
Cell Line Models for Acquired Resistance to First-Line Osimertinib in Lung Cancers—Applications and Limitations



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Identifying Mechanisms of Resistance to EGFR-TKIs through Cell Line Models
3. Cell Line Models Used to Analyze Resistance Mechanisms to First-Line Osimertinib
3.1. Search Criteria for Published Studies
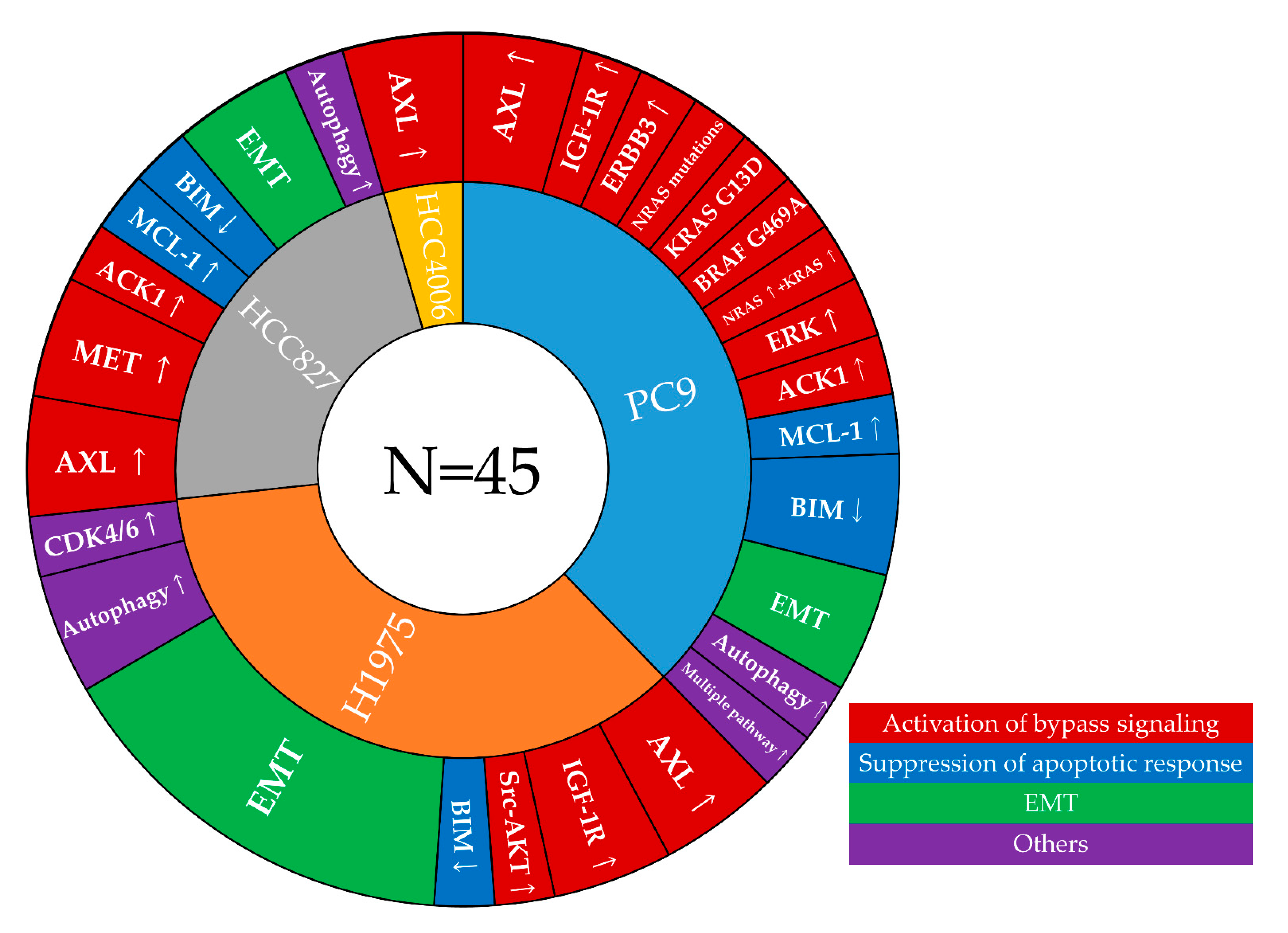
3.2. Mechanisms of Resistance to First-Line Osimertinib That Were Identified in Cell Line Models
3.2.1. Aberration of EGFR Itself—On-Target Resistance Mechanism
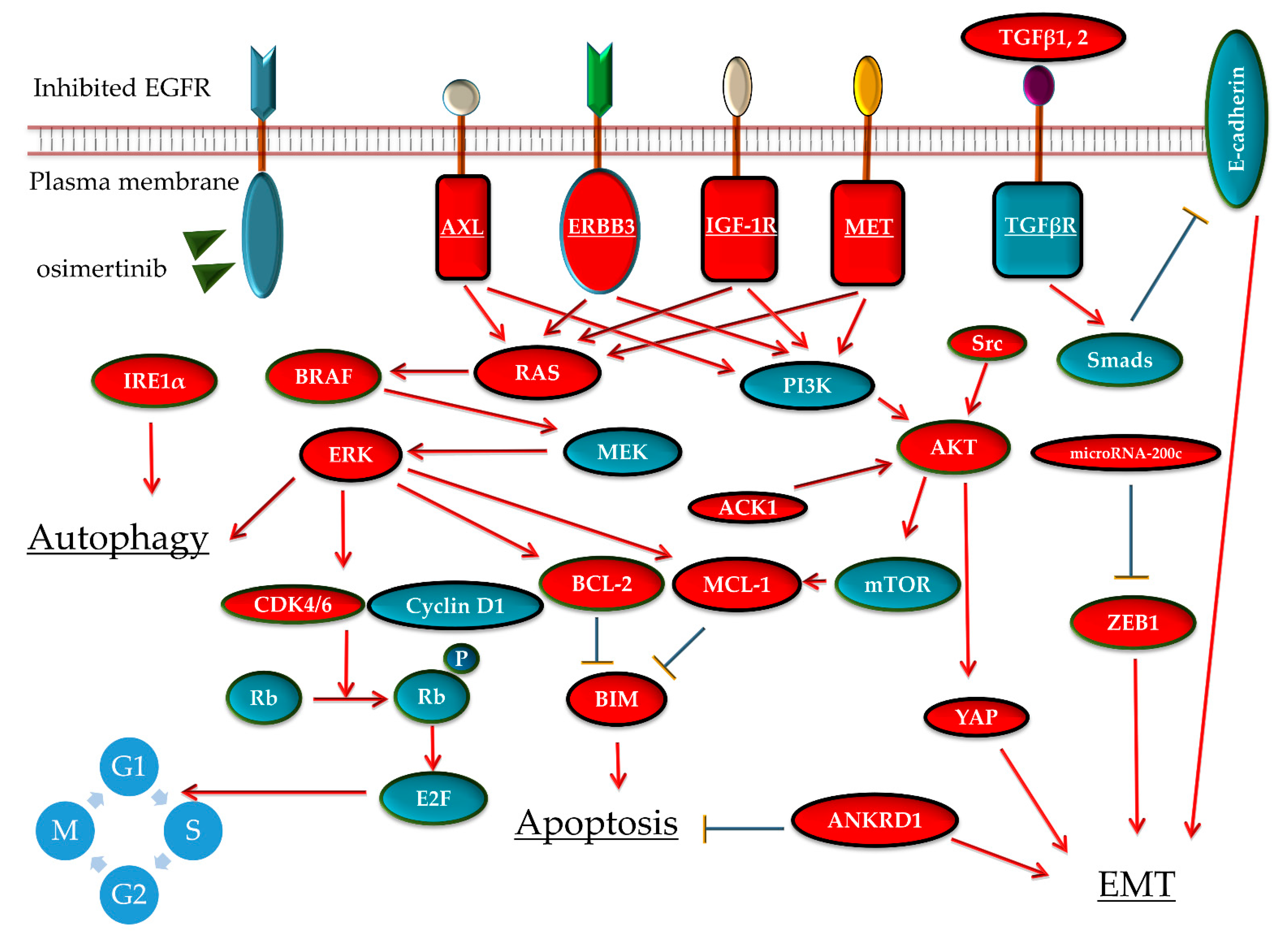
3.2.2. Activation of Bypass Signaling
MET Gene Amplification
AXL Activation
ERBB3 Activation
IGF-1R Activation
ERK or AKT Pathway Reactivation
3.2.3. Suppression of Apoptotic Response
3.2.4. EMT
3.2.5. Other Mechanisms
3.3. Correlation of Resistance Mechanisms Identified in Cell-Line Models and Clinical Specimens
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hirsch, F.R.; Suda, K.; Wiens, J.; Bunn, P.A., Jr. New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet 2016, 388, 1012–1024. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Wu, Y.L.; Yang, J.C.; Kim, D.W.; Lu, S.; Zhou, J.; Seto, T.; Yang, J.J.; Yamamoto, N.; Ahn, M.J.; Takahashi, T.; et al. Phase II Study of Crizotinib in East Asian Patients with ROS1-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1405–1411. [Google Scholar] [CrossRef]
- Marcus, L.; Donoghue, M.; Aungst, S.; Myers, C.E.; Helms, W.S.; Shen, G.; Zhao, H.; Stephens, O.; Keegan, P.; Pazdur, R. FDA Approval Summary: Entrectinib for the treatment of NTRK-gene fusion solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Souquet, P.J.; Quoix, E.; Baik, C.S.; Barlesi, F.; Kim, T.M.; Mazieres, J.; Novello, S.; et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol. 2016, 17, 984–993. [Google Scholar] [CrossRef] [Green Version]
- Fujino, T.; Suda, K.; Mitsudomi, T. Emerging MET tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Exp. Opin. Emerg. Drugs 2020, 25, 229–249. [Google Scholar] [CrossRef]
- Suda, K.; Tomizawa, K.; Mitsudomi, T. Biological and clinical significance of KRAS mutations in lung cancer: An oncogenic driver that contrasts with EGFR mutation. Cancer Metast. Rev. 2010, 29, 49–60. [Google Scholar] [CrossRef]
- Suda, K.; Bunn, P.A., Jr.; Rivard, C.J.; Mitsudomi, T.; Hirsch, F.R. Primary Double-Strike Therapy for Cancers to Overcome EGFR Kinase Inhibitor Resistance: Proposal from the Bench. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2017, 12, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. New Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Murakami, I.; Katayama, T.; Tomizawa, K.; Osada, H.; Sekido, Y.; Maehara, Y.; Yatabe, Y.; Mitsudomi, T. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5489–5498. [Google Scholar] [CrossRef] [Green Version]
- Suda, K.; Tomizawa, K.; Fujii, M.; Murakami, H.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudomi, T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2011, 6, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Yang, H.P.; Zhou, X.D.; Dai, X.T.; Chen, Y.F.; Xiong, W. CA916798 regulates multidrug resistance of lung cancer cells. Asian Pac. J. Cancer Prevent. 2011, 12, 3403–3408. [Google Scholar]
- Wang, H.J.; Yang, Z.X.; Dai, X.T.; Chen, Y.F.; Yang, H.P.; Zhou, X.D. Bisdemethoxycurcumin sensitizes cisplatin-resistant lung cancer cells to chemotherapy by inhibition of CA916798 and PI3K/AKT signaling. Apoptosis Int. J. Program. Cell Death 2017, 22, 1157–1168. [Google Scholar] [CrossRef]
- Qi, Z.; Wang, Y.; Zhou, X. CA916798 gene participates in cisplatin resistance of human lung adenocarcinoma A549 cells through PI3K/AKT/mTOR pathway. Nan Fang Yi Ke da Xue Xue Bao J. South. Med. Univ. 2012, 32, 1290–1293. [Google Scholar]
- Hanke, N.T.; Imler, E.; Marron, M.T.; Seligmann, B.E.; Garland, L.L.; Baker, A.F. Characterization of carfilzomib-resistant non-small cell lung cancer cell lines. J. Cancer Res. Clin. Oncol. 2018, 144, 1317–1327. [Google Scholar] [CrossRef]
- El-Awady, R.A.; Hersi, F.; Al-Tunaiji, H.; Saleh, E.M.; Abdel-Wahab, A.H.; Al Homssi, A.; Suhail, M.; El-Serafi, A.; Al-Tel, T. Epigenetics and miRNA as predictive markers and targets for lung cancer chemotherapy. Cancer Biol. Ther. 2015, 16, 1056–1070. [Google Scholar] [CrossRef] [Green Version]
- Cirigliano, S.M.; Díaz Bessone, M.I.; Berardi, D.E.; Flumian, C.; Bal de Kier Joffé, E.D.; Perea, S.E.; Farina, H.G.; Todaro, L.B.; Urtreger, A.J. The synthetic peptide CIGB-300 modulates CK2-dependent signaling pathways affecting the survival and chemoresistance of non-small cell lung cancer cell lines. Cancer Cell Int. 2017, 17, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, K.; Mizuuchi, H.; Maehara, Y.; Mitsudomi, T. Acquired resistance mechanisms to tyrosine kinase inhibitors in lung cancer with activating epidermal growth factor receptor mutation--diversity, ductility, and destiny. Cancer Metastasis Rev. 2012, 31, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Sesumi, Y.; Suda, K.; Mizuuchi, H.; Kobayashi, Y.; Sato, K.; Chiba, M.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; Mitsudomi, T. Effect of dasatinib on EMT-mediated-mechanism of resistance against EGFR inhibitors in lung cancer cells. Lung Cancer 2017, 104, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Uramoto, H.; Iwata, T.; Onitsuka, T.; Shimokawa, H.; Hanagiri, T.; Oyama, T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010, 30, 2513–2517. [Google Scholar] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Gu, P.; Zhou, C.; Liang, A.; Ren, S.; Liu, F.; Zeng, Y.; Wu, Y.; Zhao, Y.; Huang, B.; et al. β-Catenin overexpression is associated with gefitinib resistance in non-small cell lung cancer cells. Pulmonary Pharmacol. Ther. 2014, 28, 41–48. [Google Scholar] [CrossRef]
- Suda, K.; Mizuuchi, H.; Sato, K.; Takemoto, T.; Iwasaki, T.; Mitsudomi, T. The insulin-like growth factor 1 receptor causes acquired resistance to erlotinib in lung cancer cells with the wild-type epidermal growth factor receptor. Int. J. Cancer 2014, 135, 1002–1006. [Google Scholar] [CrossRef]
- Guix, M.; Faber, A.C.; Wang, S.E.; Olivares, M.G.; Song, Y.; Qu, S.; Rinehart, C.; Seidel, B.; Yee, D.; Arteaga, C.L.; et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J. Clin. Investig. 2008, 118, 2609–2619. [Google Scholar] [CrossRef]
- Cortot, A.B.; Repellin, C.E.; Shimamura, T.; Capelletti, M.; Zejnullahu, K.; Ercan, D.; Christensen, J.G.; Wong, K.K.; Gray, N.S.; Jänne, P.A. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 2013, 73, 834–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Martin, A.; Cufí, S.; Oliveras-Ferraros, C.; Torres-Garcia, V.Z.; Corominas-Faja, B.; Cuyàs, E.; Bonavia, R.; Visa, J.; Martin-Castillo, B.; Barrajón-Catalán, E.; et al. IGF-1R/epithelial-to-mesenchymal transition (EMT) crosstalk suppresses the erlotinib-sensitizing effect of EGFR exon 19 deletion mutations. Sci. Rep. 2013, 3, 2560. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef] [Green Version]
- Shi, P.; Oh, Y.T.; Zhang, G.; Yao, W.; Yue, P.; Li, Y.; Kanteti, R.; Riehm, J.; Salgia, R.; Owonikoko, T.K.; et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016, 380, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Oh, Y.T.; Deng, L.; Zhang, G.; Qian, G.; Zhang, S.; Ren, H.; Wu, G.; Legendre, B., Jr.; Anderson, E.; et al. Overcoming Acquired Resistance to AZD9291, A Third-Generation EGFR Inhibitor, through Modulation of MEK/ERK-Dependent Bim and Mcl-1 Degradation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6567–6579. [Google Scholar] [CrossRef] [Green Version]
- Nukaga, S.; Yasuda, H.; Tsuchihara, K.; Hamamoto, J.; Masuzawa, K.; Kawada, I.; Naoki, K.; Matsumoto, S.; Mimaki, S.; Ikemura, S.; et al. Amplification of EGFR Wild-Type Alleles in Non-Small Cell Lung Cancer Cells Confers Acquired Resistance to Mutation-Selective EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2017, 77, 2078–2089. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Seike, M.; Chiba, M.; Takahashi, S.; Nakamichi, S.; Matsumoto, M.; Takeuchi, S.; Minegishi, Y.; Noro, R.; Kunugi, S.; et al. Ankyrin Repeat Domain 1 Overexpression is Associated with Common Resistance to Afatinib and Osimertinib in EGFR-mutant Lung Cancer. Sci. Rep. 2018, 8, 14896. [Google Scholar] [CrossRef]
- Ku, B.M.; Choi, M.K.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; Ahn, M.J. Acquired resistance to AZD9291 as an upfront treatment is dependent on ERK signaling in a preclinical model. PLoS ONE 2018, 13, e0194730. [Google Scholar] [CrossRef]
- Tang, Z.H.; Su, M.X.; Guo, X.; Jiang, X.M.; Jia, L.; Chen, X.; Lu, J.J. Increased Expression of IRE1α Associates with the Resistant Mechanism of Osimertinib (AZD9291)-resistant non-small Cell Lung Cancer HCC827/OSIR Cells. Anti-Cancer Agents Med. Chem. 2018, 18, 550–555. [Google Scholar] [CrossRef]
- Namba, K.; Shien, K.; Takahashi, Y.; Torigoe, H.; Sato, H.; Yoshioka, T.; Takeda, T.; Kurihara, E.; Ogoshi, Y.; Yamamoto, H.; et al. Activation of AXL as a Preclinical Acquired Resistance Mechanism Against Osimertinib Treatment in EGFR-Mutant Non-Small Cell Lung Cancer Cells. Mol. Cancer Res. 2019, 17, 499–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wang, T.; Lv, D.; Li, L.; Yue, J.; Chen, H.Z.; Xu, L. Acquired Resistance to EGFR TKIs Mediated by TGFβ1/Integrin β3 Signaling in EGFR-Mutant Lung Cancer. Mol. Cancer Ther. 2019, 18, 2357–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.N.; Tsai, M.F.; Wu, S.G.; Chang, T.H.; Tsai, T.H.; Gow, C.H.; Chang, Y.L.; Shih, J.Y. Acquired resistance to EGFR tyrosine kinase inhibitors is mediated by the reactivation of STC2/JUN/AXL signaling in lung cancer. Int. J. Cancer 2019, 145, 1609–1624. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y.; Jiao, L.; Lin, C.; Lu, C.; Zhang, K.; Hu, C.; Ye, J.; Zhang, D.; Wu, H.; et al. Protective autophagy decreases osimertinib cytotoxicity through regulation of stem cell-like properties in lung cancer. Cancer Lett. 2019, 452, 191–202. [Google Scholar] [CrossRef] [PubMed]
- La Monica, S.; Minari, R.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Galetti, M.; Digiacomo, G.; Riccardi, F.; Petronini, P.G.; et al. Acquired BRAF G469A Mutation as a Resistance Mechanism to First-Line Osimertinib Treatment in NSCLC Cell Lines Harboring an EGFR Exon 19 Deletion. Targ. Oncol. 2019, 14, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Codony-Servat, J.; Viteri, S.; Codony-Servat, C.; Ito, M.; Bracht, J.W.P.; Berenguer, J.; Chaib, I.; Molina-Vila, M.A.; Karachaliou, N.; Rosell, R. Hsp90 inhibitors enhance the antitumoral effect of osimertinib in parental and osimertinib-resistant non-small cell lung cancer cell lines. Transl. Lung Cancer Res. 2019, 8, 340–351. [Google Scholar] [CrossRef]
- Yonesaka, K.; Takegawa, N.; Watanabe, S.; Haratani, K.; Kawakami, H.; Sakai, K.; Chiba, Y.; Maeda, N.; Kagari, T.; Hirotani, K.; et al. An HER3-targeting antibody-drug conjugate incorporating a DNA topoisomerase I inhibitor U3-1402 conquers EGFR tyrosine kinase inhibitor-resistant NSCLC. Oncogene 2019, 38, 1398–1409. [Google Scholar] [CrossRef]
- Fukuda, K.; Takeuchi, S.; Arai, S.; Kita, K.; Tanimoto, A.; Nishiyama, A.; Yano, S. Glycogen synthase kinase-3 inhibition overcomes epithelial-mesenchymal transition-associated resistance to osimertinib in EGFR-mutant lung cancer. Cancer Sci. 2020, 111, 2374–2384. [Google Scholar] [CrossRef]
- Zang, H.; Qian, G.; Arbiser, J.; Owonikoko, T.K.; Ramalingam, S.S.; Fan, S.; Sun, S.Y. Overcoming acquired resistance of EGFR-mutant NSCLC cells to the third generation EGFR inhibitor, osimertinib, with the natural product honokiol. Mol. Oncol. 2020, 14, 882–895. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Qian, L.; Zhang, G.; Mahajan, N.P.; Owonikoko, T.K.; Ramalingam, S.S.; Sun, S.Y. Inhibition of ACK1 delays and overcomes acquired resistance of EGFR mutant NSCLC cells to the third generation EGFR inhibitor, osimertinib. Lung Cancer 2020, 150, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Zang, H.; Qian, G.; Zong, D.; Fan, S.; Owonikoko, T.K.; Ramalingam, S.S.; Sun, S.Y. Overcoming acquired resistance of epidermal growth factor receptor-mutant non-small cell lung cancer cells to osimertinib by combining osimertinib with the histone deacetylase inhibitor panobinostat (LBH589). Cancer 2020, 126, 2024–2033. [Google Scholar] [CrossRef]
- Han, R.; Hao, S.; Lu, C.; Zhang, C.; Lin, C.; Li, L.; Wang, Y.; Hu, C.; He, Y. Aspirin sensitizes osimertinib-resistant NSCLC cells in vitro and in vivo via Bim-dependent apoptosis induction. Mol. Oncol. 2020, 14, 1152–1169. [Google Scholar] [CrossRef]
- Manabe, T.; Yasuda, H.; Terai, H.; Kagiwada, H.; Hamamoto, J.; Ebisudani, T.; Kobayashi, K.; Masuzawa, K.; Ikemura, S.; Kawada, I.; et al. IGF2 Autocrine-Mediated IGF1R Activation Is a Clinically Relevant Mechanism of Osimertinib Resistance in Lung Cancer. Mol. Cancer. Res. 2020, 18, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.M.; Jang, Y.; Lee, S.H.; Kang, B.; Lim, S.M. AXL/MET dual inhibitor, CB469, has activity in non-small cell lung cancer with acquired resistance to EGFR TKI with AXL or MET activation. Lung Cancer 2020, 146, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Verusingam, N.D.; Chen, Y.C.; Lin, H.F.; Liu, C.Y.; Lee, M.C.; Lu, K.H.; Cheong, S.K.; Ong, A.H.; Chiou, S.H.; Wang, M.L. Generation of Osimertinib-Resistant Cells from EGFR L858R/T790M Mutant NSCLC Cell Line. J. Chin. Med. Assoc. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Sun, H.; Robichaux, J.; Pfeifer, M.; McDermott, U.; Travers, J.; Diao, L.; Xi, Y.; Tong, P.; Shen, L.; et al. A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. CDK4/6 inhibitor palbociclib overcomes acquired resistance to third-generation EGFR inhibitor osimertinib in non-small cell lung cancer (NSCLC). Thoracic Cancer 2020, 11, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gao, W. Synergistic effects of Bcl-2 inhibitors with AZD9291 on overcoming the acquired resistance of AZD9291 in H1975 cells. Arch. Toxicol. 2020, 94, 3125–3136. [Google Scholar] [CrossRef]
- Hayakawa, D.; Takahashi, F.; Mitsuishi, Y.; Tajima, K.; Hidayat, M.; Winardi, W.; Ihara, H.; Kanamori, K.; Matsumoto, N.; Asao, T.; et al. Activation of insulin-like growth factor-1 receptor confers acquired resistance to osimertinib in non-small cell lung cancer with EGFR T790M mutation. Thoracic Cancer 2020, 11, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Lin, Y.C.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef]
- Jiang, X.M.; Xu, Y.L.; Yuan, L.W.; Zhang, L.L.; Huang, M.Y.; Ye, Z.H.; Su, M.X.; Chen, X.P.; Zhu, H.; Ye, R.D.; et al. TGFβ2-mediated epithelial-mesenchymal transition and NF-κB pathway activation contribute to osimertinib resistance. Acta Pharmacol. Sinica 2020. [Google Scholar] [CrossRef]
- Ji, W.; Choi, Y.J.; Kang, M.H.; Sung, K.J.; Kim, D.H.; Jung, S.; Choi, C.M.; Lee, J.C.; Rho, J.K. Efficacy of the CDK7 Inhibitor on EMT-Associated Resistance to 3rd Generation EGFR-TKIs in Non-Small Cell Lung Cancer Cell Lines. Cells 2020, 9, 2596. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roper, N.; Brown, A.L.; Wei, J.S.; Pack, S.; Trindade, C.; Kim, C.; Restifo, O.; Gao, S.; Sindiri, S.; Mehrabadi, F.; et al. Clonal Evolution and Heterogeneity of Osimertinib Acquired Resistance Mechanisms in EGFR Mutant Lung Cancer. Cell Rep. Med. 2020, 1. [Google Scholar] [CrossRef]
- Antony, J.; Huang, R.Y.-J. AXL-Driven EMT State as a Targetable Conduit in Cancer. J. Cancer Res. 2017, 77, 3725–3732. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Ji, W.; Choi, C.M.; Rho, J.K.; Jang, S.J.; Park, Y.S.; Chun, S.M.; Kim, W.S.; Lee, J.S.; Kim, S.W.; Lee, D.H.; et al. Mechanisms of acquired resistance to EGFR-tyrosine kinase inhibitor in Korean patients with lung cancer. BMC Cancer 2013, 13, 606. [Google Scholar] [CrossRef] [Green Version]
- Nonagase, Y.; Takeda, M.; Azuma, K.; Hayashi, H.; Haratani, K.; Tanaka, K.; Yonesaka, K.; Ishii, H.; Hoshino, T.; Nakagawa, K. Tumor tissue and plasma levels of AXL and GAS6 before and after tyrosine kinase inhibitor treatment in EGFR-mutated non-small cell lung cancer. Thoracic Cancer 2019, 10, 1928–1935. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Moasser, M.M. Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br. J. Cancer 2007, 97, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Slingerland, J.M. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003, 2, 339–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeRoith, D.; Roberts, C.T., Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003, 195, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Suda, K.; Tomizawa, K.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudomi, T. Conversion from the “oncogene addiction” to “drug addiction” by intensive inhibition of the EGFR and MET in lung cancer with activating EGFR mutation. Lung Cancer 2012, 76, 292–299. [Google Scholar] [CrossRef]
- Shen, H.; Zhu, F.; Liu, J.; Xu, T.; Pei, D.; Wang, R.; Qian, Y.; Li, Q.; Wang, L.; Shi, Z.; et al. Alteration in Mir-21/PTEN expression modulates gefitinib resistance in non-small cell lung cancer. PLoS ONE 2014, 9, e103305. [Google Scholar] [CrossRef]
- Li, B.; Ren, S.; Li, X.; Wang, Y.; Garfield, D.; Zhou, S.; Chen, X.; Su, C.; Chen, M.; Kuang, P.; et al. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer 2014, 83, 146–153. [Google Scholar] [CrossRef]
- Xu, S.H.; Huang, J.Z.; Xu, M.L.; Yu, G.; Yin, X.F.; Chen, D.; Yan, G.R. ACK1 promotes gastric cancer epithelial-mesenchymal transition and metastasis through AKT-POU2F1-ECD signalling. J. Pathol. 2015, 236, 175–185. [Google Scholar] [CrossRef]
- Barlesi, F.; Mazieres, J.; Merlio, J.P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Arcila, M.E.; Fara, M.; Sima, C.S.; Miller, V.A.; Kris, M.G.; Ladanyi, M.; Riely, G.J. Clinical Characteristics of Patients with Lung Adenocarcinomas Harboring BRAF Mutations. J. Clin. Oncol. 2011, 29, 2046–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.C.; Liao, W.Y.; Lin, C.A.; Shih, J.Y.; Yu, C.J.; Yang, J.C. Acquired BRAF V600E Mutation as Resistant Mechanism after Treatment with Osimertinib. J. Thoracic Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2017, 12, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minari, R.; Bordi, P.; La Monica, S.; Squadrilli, A.; Leonetti, A.; Bottarelli, L.; Azzoni, C.; Lagrasta, C.A.M.; Gnetti, L.; Campanini, N.; et al. Concurrent Acquired BRAF V600E Mutation and MET Amplification as Resistance Mechanism of First-Line Osimertinib Treatment in a Patient with EGFR-Mutated NSCLC. J. Thoracic Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, e89–e91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Gan, J.; Guo, K.; Deng, Y.; Fang, W. Acquired BRAF V600E Mutation Mediated Resistance to Osimertinib and Responded to Osimertinib, Dabrafenib, and Trametinib Combination Therapy. J. Thoracic Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, e236–e237. [Google Scholar] [CrossRef]
- Meng, P.; Koopman, B.; Kok, K.; Ter Elst, A.; Schuuring, E.; van Kempen, L.C.; Timens, W.; Hiltermann, T.J.N.; Groen, H.J.M.; van den Berg, A.; et al. Combined osimertinib, dabrafenib and trametinib treatment for advanced non-small-cell lung cancer patients with an osimertinib-induced BRAF V600E mutation. Lung Cancer 2020, 146, 358–361. [Google Scholar] [CrossRef]
- Suda, K.; Mitsudomi, T.J.T.L.C.R. Emerging oncogenic fusions other than ALK, ROS1, RET, and NTRK in NSCLC and the role of fusions as resistance mechanisms to targeted therapy. Transl. Lung Cancer Res. 2020, 2020. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.; Wu, Y.L.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.; John, T.; Okamoto, I.; Yang, J.C.H.; Bulusu, K.; Laus, G.; et al. LBA51Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Shim, J.H.; Lee, B.; Cho, I.; Park, W.Y.; Kim, Y.; Lee, S.H.; Choi, Y.; Han, J.; Ahn, J.S.; et al. Paired genomic analysis of squamous cell carcinoma transformed from EGFR-mutated lung adenocarcinoma. Lung Cancer 2019, 134, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Thress, K.; Paweletz, C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Felip, E.; Vivancos, A.; Kuang, Y. ORAL17.07 Mechanisms of Acquired Resistance to AZD9291 in EGFR T790M Positive Lung. J. Thorac. Oncol. 2015, 10, S207. [Google Scholar]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.; Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.H.; Lou, Y.Y.; Zhou, K.L.; Pan, D.Q. Exploration of intermolecular interaction of calf thymus DNA with sulfosulfuron using multi-spectroscopic and molecular docking techniques. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 204, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.H.; Kim, M.H.; Kim, S.Y.; Heo, S.G.; Kang, H.N.; Park, C.W.; Barrett, J.C.; Stetson, D.; Chmielecki, J.; Markovets, A.; et al. Molecular landscape of osimertinib resistance revealed by targeted panel sequencing and patient-derived cancer models in non-small cell lung cancer patients. Ann. Oncology 2018, 29, viii516. [Google Scholar] [CrossRef]
- Ng, K.P.; Hillmer, A.M.; Chuah, C.T.; Juan, W.C.; Ko, T.K.; Teo, A.S.; Ariyaratne, P.N.; Takahashi, N.; Sawada, K.; Fei, Y.; et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med. 2012, 18, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, D.; Li, B.; Zou, B.; Wang, S.; Fan, B.; Li, W.; Yu, J.; Wang, L. Clinical implications of germline BCL2L11 deletion polymorphism in pretreated advanced NSCLC patients with osimertinib therapy. Lung Cancer 2020, 151, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Murakami, I.; Yu, H.; Kim, J.; Tan, A.C.; Mizuuchi, H.; Rozeboom, L.; Ellison, K.; Rivard, C.J.; Mitsudomi, T.; et al. CD44 Facilitates Epithelial-to-Mesenchymal Transition Phenotypic Change at Acquisition of Resistance to EGFR Kinase Inhibitors in Lung Cancer. Mol. Cancer Ther. 2018, 17, 2257–2265. [Google Scholar] [CrossRef] [Green Version]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- Moretti, L.; Yang, E.S.; Kim, K.W.; Lu, B. Autophagy signaling in cancer and its potential as novel target to improve anticancer therapy. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2007, 10, 135–143. [Google Scholar] [CrossRef]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 2014, 3. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohara, S.; Suda, K.; Mitsudomi, T. Cell Line Models for Acquired Resistance to First-Line Osimertinib in Lung Cancers—Applications and Limitations. Cells 2021, 10, 354. https://doi.org/10.3390/cells10020354
Ohara S, Suda K, Mitsudomi T. Cell Line Models for Acquired Resistance to First-Line Osimertinib in Lung Cancers—Applications and Limitations. Cells. 2021; 10(2):354. https://doi.org/10.3390/cells10020354
Chicago/Turabian StyleOhara, Shuta, Kenichi Suda, and Tetsuya Mitsudomi. 2021. "Cell Line Models for Acquired Resistance to First-Line Osimertinib in Lung Cancers—Applications and Limitations" Cells 10, no. 2: 354. https://doi.org/10.3390/cells10020354
APA StyleOhara, S., Suda, K., & Mitsudomi, T. (2021). Cell Line Models for Acquired Resistance to First-Line Osimertinib in Lung Cancers—Applications and Limitations. Cells, 10(2), 354. https://doi.org/10.3390/cells10020354