed. Res. Int. 2015, 2015, 817914. [Google Scholar] [CrossRef] [Green Version]
- Egami, Y.; Fujii, M.; Kawai, K.; Ishikawa, Y.; Fukuda, M.; Araki, N. Activation-inactivation cycling of Rab35 and ARF6 is required for phagocytosis of zymosan in RAW264 macrophages. J. Immunol. Res. 2015, 2015, 429439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, D.; Fefeu, M.; Besnard, A.; Briand, D.; Gasse, P.; Arenzana-Seisdedos, F.; Rocheteau, P.; Chrétien, F. Defective angiogenesis in CXCL12 mutant mice impairs skeletal muscle regeneration. Skelet. Muscle 2019, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Hardy, D.; Besnard, A.; Latil, M.; Jouvion, G.; Briand, D.; Thépenier, C.; Pascal, Q.; Guguin, A.; Gayraud-Morel, B.; Cavaillon, J.-M. Comparative study of injury models for studying muscle regeneration in mice. PLoS ONE 2016, 11, e0147198. [Google Scholar] [CrossRef] [PubMed]
- Allagnat, F.; Fukaya, M.; Nogueira, T.C.; Delaroche, D.; Welsh, N.; Marselli, L.; Marchetti, P.; Haefliger, J.; Eizirik, D.L.; Cardozo, A.K. C/EBP homologous protein contributes to cytokine-induced pro-inflammatory responses and apoptosis in β-cells. Cell Death Differ. 2012, 19, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Stenina, M.; Krivov, L.; Voevodin, D.; Yarygin, V. Phenotypic differences between mdx black mice and mdx albino mice. Comparison of cytokine levels in the blood. Bull. Exp. Biol. Med. 2013, 155, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Berardinelli, M.G.; Forcina, L.; Spelta, E.; Rizzuto, E.; Nicoletti, C.; Camilli, C.; Testa, E.; Catizone, A.; De Benedetti, F. Increased levels of interleukin-6 exacerbate the dystrophic phenotype in mdx mice. Hum. Mol. Genet. 2015, 24, 6041–6053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharraz, Y.; Guerra, J.; Mann, C.J.; Serrano, A.L.; Muñoz-Cánoves, P. Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediat. Inflamm. 2013, 2013, 491497. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, J.P.; Picchiarelli, G.; Dupuis, L.; Gonzalez De Aguilar, J.L. The role of skeletal muscle in amyotrophic lateral sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef]
- Hattori, T.; Ohoka, N.; Hayashi, H.; Onozaki, K. C/EBP homologous protein (CHOP) up-regulates IL-6 transcription by trapping negative regulating NF-IL6 isoform. FEBS Lett. 2003, 541, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Cruceriu, D.; Baldasici, O.; Balacescu, O.; Berindan-Neagoe, I. The dual role of tumor necrosis factor-alpha (TNF-α) in breast cancer: Molecular insights and therapeutic approaches. Cell. Oncol. 2020, 43, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sathe, A.A.; Smith, G.R.; Ruf-Zamojski, F.; Nair, V.; Lavine, K.J.; Xing, C.; Sealfon, S.C.; Zhou, L. Heterogeneous origins and functions of mouse skeletal muscle-resident macrophages. Proc. Natl. Acad. Sci. USA 2020, 117, 20729–20740. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Komurov, K.; Fletcher, J.S.; Jousma, E.; Cancelas, J.A.; Wu, J.; Ratner, N. An inflammatory gene signature distinguishes neurofibroma Schwann cells and macrophages from cells in the normal peripheral nervous system. Sci. Rep. 2017, 7, 43315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brozzi, F.; Nardelli, T.R.; Lopes, M.; Millard, I.; Barthson, J.; Igoillo-Esteve, M.; Grieco, F.A.; Villate, O.; Oliveira, J.M.; Casimir, M. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 2015, 58, 2307–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Kwon, D.-Y.; Kwak, J.-H.; Lee, S.; Lee, Y.-H.; Yun, J.; Son, T.G.; Jung, Y.-S. Tunicamycin-induced ER stress is accompanied with oxidative stress via abrogation of sulfur amino acids metabolism in the liver. Int. J. Mol. Sci. 2018, 19, 4114. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Park, J.-H.; Kang, U.-B.; Choi, S.-K.; Elfadl, A.; Ullah, H.A.; Chung, M.-J.; Son, J.-Y.; Yun, H.H.; Park, J.-M. Nogo-A regulates myogenesis via interacting with Filamin-C. Cell Death Discov. 2021, 7, 1–18. [Google Scholar] [CrossRef]
- Lu, A.; Guo, P.; Wang, L.; Tseng, C.; Huard, M.; Allen, C.; McCarrick-Walmsley, R.; Whitney, K.E.; Huard, J. Heterogenetic parabiosis between healthy and dystrophic mice improve the histopathology in muscular dystrophy. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| Nogo-A | CTC AGT GGA TGA GAC CCT TTT TGC | CAG TGT TAC CTG GCT GCT CCT |
| Nogo-B | TC AGT GGT TGT TGA CCT CC | GC CGT TAC ACT GAC AAT GC |
| Nogo-C | GAT CGT GGC AAG AAA TGG ACG | AGC AGG AAT AAG CTG GCA CC |
| IL-6 | GGA GAC TTC ACA GAG GAT AC | ATC TCT CTG AAG GAC TCT GG |
| TNF-α | TTC TCA TTC CTG CTT GTG GC | TTG AGA TCC ATG CCG TTG |
| IL-1β | GC ACT ACA GGC TCC GAG ATG AAC | TT GTC GTT GCT TGG TTC TCC TTG T |
| iNOS | TT CAC CCA GTT GTG CAT CGT CCT A | TC CAT GGT CAC CTC CAA CAC AAG A |
| NF-κB | GCC TAC CCG AAA CTC AAC TTC | CTC TTT GGA ACA GGT GCA GAC |
| Cxcl1 | CC GAA GTC ATA GCC ACA CTC A | GT GCC ATC AGA GCA GTC TGT |
| Cxcl2 | GAA GTC ATA GCC ACT CTC AAG G | CCT CCT TTC CAG GTC AGT TAG C |
| CHOP | CCT GAC GAC AGA GTG TTC CAG | CTC CTG CAG ATC CTC ATA CCA |
| CD206 | CA GGT GTG GGC TCA GGT AGT | TG TGG TGA GCT GAA AGG TGA |
| Arginase-1 | CT CCA AGC CAA AGT CCT TAG AG | AG GAG CTG TCA TTA GGG ACA TC |
| IL-10 | GCC TTG CAG AAA AGA GAG CT | AAA GAA AGT CTT CAC CTG GC |
| Gapdh | TCA ATG AAG GGG TCG TTG AT | CGT CCC GTA GAC AAA ATG GT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, H.M.A.; Elfadl, A.K.; Park, S.; Kim, Y.D.; Chung, M.-J.; Son, J.-Y.; Yun, H.-H.; Park, J.-M.; Yim, J.-H.; Jung, S.-J.; et al. Nogo-A Is Critical for Pro-Inflammatory Gene Regulation in Myocytes and Macrophages. Cells 2021, 10, 282. https://doi.org/10.3390/cells10020282
Ullah HMA, Elfadl AK, Park S, Kim YD, Chung M-J, Son J-Y, Yun H-H, Park J-M, Yim J-H, Jung S-J, et al. Nogo-A Is Critical for Pro-Inflammatory Gene Regulation in Myocytes and Macrophages. Cells. 2021; 10(2):282. https://doi.org/10.3390/cells10020282
Chicago/Turabian StyleUllah, H. M. Arif, A. K. Elfadl, SunYoung Park, Yong Deuk Kim, Myung-Jin Chung, Ji-Yoon Son, Hyun-Ho Yun, Jae-Min Park, Jae-Hyuk Yim, Seung-Jun Jung, and et al. 2021. "Nogo-A Is Critical for Pro-Inflammatory Gene Regulation in Myocytes and Macrophages" Cells 10, no. 2: 282. https://doi.org/10.3390/cells10020282
APA StyleUllah, H. M. A., Elfadl, A. K., Park, S., Kim, Y. D., Chung, M.-J., Son, J.-Y., Yun, H.-H., Park, J.-M., Yim, J.-H., Jung, S.-J., Choi, Y.-C., Shin, J.-H., Kim, D.-S., Park, J.-K., & Jeong, K.-S. (2021). Nogo-A Is Critical for Pro-Inflammatory Gene Regulation in Myocytes and Macrophages. Cells, 10(2), 282. https://doi.org/10.3390/cells10020282