
Chronic Intermittent Mild Whole-Body Hypothermia Is Therapeutic in a Mouse Model of ALS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Tg Mice and Cold-Acclimation Protocol, Testing, and Triage for Endpoints
2.3. Spinal Cord and Neuromuscular Pathology
2.4. Immunoblotting and Immunoprecipitation (IP)
2.5. Data Analysis
2.6. Photography and Figure Construction
3. Results
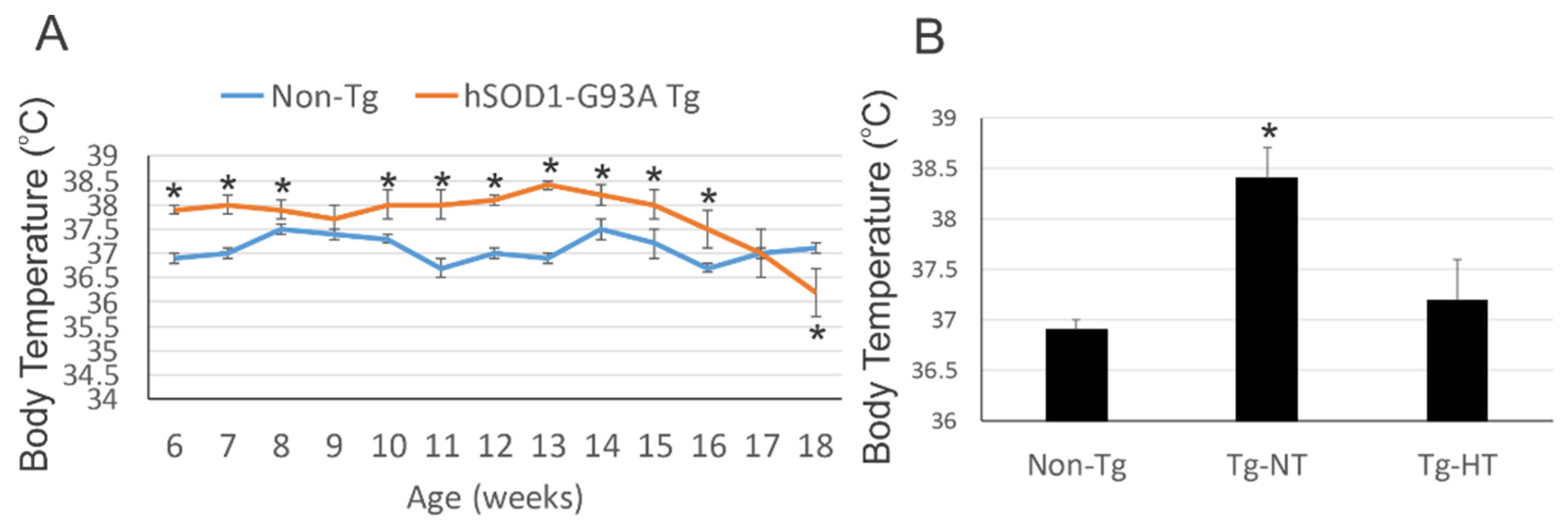
3.1. hSOD1-G93A Tg Mice Are Chronically Febrile
3.2. Environmental Temperature Management Can Induce Mild Hypothermia in hSOD1-G93A Tg Mice
3.3. Whole-Body Cold Acclimation Improves Neurological Outcome and Survival of hSOD1-G93A Tg Mice
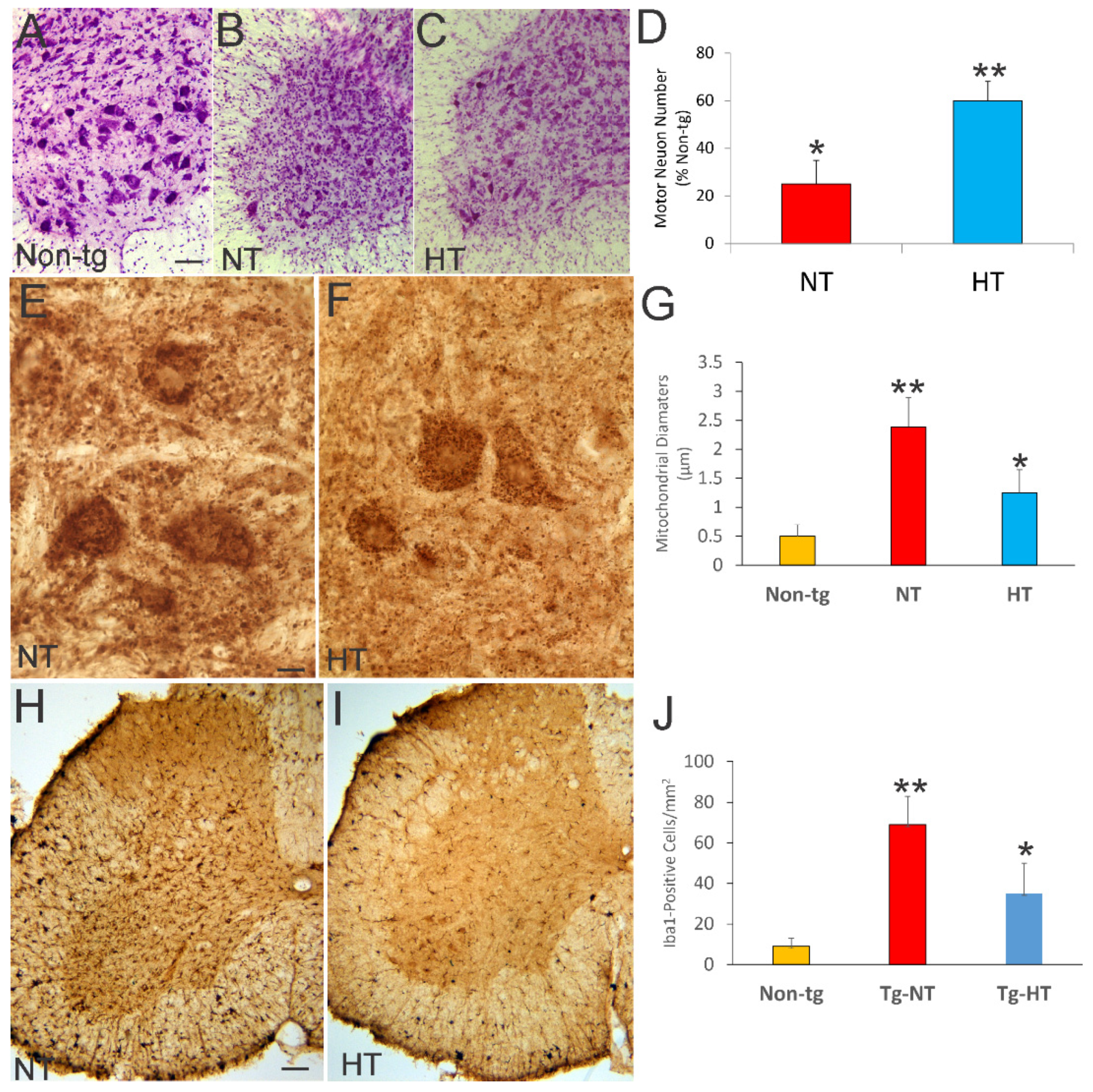
3.4. Whole-Body Cold Cooling Protects Spinal Cord MNs and Their Mitochondria and Mitigates Inflammation in hSOD1-G93A Tg Mice
3.5. Whole-Body Cold Cooling Protects the NMJ
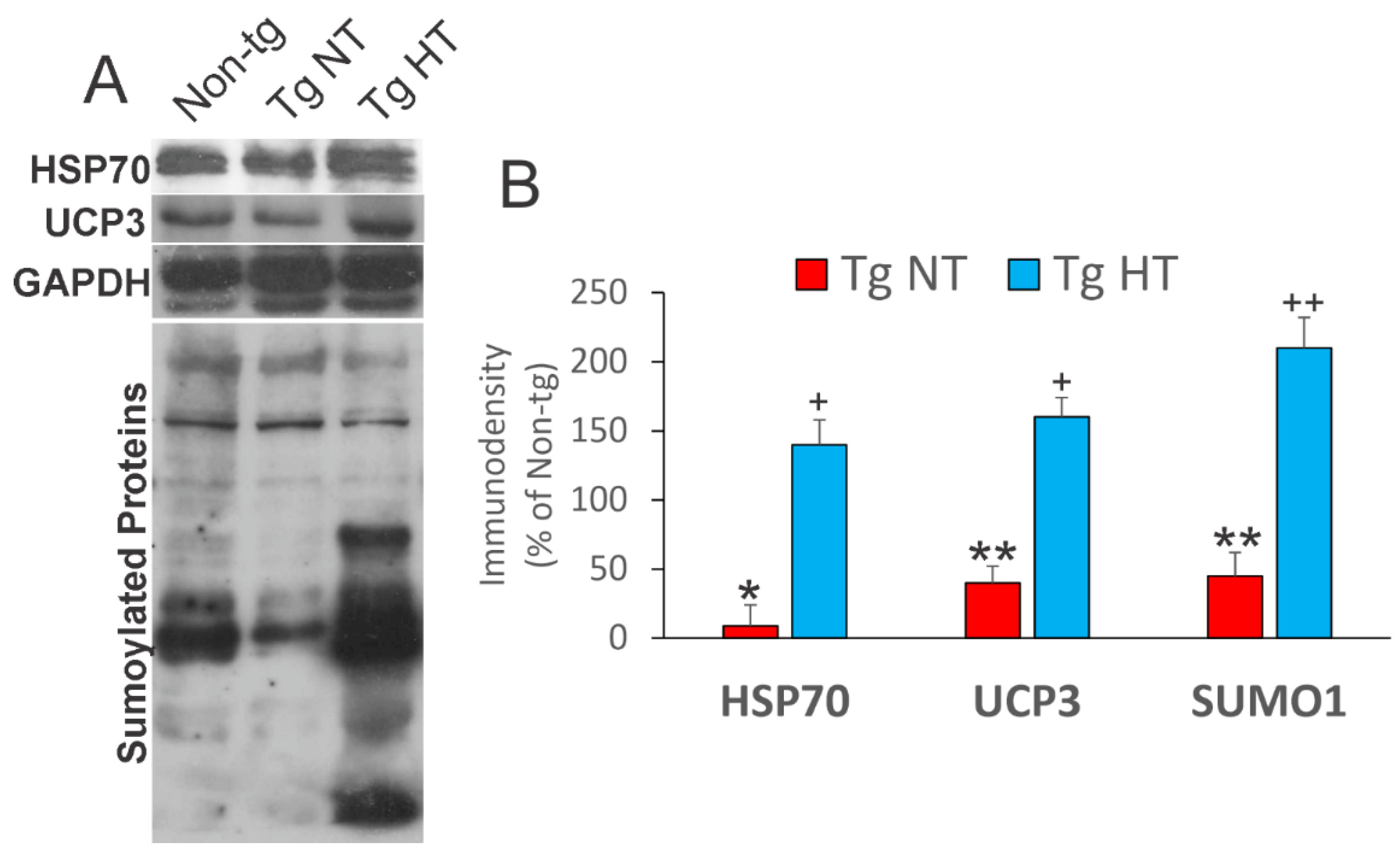
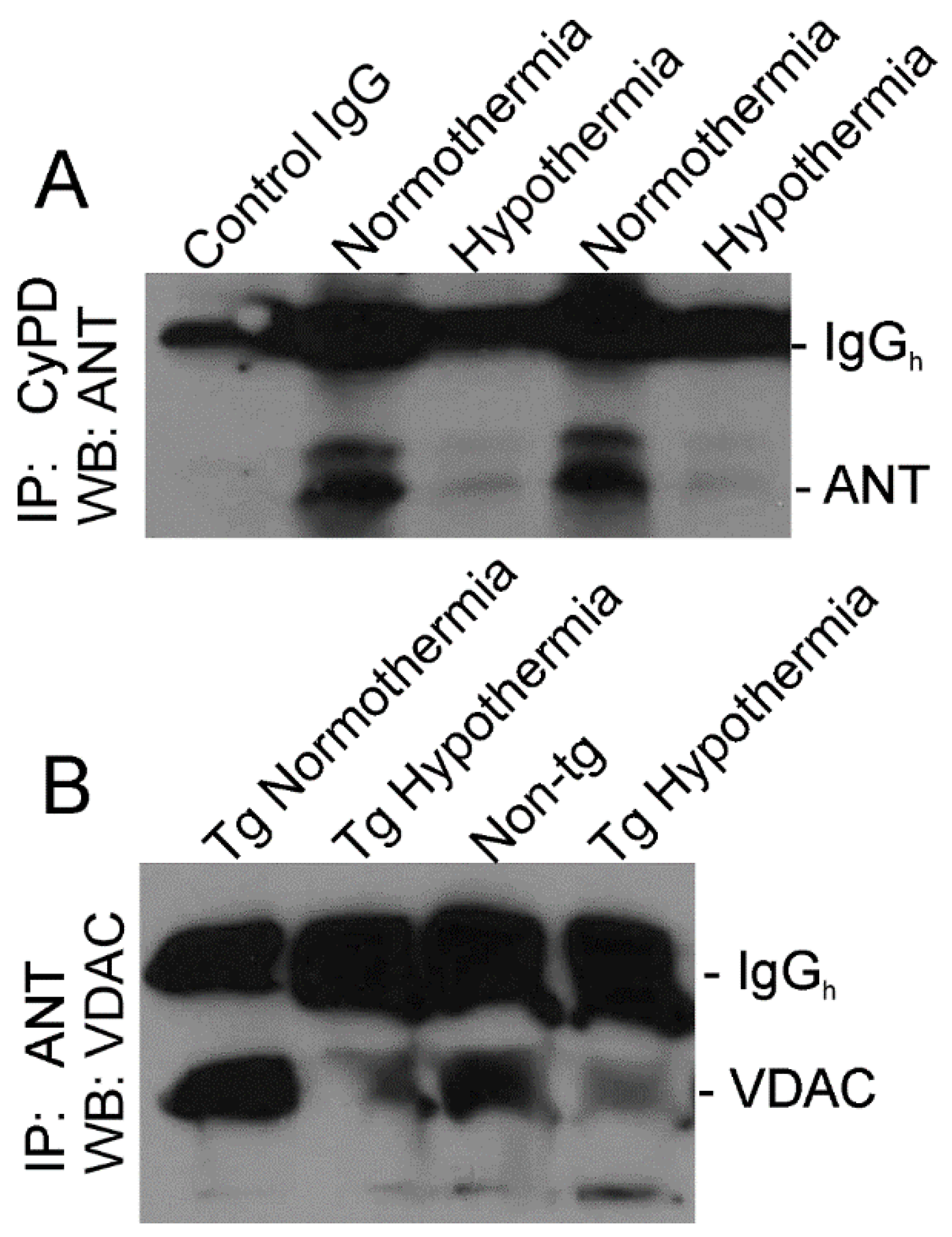
3.6. Whole-Body Cold Acclimation Induces Cytoprotection Mechanisms and Disengages the Mitochondrial Permeability Transition Pore (mPTP) in Skeletal Muscle of hSOD1-G93A Tg Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J. Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases. Pharmaceuticals 2010, 3, 839–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, T.C.; Kochanek, P.M. A New Vision for Therapeutic Hypothermia in the Era of Targeted Temperature Management: A Speculative Synthesis. Ther. Hypothermia Temp. Manag. 2019, 9, 13–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouteloup, C.; Desport, J.-C.; Clavelou, P.; Guy, N.; Derumeaux-Burel, H.; Ferrier, A.; Couratier, P. Hypermetabolism in ALS patients: An early and persistent phenomenon. J. Neurol. 2009, 256, 1236–1242. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Martin, L.J.; Liu, Z.; Chen, K.; Price, A.C.; Pan, Y.; Swaby, J.A.; Golden, W.C. Motor neuron degeneration in ALS mutant su-peroxide dismutase-1 transgenic mice: Mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 2007, 500, 2046. [Google Scholar] [CrossRef]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Northington, F.J.; Martin, L.J. Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain Struct. Funct. 2009, 214, 219–234. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J.; Wong, M.; Hanaford, A. Neonatal Brain Injury and Genetic Causes of Adult-Onset Neurodegenerative Disease in Mice Interact With Effects on Acute and Late Outcomes. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- Clerc, P.; Lipnick, S.; Willett, C. A look into the future of ALS research. Drug Discov. Today 2016, 21, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J.; Wong, M. Skeletal Muscle-Restricted Expression of Human SOD1 in Transgenic Mice Causes a Fatal ALS-Like Syndrome. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef]
- Comerford, P.T.; Fitzgerald, M.J.T. Motor innervation of rodent diaphragm. J. Anat. 1986, 149, 171–175. [Google Scholar]
- Martin, L.J.; Liu, Z. Adult olfactory bulb neural precursor cell grafts provide temporary protection from motor neuron de-generation, improve motor function, and extend survival in amyotrophic lateral sclerosis mice. J. Neuropathol. Exp. Neurol. 2007, 66, 1002–1018. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J. An Approach to Experimental Synaptic Pathology Using Green Fluorescent Protein-Transgenic Mice and Gene Knockout Mice to Show Mitochondrial Permeability Transition Pore-Driven Excitotoxicity in Interneurons and Motoneurons. Toxicol. Pathol. 2010, 39, 220–233. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J. p53 is Abnormally Elevated and Active in the CNS of Patients with Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2000, 7, 613–622. [Google Scholar] [CrossRef]
- Martin, L.J.; Price, A.C.; McClendon, K.B.; Al-Abdulla, N.A.; Subramaniam, J.R.; Wong, P.C.; Liu, Z. Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: Oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J. Neurochem. 2003, 85, 234–247. [Google Scholar] [CrossRef]
- Porter, G.A., Jr.; Beutner, G.; Cyclophilin, D. somehow a master regulator to mitochondrial function. Biomolecules 2018, 8, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, C.P. The molecular composition of the mitochondrial permeability transition pore. J. Mol. Cell. Cardiol. 2009, 46, 850–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankaran, S.; Laptook, A.R.; Ehrenkranz, R.A.; Tyson, J.E.; McDonald, S.A.; Donovan, E.F.; Fanaroff, A.A.; Poole, W.K.; Wright, L.L.; Higgins, R.D.; et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N. Engl. J. Med. 2005, 353, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, S.; Pappas, A.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Yolton, K.; Gustafson, K.E.; Leach, T.M.; Green, C.; Bara, R.; et al. Childhood Outcomes after Hypothermia for Neonatal Encephalopathy. New Engl. J. Med. 2012, 366, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, D.; Strohm, B.; Marlow, N.; Brocklehurst, P.; Deierl, A.; Eddama, O.; Goodwin, J.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; et al. Effects of Hypothermia for Perinatal Asphyxia on Childhood Outcomes. New Engl. J. Med. 2014, 371, 140–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Wang, B.; Lee, J.-H.; Armstrong, J.S.; Kulikowicz, E.; Bhalala, U.S.; Martin, L.J.; Koehler, R.C.; Yang, Z.-J. Additive Neuroprotection of a 20-HETE Inhibitor with Delayed Therapeutic Hypothermia after Hypoxia-Ischemia in Neonatal Piglets. Dev. Neurosci. 2015, 37, 376–389. [Google Scholar] [CrossRef] [Green Version]
- Fendrick, S.E.; Xue, Q.-S.; Streit, W.J. Formation of multinucleated giant cells and microglial degeneration in rats expressing a mutant Cu/Zn superoxide dismutase gene. J. Neuroinflammation 2007, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Martinex, L.; de la Torre, M.; Toivonen, J.M.; Zaragoza, P.; Garcia-Redondo, A.; Calva, A.C.; Osta, R. Circulating cytokines could not be good prognostic biomarkers in a mouse model of amyotrophic lateral sclerosis. Front. Immunol. 2019, 10, 801. [Google Scholar] [CrossRef]
- Netea, M.G.; Kullberg, B.J.; Van Der Meer, J.W.M. Circulating Cytokines as Mediators of Fever. Clin. Infect. Dis. 2000, 31, S178–S184. [Google Scholar] [CrossRef]
- Tortarolo, M.; Coco, D.L.; Veglianese, P.; Vallarola, A.; Giordana, M.T.; Marcon, G.; Beghi, E.; Poloni, M.; Strong, M.J.; Iyer, A.M.; et al. Amyotrophic Lateral Sclerosis, a Multisystem Pathology: Insights into the Role of TNFα. Mediat. Inflamm. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Duan, K.; Gao, X.; Zhu, D. The clinical relevance and mechanism of skeletal muscle wasting. Clin. Nutr. 2021, 40, 27–37. [Google Scholar] [CrossRef]
- Sakuma, K.; Aoi, W.; Yamaguchi, A. Molecular mechanism of sarcopenia and cachexia: Recent research advances. Pflügers Archiv – Eur. J. Physiol. 2017, 469, 573–591. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Bonconpagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal Muscle Is a Primary Target of SOD1G93A-Mediated Toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef]
- Dupuis, L.; Petersen, A.; Wedt, P. Thermoregulation in amyotrophic lateral sclerosis. In Thermoregulation: From Basic Neuroscience to Clinical Neurology, Part II; Romanovsky, A.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 157, pp. 749–760. [Google Scholar]
- Dupuis, L.; Oudart, H.; René, F.; Gonzalez de Aguilar, J.L.; Loeffler, J.P. Evidence for defective energy homeostasis in amyo-trophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef] [Green Version]
- Nolano, M.; Provitera, V.; Manganelli, F.; Iodice, R.; Caporaso, G.; Stancanelli, A.; Marinou, K.; Lanzillo, B.; Santoro, L.; Mora, G. Non-motor involvement in amyotrophic lateral sclerosis: New insight from nerve and vessel analysis in skin biopsy. Neuropathol. Appl. Neurobiol. 2017, 43, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Weis, J.; Katona, I.; Müller-Newen, G.; Sommer, C.; Necula, G.; Hendrich, C.; Ludolph, A.C.; Sperfeld, A.-D. Small-fiber neuropathy in patients with ALS. Neurology 2011, 76, 2024–2029. [Google Scholar] [CrossRef] [PubMed]
- Bella, E.D.; Lombardi, R.; Porretta-Serapiglia, C.; Ciano, C.; Gellera, C.; Pensato, V.; Cazzato, D.; Lauria, G. Amyotrophic lateral sclerosis causes small fiber pathology. Eur. J. Neurol. 2016, 23, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Gorges, M.; Vercruysse, P.; Müller, H.-P.; Huppertz, H.-J.; Rosenbohm, A.; Nagel, G.; Weydt, P.; Petersén, Å.; Ludolph, A.C.; Kassubek, J.; et al. Hypothalamic atrophy is related to body mass index and age at onset in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gampert, L.; Nething, K.; Steinacker, J.M. Response and function of skeletal muscle heat shock protein 70. Front. Biosci. 2006, 11, 2802–2827. [Google Scholar] [CrossRef] [Green Version]
- Gifondorwa, D.J.; Robinson, M.B.; Hayes, C.D.; Taylor, A.; Prevette, D.M.; Oppenheim, R.W.; Caress, J.; Milligan, C.E. Exogenous Delivery of Heat Shock Protein 70 Increases Lifespan in a Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2007, 27, 13173–13180. [Google Scholar] [CrossRef]
- Wang, L.; Ma, Q.; Yang, W.; Mackensen, G.B.; Paschen, W. Moderate hypothermia induces marked increase in levels and nuclear accumulation of SUMO2/3-conjugated proteins in neurons. J. Neurochem. 2012, 123, 349–359. [Google Scholar] [CrossRef]
- Jalal, D.; Chalissery, J.; Hassan, A.H. Genome maintenance in Saccharomyces cerevisiae: The role of SUMO and SUMO-targeted ubiquitin ligases. Nucleic Acid Res. 2017, 45, 2242–2261. [Google Scholar]
- Mann, D.M.A.; Yates, P.O. Motor neurone disease: The nature of the pathogenic mechanism. J. Neurol. Neurosurg. Psychiatry 1974, 37, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Bradley, W.G.; Krasin, F. A New Hypothesis of the Etiology of Amyotrophic Lateral Sclerosis. Arch. Neurol. 1982, 39, 677–680. [Google Scholar] [CrossRef]
- Fitzmaurice, P.S.; Shaw, I.C.; E Kleiner, H.; Miller, R.T.; Monks, T.J.; Lau, S.S.; Mitchell, J.D.; Lynch, P.G. Evidence for DNA damage in amyotrophic lateral sclerosis. Muscle Nerve 1996, 19, 797–798. [Google Scholar]
- Martin, L.J. DNA damage and repair: Relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Commun. 2020, 8, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Wong, M. Enforced DNA repair enzymes rescue neurons from apoptosis induced by target deprivation and axotomy in mouse models of neurodegeneration. Mech. Ageing Dev. 2017, 161, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, V.; Gane, B.D.; Nandhakumar, S.; Rao, R. Effect of therapeutic hypothermia on chromosomal aberration in perinatal asphyxia. J. Pediatr. Neurosci. 2016, 11, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M. Enhanced Uncoupling of the Mitochondrial Respiratory Chain as a Potential Source for Amyotrophic Lateral Sclerosis. Front. Neurol. 2013, 4, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal-Puig, A.J.; Grujic, D.; Zhang, C.-Y.; Hagen, T.; Boss, O.; Ido, Y.; Szczepanik, A.; Wade, J.; Mootha, V.; Cortright, R.; et al. Energy Metabolism in Uncoupling Protein 3 Gene Knockout Mice. J. Biol. Chem. 2000, 275, 16258–16266. [Google Scholar] [CrossRef] [Green Version]
- Rousset, S.; Alves-Guerra, M.C.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.M.; Bouillaud, F.; Ricquier, D. The biology of mi-tochondrial uncoupling proteins. Diabetes 2004, 53 (Suppl. S1), S130–S135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabalina, I.G.; Hoeks, J.; Kramarova, T.V.; Schrauwen, P.; Cannon, B.; Nedergaard, J. Cold tolerance of UCP1-ablated mice: A skeletal muscle mitochondria switch toward lipid oxidation with marked UCP3 up-regulation not associated with increased basal, fatty acid- or ROS-induced uncoupling or enhanced GDP effects. Biochim. et Biophys. Acta (BBA)—Bioenerg. 2010, 1797, 968–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in mitochondrial pathobi-ology. Biochim. Biophys. Acta 2010, 1997, 1113–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J.; Fancelli, D.; Wong, M.; Niedzwiecki, M.; Ballarini, M.; Plyte, S.; Chang, Q. GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 433. [Google Scholar] [CrossRef] [Green Version]
- Jahandiez, V.; Cour, M.; Bochaton, T.; Abrial, M.; Loufouat, J.; Gharib, A.; Varennes, A.; Ovize, M.; Argaud, L. Fast therapeutic hypothermia prevents post-cardiac arrest syndrome through cyclophilin D-mediated mitochondrial permeability transition inhibition. Basic Res. Cardiol. 2017, 112, 35. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, L.J.; Niedzwiecki, M.V.; Wong, M. Chronic Intermittent Mild Whole-Body Hypothermia Is Therapeutic in a Mouse Model of ALS. Cells 2021, 10, 320. https://doi.org/10.3390/cells10020320
Martin LJ, Niedzwiecki MV, Wong M. Chronic Intermittent Mild Whole-Body Hypothermia Is Therapeutic in a Mouse Model of ALS. Cells. 2021; 10(2):320. https://doi.org/10.3390/cells10020320
Chicago/Turabian StyleMartin, Lee J., Mark V. Niedzwiecki, and Margaret Wong. 2021. "Chronic Intermittent Mild Whole-Body Hypothermia Is Therapeutic in a Mouse Model of ALS" Cells 10, no. 2: 320. https://doi.org/10.3390/cells10020320